

Abstract
Summary
We developed annoPeak, a web application to annotate, visualize and compare predicted protein-binding regions derived from ChIP-seq/ChIP-exo-seq experiments using human and mouse cells. Users can upload peak regions from multiple experiments onto the annoPeak server to annotate them with biological context, identify associated target genes and categorize binding sites with respect to gene structure. Users can also compare multiple binding profiles intuitively with the help of visualization tools and tables provided by annoPeak. In general, annoPeak will help users identify patterns of genome wide transcription factor binding profiles, assess binding profiles in different biological contexts and generate new hypotheses.
Availability and Implementation
The web service is freely accessible through URL: https://apps.medgen.iupui.edu/rsc/content/19/. Source code is available at https://github.com/XingTang2014/annoPeak.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
ChIP-seq and ChIP-exo-seq are widely used techniques to study protein-DNA-binding interactions on a genome-wide scale (Furey, 2012). Binding profiles of different proteins under various biological conditions are often generated and compared with answer specific biological questions (Furey, 2012; Heinz, 2010; Rosenbloom et al., 2013). To help biologists annotate binding sites and compare binding profiles from multiple experiments, we developed a user friendly web tool named annoPeak (https://apps.medgen.iupui.edu/rsc/content/19/). annoPeak accepts peak data files that describe protein-binding regions in BED format and provides five analytical modules. These include analytical tools to identify and illustrate binding profiles associated with different gene structures (promoter, enhancer, gene body, 5′ region, 3′ region), to annotate peak size distribution, Peak to nearest peak distance, overlapping peaks, and overlapping peak-associated genes. In addition, annoPeak can be used to identify functional classes of selected subsets of peak-associated genes. Other specific features are revealed in our teaching video (https://www.youtube.com/watch?v=yzV4k4boplQ). The application can also be easily deployed on your own laptop from the source code (https://github.com/XingTang2014/annoPeak).
annopeak provides a one click solution to address the challenges that high throughput data presents. It’s easy to use and requires no programming proficiency. A summarized table for features compared with similar tools can be found in Supplementary Table S1.
2 Implementation
The annoPeak application is developed in R (Team, 2015) and relies on multiple R/Bioconductor packages (Supplementary Materials). The web interface is developed using the R package shiny (Winston, 2015). The web service is deployed on a server with Redhat 7 system. The maximum allowed file size to upload is 50 M. The efficiency was benchmarked by utilizing simulated data sets and the entire analysis was completed in a few minutes for a dataset with 250 K peak regions (Supplementary Table S2).
3 Results
To test annoPeak performance, peak sets for SUZ12, EZH2, RING1B and PCGF2 chromatin-binding profiles derived from mouse embryonic stem (mES) cells and neural progenitor cells (NPCs) were downloaded from NCBI GEO with series ID GSE74330 (Kloet et al., 2016).
3.1 Peak associated gene structures
Binding bias towards specific genomic region with respect to gene position (5′ distal region, enhancer, promoter, gene body and 3′ distal region) indicates specific regulatory mechanisms of DNA binding proteins (Chen et al., 2009; Lee and Iyer, 2012). To investigate this, peaks were assigned to specific genes based on the nearest transcription start site and categorized into five specific regions (Supplementary Materials). The gene annotations used are from Bioconductor TxDb.* family of packages. Peaks associated with a specific gene structure could be selected and analyzed separately in annoPeak. In the example dataset, all the four proteins bind preferentially to promoter regions in both mES and NPC (Fig. 1A and B). The relative binding of SUZ12, RING1B and EZH2 to promoter regions was lower in mES than in NPC but for PCGF2 it was higher in mES than in NPC.
Fig. 1.
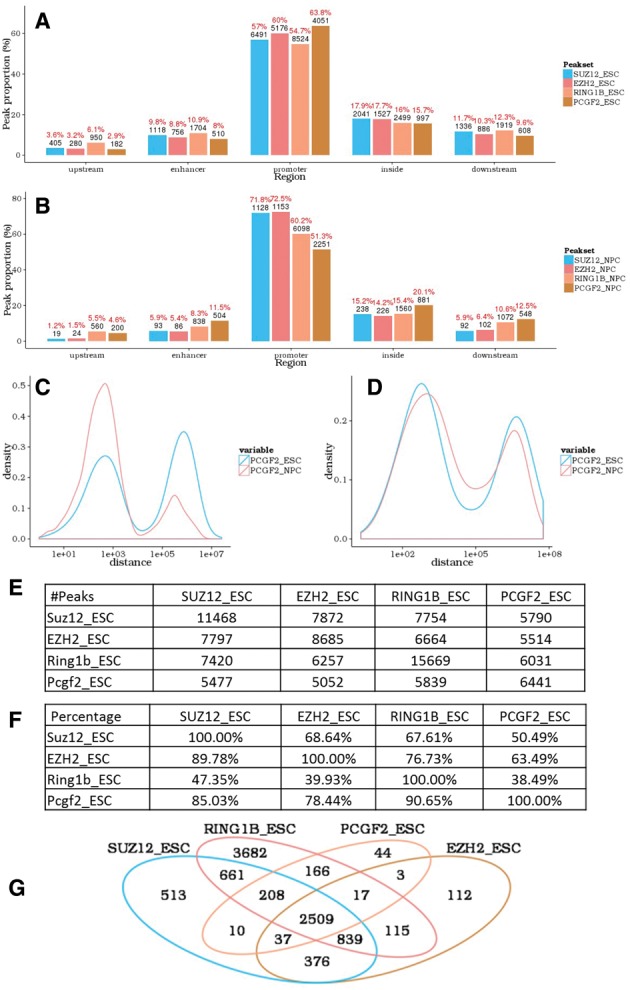
annoPeak results for example ChIP-seq datasets (NCBI GEO with series ID GSE74330). (A,B) Peak-associated gene structure; distribution of SUZ12, EZH2, RING1B and PCGF2 binding to gene structures in mES (A) and NPC (B). (C,D) Peak to nearest peak distance; distribution plots comparing binding profiles of PCGF2 from mES to NPC in promoter regions (C) and enhancer regions (D). (E,F) Tables with the number (E) and percentage (F) of overlapping peaks between the four proteins in mES; setting a maximum allowed distance between peaks of 1 kb. (G). Venn diagram depicting overlapping peak-associated gene targets for the four proteins in mES
3.2 Peak size distribution
At a single glance, this tool allows visualization of preferential binding and may assist in the validation of the experimental data depending on previous knowledge about the pattern of the binding profile.
3.3 Peak to nearest peak distance
For each peak in set A, we find the nearest peak in set B and calculate the distance between the related peaks in sets A and B, and vice versa. The distribution of the two distances (A to B and B to A) are plotted in the same figure with different colors. The frequency of potential concurrent binding may be estimated by comparing the relative area under the curves at each distance cutoff. Figure 1C (promoter region specifically) and D (enhancer region specifically) show the relative positions of PCGF2 binding in mES and NPC.
3.4 Overlapping peak identifications
This module allows user to calculate overlaps across multiple peak sets with user defined distance cutoffs (Fig. 1E and F).
3.5 Overlapping peak-associated genes
We provide tables and a venn diagram to show the overlaps of target genes across multiple experiments (Fig. 1G). Subset of genes could be selected to perform functional enrichment analysis. Functional categories defined by Gene Otology and GeneSigDB (Culhane et al., 2010) are both incorporated. Tests against both of the two databases are performed simultaneously and results are listed separately.
Funding
This work was supported by Ohio Supercomputer Center and also supported by NIH grants to G.L. (R01CA121275 and P01 CA97189) and K.H. was supported by NCI ITCR 1U01CA188547-01A1.
Conflict of Interest: none declared.
Supplementary Material
References
- Chen H.Z. et al. (2009) Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat. Rev. Cancer, 9, 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culhane A.C. et al. (2010) GeneSigDB—a curated database of gene expression signatures. Nucleic Acids Res., 38(suppl 1): D716–D725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey T.S. (2012) ChIP–seq and beyond: new and improved methodologies to detect and characterize protein–DNA interactions. Nat. Rev. Genet., 13, 840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S. et al. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell, 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloet S.L. et al. (2016) The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat. Struct. Mol. Biol., 23, 682–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B.K., Iyer V.R. (2012) Genome-wide studies of CCCTC-binding factor (CTCF) and cohesin provide insight into chromatin structure and regulation. J. Biol. Chem., 287, 30906–30913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom K.R. et al. (2013) ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res., 41, D56–D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team,R.C. (2015) R: A Language and Environment for Statistical Computing. Vienna, Austria, 2014. [Google Scholar]
- Winston W.C. et al. (2015) shiny: Web Application Framework for R. R package version 0.12.2. https://CRAN.R-project.org/package=shiny.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.