

Abstract
Motivation
Biological data and knowledge bases increasingly rely on Semantic Web technologies and the use of knowledge graphs for data integration, retrieval and federated queries. In the past years, feature learning methods that are applicable to graph-structured data are becoming available, but have not yet widely been applied and evaluated on structured biological knowledge. Results: We develop a novel method for feature learning on biological knowledge graphs. Our method combines symbolic methods, in particular knowledge representation using symbolic logic and automated reasoning, with neural networks to generate embeddings of nodes that encode for related information within knowledge graphs. Through the use of symbolic logic, these embeddings contain both explicit and implicit information. We apply these embeddings to the prediction of edges in the knowledge graph representing problems of function prediction, finding candidate genes of diseases, protein-protein interactions, or drug target relations, and demonstrate performance that matches and sometimes outperforms traditional approaches based on manually crafted features. Our method can be applied to any biological knowledge graph, and will thereby open up the increasing amount of Semantic Web based knowledge bases in biology to use in machine learning and data analytics.
Availability and implementation
https://github.com/bio-ontology-research-group/walking-rdf-and-owl
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The Semantic Web (Berners-Lee et al., 2001), a project with the stated purpose of forming a consistent logical and meaningful web of data using semantic technologies to make data machine-understandable and processable, has been highly successful in biology and biomedicine (Katayama et al., 2014). Many major bioinformatics databases now make their data available as Linked Data in which both biological entities and connections between them are identified through a unique identifier (an Internationalized Resource Identifier or IRI) and the connections between them are expressed through standardized relations (Smith et al., 2005; Wood et al., 2014). Linked Data can enable interoperability between multiple databases simply by reusing identifiers and utilizing no-SQL query languages such as SPARQL (Seaborne and Prud’hommeaux, 2008) that can perform distributed queries over multiple databases. Some of the first major efforts to make life science data available as Linked Data have been the UniProt RDF initiative (The UniProt Consortium, 2015) and the Bio2RDF project (Belleau et al., 2008; Callahan et al., 2013). UniProt focuses on making data within a single database, UniProt, available as Linked Data so that information and identifiers can be reused in other databases, while Bio2RDF has the aim to combine multiple databases and demonstrate the potential of Linked Data in life sciences, in particular with regard to provenance tracking, usability and interoperability. Now, major databases, such as those provided by the European Bioinformatics Institute (EBI) and the National Center for Biotechnology Information (NCBI), are made available as Linked Data (Jupp et al., 2014; Kim et al., 2016). Additionally, community guidelines and principles for data publishing such as the FAIR principles (Wilkinson et al., 2016) require data to be made available in a way that is amenable to interoperability through linking and federation of queries.
A second major component of applications of the Semantic Web in the life sciences has been the development and use of ontologies. Ontologies are specifications of a conceptualization of a domain (Gruber, 1995), i.e. they formally and explicitly specify some of the main regularities (classes of entities) that can be found within a domain and their interconnections (Hoehndorf et al., 2015b). Ontologies are now widely used in biological datasets for the annotation and provision of metadata. They are commonly represented in formal languages with model theoretic semantics (Grau et al., 2008; Horrocks, 2007) which makes them amenable to automated reasoning. However, the large size of the ontologies and the complexity of the languages and reasoning tasks involved have somewhat limited the use of ontologies in automated reasoning. In particular, there is still a large disparity between the ontologies in biomedicine and the databases that uses them for annotation in the sense that they are rarely integrated within the same data model. While inferences over the ontologies, as part of ontology development and quality assurance process, become increasingly common (Mungall et al., 2012; The Gene Ontology Consortium, 2015), they are not always applied to infer new relations between biomedical data.
Recently, several machine learning methods have become available that can be utilized to learn features from raw data (Lecun et al., 2015). Several of these methods can also be applied to graph-structured data (Perozzi et al., 2014; Yanardag and Vishwanathan, 2015). While most of these methods are developed for graphs without edge labels (in contrast to Linked Data in which edge labels represent the type of relation between entities), some methods have also been extended to incorporate edge labels (Ristoski and Paulheim, 2016). However, to be applicable to biological data, a crucial aspect is the interoperability between both the data layer (as expressed in Linked Data formats) and the annotations of data items or semantic layer (expressed through ontologies and the background knowledge they provide). This tight integration between data and knowledge, as dominantly present in biological databases, benefits from automated reasoning so that it becomes possible to consider inferred knowledge, handle data consistency and identify incompatible conceptualizations.
We developed a method to leverage the semantic layer in knowledge graphs such as the Semantic Web or Wikidata by combining automated reasoning over ontologies and feature learning with neural networks, to generate vector representations of nodes in these graphs (node embeddings). We demonstrate that these representations can be used to predict edges with biological meaning. In particular, we demonstrate that our approach can predict disease genes, drug targets, drug indications, gene functions and other associations with high accuracy, in many cases matching or outperforming state of the art methods.
Our results demonstrate how Linked Data and ontologies can be used to form biological knowledge graphs in which heterogeneous biological data and knowledge are combined within a formal framework, and that these graphs can not only be used for data retrieval and search, but provide a powerful means for data analysis and discovery of novel biological knowledge.
2 Materials and methods
2.1 Data description
In our experiments, we build a knowledge graph based on three ontologies: the Gene Ontology (GO) (Ashburner et al., 2000) downloaded on 18 July 2016, the Human Phenotype Ontology (Köhler et al., 2014) downloaded on 18 July 2016, and the Disease Ontology (Kibbe et al., 2014) downloaded on 19 August 2016. We also use the following biological databases in our knowledge graph:
Human GO annotations from SwissProt (The UniProt Consortium, 2015), and phenotype annotations from the HPO databases (Köhler et al., 2014), downloaded on 23 July 2016. We include a total of 212 078 GO annotations and 153 575 phenotype annotations.
Human Proteins interactions from the STRING database (Szklarczyk et al., 2011) downloaded on 18 July 2016. We filter proteins by their interactions confidence score and choose those above 700. The total number of interactions in this dataset is 188 424.
Human chemical–protein interactions downloaded from the STITCH database (Kuhn et al., 2012), on 28 August 2016, filtered for confidence score of 700. The total number of drug-target interactions present in the graph is 335 780.
Genes and disease associations from DisGeNET (Piñero et al., 2015), downloaded on 28 August 2016, consisting of 236 259 associations.
Drug side effects and indications from SIDER (Kuhn et al., 2010), downloaded on 15 August 2016. We include a total of 54 806 drug–side effect pairs and 6159 drug–indication pairs in our graph.
Diseases and their phenotypes from the HPO database (Köhler et al., 2014) and text mining (Hoehndorf et al., 2015a). We include a total of 84 508 phenotype annotations of diseases.
We map all protein identifiers to Entrez gene identifiers and use these to represent both genes and proteins. We use PubChem identifiers to represent chemicals and we map UMLS identifiers associated with diseases in DisGeNET and indications in SIDER to the Disease Ontology using mappings provided by Disease Ontology. We further map UMLS identifiers associated with side effects in SIDER to HPO identifiers using mapping between UMLS and HPO (Hoehndorf et al., 2014).
A knowledge graph is a graph-based representation of entities in the world and their interrelations. Knowledge graphs are widely used to facilitate and improve search, and they are increasingly being developed and used through Semantic Web technologies such as the Resource Description Framework (RDF) (Candan et al., 2001). Here, we focus on knowledge graphs centered around biological entities and their interactions, ignoring all meta-data including labels or provenance. The knowledge graphs we consider have two distinct types of entities: biological entities, and classes from biomedical ontologies that provide background knowledge about a domain. The aim of building a biological knowledge graph is to represent, within a single formal structure, biological relations between entities, their annotations with biological ontologies, and the background knowledge in ontologies.
We make a clear distinction between instances and classes. While there is some debate about which kinds of biological entities should be treated as instances and which as classes (Smith et al., 2005), similarly to other Linked Data approaches (The UniProt Consortium, 2015), we treat biological entities such as types of proteins, diseases, or chemicals, as instances in the knowledge graph. In our case, classes from the Disease Ontology are also treated as instances. On the level of instances, we can integrate existing graph-based representations used in biology and biomedicine, in particular biological networks such as protein-protein interaction networks, genetic interaction networks, metabolic interactions or pathways.
Ontology-based annotations are expressed by asserting a relation between the instance (e.g. a disease or protein) and an instance of the ontology class. For example, we express the information that the protein Foxp2 has the function transcription factor binding (GO:0003700) by the two axioms and where foxp2 and f1 are instances, the class http://purl.obolibrary.org/obo/GO_0003700 in GO, hasFunction an object property, and instanceOf the rdf:type property specified in the OWL standard (OWL Working Group, 2009) as expressing an instantiation relation. The instance f1 can be expressed as an anonymous instance (i.e. a blank node in the RDF representation) or be assigned a unique new IRI. In our knowledge graph, we create a new IRI (i.e. an IRI that does not occur anywhere else in the graph) for each of these instances.
2.2 Ontology-based classification
Due to the large size of the knowledge graphs we process, we rely on polynomial-time automated reasoning methods. OWL provides three profiles (Motik et al., 2009) that facilitate polynomial time inferences, and multiple RDF stores implement different subsets of OWL to facilitate inferences and improve querying. For example, the OWL-Horst subset (ter Horst, 2005) is used by several RDF stores and is useful in data management and querying. In biological and biomedical ontologies, the OWL 2 EL profile is widely used to develop the large ontologies that are in use in the domain, and has been found to be useful and sufficient for a large number of tasks (Hoehndorf et al., 2011; Mungall et al., 2012; Suntisrivaraporn et al., 2007).
OWL 2 EL supports basic inferences over ontologies’ class hierarchies (including intersection, existential quantification and disjointness between named classes), supports inferences over object properties (transitivity, reflexivity and object property composition) and can infer the classification of instances. We make use of OWL 2 EL for representing the knowledge graphs we generate and utilize the ELK reasoner (Kazakov et al., 2014) for automated reasoning over them. In principle, other profiles of OWL can also be used following a similar approach, but may not be feasible due to the high computational complexity of generating inferences (Baader et al., 2003). OWL 2 EL supports the following class descriptions, class and object property axioms (using capital letters for classes, lower case letters for object properties, and for instances):
Class description: class intersection (), existential quantification (), limited enumeration using a single instance ()
Class axioms: subclass (), equivalent class (), disjointness ()
Object property axioms: sub-property (), property chains (), equivalent property (), transitive properties (), reflexive properties
We deductively close the knowledge graph with respect to the OWL 2 EL profile, using an OWL 2 EL reasoner (Kazakov et al., 2014). A knowledge graph is deductively closed if and only if for all such that . In general, the deductive closure of a knowledge is countably infinite. Therefore, we only add inferences that can be represented explicitly as edges between named individuals and classes in , i.e. between entities that are explicitly named in . In particular, for all instances and object properties , if , then . Furthermore, for all named classes and instances , if , then . Finally, we also infer relations between classes, in particular subclass axioms, and add them to the inferred graph: for any class , if , then .
We use the OWL API version 4 (Horridge et al., 2007) to classify the input knowledge graph and add all inferences obtained by using the ELK reasoner as new edges to the knowledge graph to generate . We use this fully inferred graph as a basis for generating the node embeddings through our method.
2.3 Walking RDF and OWL
To generate node embeddings, we use a modified version of the DeepWalk algorithm (Perozzi et al., 2014) in which we consider edge labels as part of the walk. A random walk of length n over a graph and start vertex is an ordered sequence of vertices , and each vi (i > 0) is determined by randomly selecting an adjacent node of . As knowledge graphs generated by our method additionally have edges of different types (i.e. edge labels, ), we extend this notion to edge-labeled random walks. An edge-labeled random walk of length n over the graph , edge labels in the label space L (i.e. the set of object properties in the knowledge graph underlying G), and start vertex is a sequence such that , and, starting with v0 and for all vi (i < n), a random outgoing edge of vi, ending in is chosen to generate from and .
We implement this algorithm as an extension of the DeepWalk (Perozzi et al., 2014). The algorithm takes a knowledge graph as input and generates a corpus consisting of a set of edge-labeled random walks, starting either from all vertices , or all vertices of a specified subset of . Parameters of the algorithm are the length of the walks and the number of walks per node. Source code of the algorithm and documentation are freely available at https://github.com/bio-ontology-research-group/walking-rdf-and-owl.
2.4 Learning embeddings
We use the corpus of edge-labeled random walks as an input for learning embeddings of each node. We follow the skip-gram model (Mikolov et al., 2013) to generate these embeddings. Given a sequence of words, in , a skip-gram model aims to maximize the average log probability
| (1) |
in which c represents a context or window size. To define , we use negative sampling, following (Mikolov et al., 2013), i.e. replacing above with a function to discriminate target words (wO) from a noise distribution (Mikolov et al., 2013), drawing k words from :
| (2) |
The vector representation (embedding) of a word s occurring in corpus C is the vector vs in Eq. 2 derived by maximizing Eq. 1. The dimension of this vector is a parameter of the method.
Since our corpus consists of often repeated edge labels (due to the relatively small size of the label space ), we further use sub-sampling of frequent words (Mikolov et al., 2013) (which mainly represent edge labels in the corpora we generate) to improve the quality of node embeddings. We follow (Mikolov et al., 2013) and discard, during training, each word wi (i.e. node or edge) with a probability where t is a threshold parameter.
It is obvious from this formulation that the parameters for learning the representation of nodes in a knowledge graph include the number of walks to perform for each vertex, the length of each individual walk, a subset U of vertices from which to start walks, the size of the vector representations learned by the skip-gram model, the window or context size employed in the skip-gram model, the parameter t used to sub-sample frequent words (we use for all our experiments), and the number of words to draw from the noise distribution (we fix this parameter to 5 in our experiments). There are several additional parameters for training a skip-gram model, including learning rate and certain processing steps on the corpus, for which we chose default values in the gensim (https://radimrehurek.com/gensim/) skip-gram implementation.
2.5 Prediction
The embeddings can be used as features in machine learning tasks that should encode for the local neighborhood of each node, thereby encoding for the (local) information contained in a knowledge graph about a certain vertex. We apply these features to the task of edge prediction, in which we aim to estimate the probability that an edge with label l exists between vertices v1 and v2 given their vector representation, and : . We use the logistic regression classifier implemented in the sklearn library (Pedregosa et al., 2011) to train logistic regression models.
We build separate binary prediction models for each object property in the knowledge graphs. For model building and testing, we employ 5-fold cross-validation. For each object property representing edge label l, cross-validation folds are built by randomly removing 20% of edges with label l in the knowledge graph, then applying deductive inference, corpus generation through edge-labeled random walks, learning of vector representations of nodes, and building of a binary logistic regression model. The degree distribution in our knowledge graph before and after removing 20% of edges is available as Supplementary Material. A model for edges with label l is trained using as positive instances all pairs of vertices for which an edge with label l exists in the modified knowledge graph (in which 20% of edges with label l have previously been removed), and using as negatives a random subset of all pairs of vertices such that is of the same type (i.e. an instance of the same class in the knowledge graph) as all sources of edges with label l, and is of the same type as all targets of edges with label l. For example, if edges with label l are all between instances of Drug and Disease in a knowledge graph, then we sub-sample negative instances among all pairs of instances of Drug and Disease for which no edge exists in the original knowledge graph. The constraint of choosing negatives from the same general types of entities is necessary because instances of different types will be clearly separable within the embeddings, and evaluation using those would therefore bias the results. We randomly generate a set of negative samples with the same cardinality as the set of positive samples, both for model training and prediction. A limitation of our choice of negatives is that some edges we consider as negatives may not be true negatives due to the likely incompleteness of the knowledge graph and the sources we used for its generation.
The embeddings can also be used for findings similar nodes using a measure of similarity. We use cosine similarity to compute the similarity between two vectors:
2.6 Parameter optimization
Using the performance on the final prediction model, we perform parameter optimization through a limited grid search. We only optimize embedding size, number of walks, walk length and context size for the skip-gram model through a grid search since an exhaustive optimization would be too computationally expensive. Furthermore, we only use a single object property to test how results change with each choice of parameter, due to computational constraints. We tested the following 625 parameters: embedding sizes of 32, 64, 128, 256 and 512, number of walks 50, 100, 200, 300 and 500, walk length 5, 10, 15, 20 and 30, and skip-gram context sizes 5, 10, 15, 20 and 30. We found the best performing parameters to be 512 for the embedding size, 100 for the number of walks, 20 for the walks length and 10 for the skip-gram context size, and we fix these parameters throughout our experiments.
3 Results
3.1 Neuro-symbolic feature learning using Semantic Web technologies
We build a knowledge graph using Semantic Web technologies centered on human biomedical data. The graph incorporates several biological and biomedical datasets and is split in two layers, instances and classes. On the level of instances in the knowledge graph, we combine protein-protein interactions (PPIs) (Szklarczyk et al., 2011), chemicals (drugs) and their protein targets (Kuhn et al., 2012), drugs and their indications (Kuhn et al., 2010), and genes and the diseases they are involved in (Piñero et al., 2015). On the level of classes, we include the Human Phenotype Ontology (Köhler et al., 2014), and the Gene Ontology (Ashburner et al., 2000), and we include annotations of diseases and their phenotypes (Hoehndorf et al., 2015a; Köhler et al., 2014), genes and their phenotypes (Köhler et al., 2014), and human protein functions and subcellular locations (The UniProt Consortium, 2015). The knowledge graph, including the data, ontologies and our formal representation of ontology-based annotations, consists of 7 855 737 triples. We use the Elk reasoner (Kazakov et al., 2014) to deductively close this graph, and through the application of ontology-based inference, we further infer 5 616 273 new triples and add them to the knowledge graph.
We utilize this knowledge graph as the input to our algorithm that can learn representations of nodes. These representations represent the neighborhood of a node as well as the kind of relations that exist to the neighboring nodes. To learn these representations, we perform random walks from each node in the knowledge graph repeatedly, use the resulting walks as sentences within a corpus, and apply the Word2Vec skip-gram model (Mikolov et al., 2013) to learn embeddings for each node.
We use the fully inferred, deductively closed knowledge graph to perform the random walks. Performing random walks on the deductively closed graph has the advantage that not only asserted axioms will be taken into consideration, but representations can also include inferred knowledge that is not present explicitly in the graph. For example, for an assertion that a gene g has a function F (where F is a class in the GO), all superclasses of F in GO will be added as annotations to g; sub-properties (such as ) asserted in an ontology or database will be resolved; transitive, reflexive object properties and property chains resolved and the inferred edges added.
We automated these steps (ontology-based classification, repeated random walk, generation of embeddings) in an algorithm that combines the steps relying on symbolic inference and the learning of embeddings using a neural network. The input of the algorithm is a knowledge graph and the parameters needed for the algorithm such as the length and number of walks and size of the resulting embeddings, the output is an embedding (of a specified size) for each node in the knowledge graph. Figure 1 illustrates our basic workflow.
Fig. 1.

Overview over the main steps in our workflow. We first build biological knowledge graphs by integrating Linked Data, biomedical ontologies and ontology-based annotations in a single, two-layered graph, then deductively close the graph using automated reasoning and apply feature learning on the inferred graph to take into account both explicitly represented data and inferred information. The two layers of the knowledge graph arise from the different semantics of linked biological data (represented in the graph-based language RDF) and the ontologies (represented in the model-theoretic language OWL); we formally connect the entities in the data layer through the rdf:type relation to ontology classes
3.2 Edge prediction
The resulting embeddings can be used in standard machine learning classifiers. We demonstrate these uses in two settings. First, we remove edges from the knowledge graph, regenerate the embeddings using the reduced graph, and train a logistic regression classifier to predict whether or not an edge exists between two nodes, given the embeddings for two nodes as input. This kind of application is intended to demonstrate how associations between two potentially different types of entities, such as a gene and disease, can be identified. We perform these experiments in 5-fold cross-validation setting for every object property in our graph except for edges that exist only between ontology classes. Table 1 summarizes the results. We performed the same experiment using a one-class support vector machine and include results as Supplementary Material.
Table 1.
Performance results for edge prediction in a biological knowledge graph
| Object property | Source type | Target type | Without reasoning | With reasoning | ||
|---|---|---|---|---|---|---|
| F-measure | AUC | F-measure | AUC | |||
| has target | Drug | Gene/Protein | 0.94 | 0.97 | 0.94 | 0.98 |
| has disease annotation | Gene/Protein | Disease | 0.89 | 0.95 | 0.89 | 0.95 |
| has side-effect* | Drug | Phenotype | 0.86 | 0.93 | 0.87 | 0.94 |
| has interaction | Gene/Protein | Gene/Protein | 0.82 | 0.88 | 0.82 | 0.88 |
| has function* | Gene/Protein | Function | 0.85 | 0.95 | 0.83 | 0.91 |
| has gene phenotype* | Gene/Protein | Phenotype | 0.84 | 0.91 | 0.82 | 0.90 |
| has indication | Drug | Disease | 0.72 | 0.79 | 0.76 | 0.83 |
| has disease phenotype* | Disease | Phenotype | 0.72 | 0.78 | 0.70 | 0.77 |
Object properties marked with an asterisk are between instances and instances of ontology classes.
We find that the performance of the prediction differs significantly by object property, but some object properties can be predicted with high F-measure. Furthermore, using the knowledge graph with reasoning improves the performance slightly when predicting edges between instances and mostly results in decreased performance when aiming to predict edges between instances and instance of an ontology class. We achieve overall highest performance on predicting has target edges with an F-measure of 0.94 and ROCAUC of 0.98, and lowest overall performance on associations between diseases and their phenotypes (has disease phenotype, ROCAUC 0.77). While our aim here is not to propose a novel method of predicting drug targets, protein functions or phenotypes, our performance is similar to state of the art approaches for related tasks (Wang et al., 2013, 2014). Some of the edges, such as has function or has phenotype, have to be predicted in a hierarchical output space (i.e. an ontology such as the Gene Ontology (Ashburner et al., 2000) and the Human Phenotype Ontology (Köhler et al., 2014)) and need to satisfy additional consistency constraints (due to formal dependencies between the labels), which may overall result in lower performance when applied to these tasks (Radivojac et al., 2013; Sokolov et al., 2013).
3.3 Drug repurposing on biological knowledge graphs
As second use case, we also test how well the node embeddings can be used to predict novel relations, i.e. relations that are not explicitly represented in the knowledge graph. Such an evaluation can provide information about how well the embeddings our algorithm generates can be reused in novel applications or as part of larger predictive systems for hypothesis generation (Gottlieb et al., 2011).
We aim to test how much information about shared mode of action is encoded in the embeddings of drug nodes generated by our method, and how the performance of our approach compares to related efforts. Using side-effect similarity alone, it is possible to identify pairs of drugs that share protein targets and indications (Campillos et al., 2008; Tatonetti et al., 2012), thereby demonstrating that side effects provide some information about drugs’ modes of action (Campillos et al., 2008). We train a logistic regression classifier to predict whether a pair of drugs (represented by the embeddings we generate) share an indication or target. To make our input data comparable to studies that compare only drugs’ side effects, and to avoid bias introduced by encoding targets and indications in the knowledge graph, we remove all has indication and has target edges from our graph and further retain only drugs contained in the SIDER database (Kuhn et al., 2010). We then train a logistic regression classifier to determine whether a pair of drugs shares an indication or a target using 80% of the drug pairs as training and keeping 20% as testing.
Figure 2 shows the resulting performance. We can achieve 0.79 ROCAUC for drugs that share targets, and 0.77 ROCAUC for drugs that share indications. In comparison, ranking drug pairs by their side effect similarity alone can achieve a ROCAUC of up to 0.75 for drugs sharing targets and 0.83 for drugs sharing indications (Tatonetti et al., 2012). Our results demonstrate that our method generates embeddings that encode for the explicit information in a knowledge graph, is capable of utilizing this for prediction and achieve comparable results to other approaches. Moreover, after removing has target and has indication edges, drugs are not directly linked to protein-protein interactions, protein functions or disease phenotypes. Nevertheless, the embeddings generated for drugs based on the corpus generated by random walks can encode some of this information, for example by linking both genes and drugs to similar phenotypes (and thereby providing information about potential drug targets), linking diseases and drugs to similar phenotypes (and thereby providing information about potential indications), as well as more complex interactions.
Fig. 2.
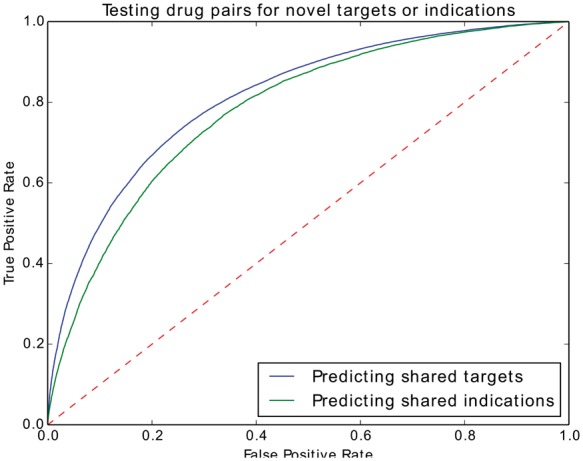
ROCAUC test scores of SIDER drug pairs the for predicting novel indications or targets or both
Instead of using a classifier, similarity between the embeddings can also be exploited to identify biological relations. Using the full knowledge graph, we further tested whether drug-drug similarity can be used to identify drugs that fall in the same indication group. We use cosine similarity to determine how similar two drugs are and evaluate whether drugs that share the same top-level Anatomical Therapeutic Chemical Classification System (ATC) code are more similar than drugs that do not share codes. We find drugs in the same ATC top-level category are significantly (, Mann-Whitney U test) more similar than drugs that do not fall in the same ATC top-level category.
4 Discussion
We present an approach for feature learning on biological knowledge graphs, and demonstrate that these features are predictive of relations between biological entities. Our approach has several advantages over traditional machine learning approaches based on hand-crafted features. First, we reuse existing Linked Data representations of biological databases as well as the OWL ontologies that were developed to characterize their content, and our approach is therefore widely applicable to any kind of biological data represented through RDF and OWL. In the past decades, there have been significant resources committed to the development of linked datasets in biology and biomedicine as well as the development of high-quality ontologies (Jupp et al., 2014; Katayama et al., 2014; Smith et al., 2007), and our work can be applied to these resources and enable or improve data analytics. Our approach also utilizes structured data as well as the ontologies used to capture background knowledge, and through the application of automated reasoning it will therefore not only encode associations between biological entities represented in databases but also their ontology-based classifications, even when these associations are not explicitly stated but inferred. Furthermore, our approach encodes information about network connectivity and communities within a node’s neighborhood in a knowledge graph. The features learned from this information may be used to build prediction models where such information is important, such as gene–disease associations based on the structure of the interactome (Köhler et al., 2008).
We do not demonstrate that we significantly outperform the state of the art in predicting certain biological relations. Our approach has several limitation that affect its performance when used on its own for predicting biological relations. First, machine learning models built using manually crafted features will be able to utilize more specific features that are directly relevant for predicting a particular type of relation. They will also be able to utilize these features better, for example by combining or transforming them so that they can be utilized better to solve a particular problem. Our approach, on the other hand, is not specific to a particular application; we use the same knowledge graph and the same feature learning method for predicting multiple different types of biological relations. Second, our approach only uses qualitative information, while prediction of certain association will often use both qualitative and quantitative information. For example, to predict drug targets, differential gene expression profiles can provide a significant amount of information (Lamb et al., 2006) but we currently cannot incorporate such information in our approach. Third, our approach is approximate in the sense that the neighborhood of a node in the knowledge graph is sampled through a random walk, and in particular for nodes with a high degree of connectivity, information is lost as not all outgoing or incoming edges will be included in a random walk. Despite these limitations, the embeddings that our approach generates can be added as additional features to existing machine learning methods without spending significant effort to manually extract and represent features. The low dimensionality of the embeddings for each node makes our approach particularly suitable for such a combination.
Despite the large success of machine learning methods in the past years (Lecun et al., 2015), they have not yet widely been applied to symbolically represented biological knowledge. Symbolic representations in biology, based on Linked Data and ontologies, are relying on formal languages such as OWL and RDF, and utilize symbolic inference. The kind of inferences performed on this knowledge is either formally specified in the knowledge representation language (Baader et al., 2003) or produced by hand-crafted inference rules that are applicable within a particular database, application, or query (Callahan et al., 2013; The UniProt Consortium, 2015). Here, we use knowledge graphs built using the semantics of OWL and data is represented as instances of OWL classes, but our approach of building knowledge graphs can be replaced with, or amended by, the use of explicit inference rules. In this case, instead of applying an OWL reasoner to infer edges with respect to the OWL semantics, rules can be used to infer edges and deductively close the knowledge graph with respect to a set of inference rules.
A key difference between the knowledge graphs we use in our approach and knowledge graphs widely used in biological databases is the strong focus on representing biological entities and their relations in contrast to representing the (non-biological) meta-data about these entities and their associations, such as provenance (Belhajjame et al., 2012) and authorship. Only few knowledge graphs have been developed that employ such a clear distinction, notably the KaBOB knowledge graph (Livingston et al., 2015). While inclusion of such metadata in knowledge graphs is required for retrieval and to ensure data quality (Wilkinson et al., 2016), our method relies on the use of data models that make it possible to separate the biological content of a knowledge graph from the metadata.
We demonstrate that knowledge graphs based on Semantic Web standards and technologies can not only be used to store and query biological information, but also have the capability to be used for data analysis. The key advantage of choosing knowledge graphs as representation formats for analytical services over other representations is the inherent focus on representing heterogeneous information in contrast to single types of relations, the possibility to continuously add information, the use of inference rules, and the use of World Wide Web standards. Our method allows all these advantages to be utilized and incorporated in predictive models, and may encourage database curators and biologists to increasingly rely on knowledge graphs to represent the biological phenomena of their interest.
Supplementary Material
Acknowledgements
A prototype of the feature learning algorithm was implemented at the NBDC/DBCLS BioHackathon 2016 in Tsuruoka. We acknowledge the use of the compute resources of the Computational Bioscience Research Center at King Abdullah University of Science and Technology (KAUST).
Funding
M.A., M.A.K., O.M. and R.H. were supported by funding from King Abdullah University of Science and Technology (KAUST). A.R.K. was supported by Database Integration Coordination Program (Upgrading and Integrative Management of Protein Data Bank Japan) from the National Bioscience Database Center (NBDC) and Platform Project for Supporting in Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from Japan Agency for Medical Research and Development (AMED). N.Q.R. acknowledges support from ISCIII-FEDER (PI13/00082, CP10/00524, CPII16/00026), IMI-JU under grants agreements no. 115191 (Open PHACTS), and no. 115372 (EMIF), resources of which are composed of financial contribution from the EU-FP7 (FP7/2007-2013) and EFPIA companies in kind contribution, and the EU H2020 Programme 2014-2020 under grant agreements no. 634143 (MedBioinformatics) and no. 676559 (Elixir-Excelerate), and the Spanish Ministry of Economy and Competitiveness, through the “María de Maeztu” Programme for Units of Excellence in R&D (MDM-2014-0370). The Research Programme on Biomedical Informatics (GRIB) is a member of the Spanish National Bioinformatics Institute (INB), PRB2-ISCIII and is supported by grant PT13/0001/0023, of the PE I+D+i 2013-2016, funded by ISCIII and FEDER.
Conflict of Interest: none declared.
References
- Ashburner M. et al. (2000) Gene ontology: tool for the unification of biology. Nat. Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baader F. et al. (2003) The Description Logic Handbook: Theory, Implementation and Applications. Cambridge University Press, Cambridge, UK. [Google Scholar]
- Belhajjame K. et al. (2012). PROV-O: The PROV ontology. Technical report, W3C.
- Belleau F. et al. (2008) Bio2RDF: Towards a mashup to build bioinformatics knowledge systems. J. Biomed. Inf., 41, 706–716. [DOI] [PubMed] [Google Scholar]
- Berners-Lee T. et al. (2001) The Semantic Web. Sci. Am., 284, 28–37. [Google Scholar]
- Callahan A. et al. (2013) Bio2RDF Release 2: Improved Coverage, Interoperability and Provenance of Life Science Linked Data, Pages 200–212. Springer, Berlin, Heidelberg. [Google Scholar]
- Campillos M. et al. (2008) Drug target identification using side-effect similarity. Science, 321, 263–266. [DOI] [PubMed] [Google Scholar]
- Candan K.S. et al. (2001) Resource description framework: metadata and its applications. SIGKDD Explor. Newsl., 3, 6–19. [Google Scholar]
- Gottlieb A. et al. (2011) PREDICT: a method for inferring novel drug indications with application to personalized medicine. Mol. Syst. Biol., 7, 496.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau B. et al. (2008) OWL 2: The next step for OWL. Web Semantics Sci. Serv. Agents World Wide Web, 6, 309–322. [Google Scholar]
- Gruber T.R. (1995) Toward principles for the design of ontologies used for knowledge sharing. Int. J. Hum. Comput. Stud., 43, [Google Scholar]
- Hoehndorf R. et al. (2011) A common layer of interoperability for biomedical ontologies based on OWL EL. Bioinformatics, 27, 1001–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehndorf R. et al. (2014) Mouse model phenotypes provide information about human drug targets. Bioinformatics, 30, 719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehndorf R. et al. (2015a) Analysis of the human diseasome using phenotype similarity between common, genetic, and infectious diseases. Sci. Rep., 5, 10888.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehndorf R. et al. (2015b) The role of ontologies in biological and biomedical research: a functional perspective. Brief. Bioinf., 16, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horridge M. et al. (2007). Igniting the OWL 1.1 touch paper: The OWL API. In: Proceedings of OWLED 2007: Third International Workshop on OWL Experiences and Directions.
- Horrocks I. (2007). OBO flat file format syntax and semantics and mapping to OWL Web Ontology Language. Technical report, University of Manchester.
- Jupp S. et al. (2014) The EBI RDF platform: linked open data for the life sciences. Bioinformatics, 30, 1338–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama T. et al. (2014) Biohackathon series in 2011 and 2012: penetration of ontology and linked data in life science domains. J. Biomed. Semantics, 5, 5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazakov Y. et al. (2014) The incredible elk. J. Automated Reason., 53, 1–61. [Google Scholar]
- Kibbe W.A. et al. (2014) Disease ontology 2015 update: an expanded and updated database of human diseases for linking biomedical knowledge through disease data. Nucleic Acids Res., 43, D1071–D1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. et al. (2016) Pubchem substance and compound databases. Nucleic Acids Res., 44, D1202–D1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler S. et al. (2008) Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet., 82, 949– 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler S. et al. (2014) The human phenotype ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res., 42, D966–D974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M. et al. (2010) A side effect resource to capture phenotypic effects of drugs. Mol. Syst. Biol., 6, 343.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M. et al. (2012) STITCH 3: zooming in on protein-chemical interactions. Nucleic Acids Res., 40, D876–D880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J. et al. (2006) The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science, 313, 1929–1935. [DOI] [PubMed] [Google Scholar]
- Lecun Y. et al. (2015) Deep learning. Nature, 521, 436–444. [DOI] [PubMed] [Google Scholar]
- Livingston K.M. et al. (2015) Kabob: ontology-based semantic integration of biomedical databases. BMC Bioinformatics, 16, 126.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikolov T. et al. (2013) Distributed representations of words and phrases and their compositionality In Burges C.J.C.et al. (eds.) Advances in Neural Information Processing Systems 26. Curran Associates, Inc, Red Hook, NY, pp. 3111–3119. [Google Scholar]
- Motik B. et al. (2009) Owl 2 web ontology language: Profiles. Recommendation, World Wide Web Consortium (W3C).
- Mungall C. et al. (2012) Uberon, an integrative multi-species anatomy ontology. Genome Biol., 13, R5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OWL Working Group, W. (2009) OWL 2 Web Ontology Language: Document Overview. W3C recommendation, W3C.
- Pedregosa F. et al. (2011) Scikit-learn: machine learning in Python. J. Mach. Learn. Res., 12, 2825–2830. [Google Scholar]
- Perozzi B. et al. (2014). Deepwalk: Online learning of social representations. In: Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, KDD ’14, New York, NY, USA. ACM, pp. 701–710.
- Piñero J. et al. (2015) Disgenet: a discovery platform for the dynamical exploration of human diseases and their genes. Database, 2015, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radivojac P. et al. (2013) A large-scale evaluation of computational protein function prediction. Nat. Methods, 10, 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristoski P., Paulheim H. (2016) Rdf2vec: Rdf graph embeddings for data mining. In: The Semantic Web - ISWC 2016: 15th International Semantic Web Conference, Kobe, Japan, October 17-21, 2016, Proceedings, Part I, volume 9981, Cham. Springer International Publishing, pp. 498–514.
- Seaborne A., Prud’hommeaux E. (2008) SPARQL query language for RDF. W3C recommendation, W3C.
- Smith B. et al. (2005) Relations in biomedical ontologies. Genome Biol., 6, R46.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. et al. (2007) The OBO Foundry: coordinated evolution of ontologies to support biomedical data integration. Nat. Biotechnol., 25, 1251–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov A. et al. (2013) Combining heterogeneous data sources for accurate functional annotation of proteins. BMC Bioinformatics, 14, S10.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suntisrivaraporn B. et al. (2007) Replacing sep-triplets in snomed ct using tractable description logic operators. In: Riccardo Bellazzi,J.H. and Ameen Abu-Hanna (eds.) Proceedings of the 11th Conference on Artificial Intelligence in Medicine (AIME’07), Lecture Notes in Computer Science. Springer-Verlag.
- Szklarczyk D. et al. (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res., 39, D561–D568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatonetti N.P. et al. (2012) Data-driven prediction of drug effects and interactions. Sci. Transl. Med., 4, 125ra31.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Horst H.J. (2005). Combining RDF and part of OWL with rules: Semantics, decidability, complexity. In: The Semantic Web – ISWC 2005, 4th International Semantic Web Conference, ISWC 2005, Galway, Ireland, November 6–10, 2005, Proceedings, pp. 668–684.
- The Gene Ontology Consortium (2015) Gene ontology consortium: going forward. Nucleic Acids Res., 43, D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium (2015) Uniprot: a hub for protein information. Nucleic Acids Res., 43, D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. et al. (2013) Drug target predictions based on heterogeneous graph inference. Pac. Symp. Biocomput., 2013, 53–64. [PMC free article] [PubMed] [Google Scholar]
- Wang W. et al. (2014) Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics, 30, 2923–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson M. et al. (2016) The fair guiding principles for scientific data management and stewardship. Scientific Data, 3, 160018.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood D. et al. (2014) Linked Data. Manning Publications Co, Greenwich, CT, USA, 1st edition. [Google Scholar]
- Yanardag P., Vishwanathan S. (2015). Deep graph kernels. In: Proceedings of the 21th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, KDD ’15, New York, NY, USA. ACM, pp. 1365–1374.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.