

Abstract
Motivation
Transcription factor binding sites (TFBSs) are clustered in the human genome, forming the TFBS-clustered regions that regulate gene transcription, which requires dynamic chromatin configurations between promoters and distal regulatory elements. Here, we propose a regulatory model called spatially adjacent TFBS-clustered regions (SATs), in which TFBS-clustered regions are connected by spatial proximity as identified by high-resolution Hi-C data.
Results
TFBS-clustered regions forming SATs appeared less frequently in gene promoters than did isolated TFBS-clustered regions, whereas SATs as a whole appeared more frequently. These observations indicate that multiple distal TFBS-clustered regions combined to form SATs to regulate genes. Further examination confirmed that a substantial portion of genes regulated by SATs were located between the paired TFBS-clustered regions instead of the downstream. We reconstructed the chromosomal conformation of the H1 human embryonic stem cell line using the ShRec3D algorithm and proposed the SAT regulatory model.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
In eukaryotes, multiple transcription factors (TFs) cooperatively bind to regulatory DNA elements to control gene expression temporally and spatially. Therefore, a full understanding of how TFs contribute to the control of cellular transcriptional regulation requires an in-depth analysis of the complete ensemble of TF binding events in a cell. Recent studies have revealed that TF binding events are highly clustered in the worm (Gerstein et al., 2010), fruit fly (Negre et al., 2011; Roy et al., 2010) and human genomes (Boyer et al., 2005; Yan et al., 2013). The broad presence of clustered transcription factor binding sites (TFBSs) may suggest a general regulatory model of gene transcription.
To discover how hundreds of TFs coordinate their binding sites in clusters across cell types and tissues, we previously developed a computational method for the genome-wide mapping of TFBS-clustered regions (Chen et al., 2015). We identified a large set of human TFBS-clustered regions in 133 human cell types and revealed new models of transcriptional regulation through an integrative analysis of these regions. These previous analyses assumed a linear genome structure; however, transcriptional regulation relies strongly on the three-dimensional (3D) conformations of chromosomes (Li et al., 2012; Spitz 2016). The emergence of unbiased genome-wide chromosomal conformation analysis technology, such as Hi-C, has significantly facilitated the study of 3D genomic structures (Lieberman-Aiden et al., 2009) and provided great opportunities to survey the spatial relationships between TFBS-clustered regions and genes. High-resolution Hi-C data are now accessible at the kilobase-level (Dixon et al., 2012), enabling detailed studies of the mechanisms of long-range transcriptional regulation.
Recently, Jin et al. determined over one million long-range chromatin interactions at 5- to 10-kb resolution (Jin et al., 2013). Based on their high-resolution Hi-C data and our TFBS-clustered regions, we were able to discover spatially adjacent TFBS-clustered regions (SATs). Furthermore, by reconstructing the 3D chromatin structure, we discovered different models of long-range transcriptional regulation and different models corresponding to different functions.
2 Materials and methods
2.1 Dataset
TFBS-clustered regions in this study were obtained from our former research (Chen et al., 2015). Download link for GEO Series GSE59016 data: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE59016. HiC data is from Dr. Bing Ren Laboratory (Jin et al., 2013). (GEO Series GSE43070: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE43070). Gene annotations were obtained from the GENCODE data (V17) . Transcription factors CTCF by ChIP-seq data were obtained from the HAIB TFBS ENCODE groups. All of these data were provided through ENCODE Project19. Data use strictly adhered to the ENCODE Consortium Data Release Policy.
2.2 Definition of SATs
In our previous research, we described the transcriptional regulation model named TFBS-clustered regions to characterize the genomic feature that TF binding is highly clustered in the human genome. We identified TFBS-clustered regions by using a Gaussian kernel density estimation with a bandwidth of 300 bp to assay the binding profiles of 542 TFs (Fig. 1A). We used the homer algorithm (Heinz et al., 2010, http://homer.salk.edu/homer/interactions/HiCinteractions.html) to identify significant interactions. First, we generated a contact matrix for the counts of Hi-C reads of any two 5-kb-long genomic regions. Any two regions with significantly larger-than-expected numbers of Hi-C reads were selected as significant interactions based on the background model. We used a default P-value of 0.001 and resolution of 5000 for the computation of Hi-C data. A significant interaction consists of 2 regions of 5 kb in length that are considered spacial adjacent. The identified significant interactions were deposited in the GEO under accession ID GSE93834. An SAT was identified by overlapping significant interaction regions and TFBS-clustered regions: When two TFBS-clustered regions overlapped each 5-kb region, respectively, they were defined as an SAT (Fig. 1B). Here, an overlap was designated only when every base pair of the TFBS-clustered region was contained within the 5-kb region. When a group of three or more TFBS-clustered regions satisfied the condition that any two of them were spatially adjacent, we defined this group of TFBS-clustered regions together as a multiple spatially adjacent TFBS-clustered region (mSAT) (Fig. 1C). If a TFBS-clustered region was classified as part of an mSAT, then we no longer used it to construct SATs. In this way, SATs and mSATs did not share any common TFBS-clustered regions. The average one-dimensional (1D) distance between 2 TFBS-clustered regions (defined as the number of base pairs separating the midpoints of the two regions) that are space adjacent was 132 kb in the H1 cell line.
Fig. 1.
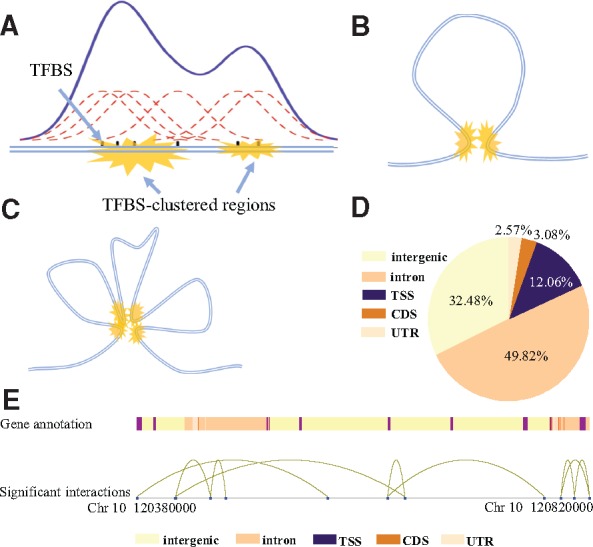
Identification of TFBS-clustered regions and SATs. (A) Identification of TFBS-clustered regions. Gaussian kernel density estimation was used to identify regions where TFBSs were clustered. (B) Typical structure of an SAT. Yellow markings represent pairs of spatially adjacent TFBS-clustered regions. (C) Typical structure of mSATs. Four yellow markings represent multiple TFBS-clustered regions spatially adjacent to each other. (D) Illustration of relative genome positions between SATs and genes. Different colors represent distinct gene annotations. Distant TFBS-clustered regions (blue dots) near gene promoters connected by curves represent SATs. (E) Genome-wide distribution of TFBS-clustered regions in proximal promoters (defined as 1-kb upstream and downstream of TSS), exons, introns, CDSs and intergenic regions for the H1 cell line
2.3 Reconstruction of the 3D chromatin structure
ShRec3D (Lesne et al., 2014) was used to reconstruct the conformation of the chromosome, taking the interaction matrix derived from raw Hi-C data as input. The length of a link was the inverse contact frequency. The Floyd–Warshal algorithm was applied to get the length of the shortest path representing the distance between any two nodes. The 3D chromosomal structure was generated by using classical multidimensional scaling.
3 Results
3.1 Distribution of SATs
Using 183 391 TFBS-clustered regions and 261 758 significant interactions at 5-kb resolution in the H1 human embryonic stem cell line, we identified 28 035 SATs and 2960 mSATs, which comprised 36 730 TFBS-clustered regions representing 20.03% of all TFBS-clustered regions (Supplementary Fig. S1). We identified 2707 isolated SATs, defined as an SAT with two TFBS-clustered regions that connect only to each other and not to any TFBS-clustered region from other SATs. Among the 146 661 TFBS-clustered regions that did not belong to any SAT or mSAT, 28 996 of them overlapped with a 5-kb distal region of a significant interaction. However, the other distal region (each significant interaction contains two 5-kb distal regions) was not found to overlap with any TFBS-clustered region, and no SAT or mSAT was formed. The number of TFBS-clustered regions that did not overlap with any distal region was 117 665 (146 661 minus 28 996). Most of the TFBS-clustered regions involved in SATs were intrachromosomal; only a small fraction of interactions between TFBS-clustered regions (722, 1.81%) were interchromosomal. The relative position between SATs and genes is demonstrated in Figure 1D.
3.2 Relationship between SATs and genes
The TFBS-clustered regions were annotated by overlapping with functional genomic regions. Among all 183 391 TFBS-clustered regions, 4706 (2.57%) were located in untranslated regions (UTRs), 22 108 (12.06%) in promoter domains, 5641 (3.08%) in exonic domains, 91 372 (49.82%) in intronic domains and the remaining 59 564 (32.48%) in intergenic domains (Fig. 1E).
As the TFBSs are preferentially clustered at the promoter domains of genes, we focused our analyses mainly on these regions. A promoter region was identified as a 2-kb region whose midpoint was the corresponding transcription start site (TSS) of a gene. Moreover, 12.92% of TFBS-clustered regions that were not in SATs or mSATs were in promoter regions, compared to 10.35% of TFBS-clustered regions that were in SATs (15202/117665 versus 3420/33029, hypergeometric test P-value = 7.04 × 10−63) and only 9.83% of TFBS-clustered regions that were in mSATs (15202/117665 versus 364/3701, hypergeometric test P-value = 2.30 × 10−9) (Fig. 2A).
Fig. 2.

Spatial and regulatory characteristics of SATs. (A) Proportions of TFBS-clustered regions in different regulatory models located in promoter domains. (B) Proportions of different types of regulatory models located in promoter domains. (C) The previous regulatory model. An upstream SAT (yellow marks) regulates one gene. (D) Reconstructed chromosomal conformation showing spatial relationship between an mSAT (yellow dots) and the regulated gene (red dot). Blue lines represent chromatin structure. (E) SAT model where the regulated gene is located between two TFBS-clustered regions. (F) SAT model where multiple regulated genes are located between two TFBS-clustered regions. (G) mSAT model where multiple genes are regulated by multiple TFBS-clustered regions combined
Considering SATs and mSATs as a whole, we found that 18.77% of SATs (15202/117665 versus 4830/28035, Fisher's exact test P-value < 2.20 × 10−16) and 27.20% of mSATs (15202/117665 versus 698/2960, Fisher's exact test P-value < 2.20 × 10−16) were in promoter regions (Fig. 2B).
Multiple TFBS-clustered regions in the same SAT or mSAT ‘share’ their common promoter. Thus, we expected that the TFBS-clustered regions in SATs/mSATs would be less likely to be located in promoter regions than other TFBS-clustered regions that regulate target genes on their own. The downward trend of independent TFBS-clustered regions and upward trend of whole SATs/mSATs indicated that multiple distal TFBS-clustered regions might function together to regulate the same gene, resulting in a decline of the proportion of ‘promoter proxy’ TFBS-clustered regions. The results suggest that multiple upstream cis-elements regulate gene expression in a cooperative manner, consistent with regulatory models proposed in the article providing the Hi-C data (Jin et al., 2013) (Fig. 2C) and in the article by Li et al. (Li et al., 2012).
We obtained the same results with a similar analysis in the human IMR90 cell line, for which high-resolution Hi-C and TFBS-clustered region data were available (Supplementary Fig. S2). These results further support our conclusion that multiple distal TFBS-clustered regions combine to form SATs/mSATs to regulate genes. We also repeated the analysis using non-TFBS-clustered regions generated by the ‘bedtools shuffle’ command, which samples a set of regions with random position and the same size distribution as that of TFBS-clustered regions in the H1 cell line. We observed no downward trend (Supplementary Fig. S3), indicating that our conclusion was specific to TFBS-clustered regions.
3.3 A new regulatory model: SATs regulate genes
According to the previous model (Fig. 2C), SATs are located upstream of their regulated genes. However, the results of genome-wide analyses (Table 1) showed that this model can explain only 10.28% of SATs and 10.10% of mSATs. These proportions are lower than expected. Thus, other regulatory models must exist among the remaining SATs/mSATs. We categorized them based on two factors: the relative linear position between the TFBS-clustered regions of an SAT/mSAT, and the number of its regulated genes. For the relative position, we considered a gene to be ‘inside’ an SAT/mSAT when the TSS of the gene was downstream of one TFBS-clustered region and upstream of another, and the gene promoter overlapped a midpoint of any TFBS-clustered region of the SAT/mSAT. We consider the ‘distal’ SATs/mSATs when none of their TFBS-clustered regions overlapped any promoter. We identified 2145 (7.65%) SATs that regulated a single gene inside, 235 (0.84%) that regulated multiple genes inside, and more than 10% of mSATs that regulated one or multiple genes inside.
Table 1.
Positional relationship between genes and SATs/mSATs
| Distal | Upstream | One gene | Multiple genes | |
|---|---|---|---|---|
| 1 kb | inside | inside | ||
| SAT | 22773 | 2882 | 2145 | 235 |
| (81.23%) | (10.28%) | (7.65%) | (0.84%) | |
| mSAT | 2155 | 299 | 412 | 94 |
| (72.80%) | (10.10%) | (13.91%) | (3.17%) |
For better understanding of the structure of the ‘wrap around’ model, we reconstructed the chromosomal structure of the H1 cell line near an mSAT regulating protein-coding gene FGF18 on chromosome 5 using ShRec3D (Fig. 2D). The findings showed that spatially adjacent TFBS-clustered regions wrapped around the regulated gene.
We proposed three novel SAT regulatory models categorized by the relative position between SATs and genes and the reconstructed 3D chromosomal conformation, based on the original model. Figure 2 shows the cases in which two TFBS-clustered regions regulate one gene cooperatively (Fig. 2E), two TFBS-clustered regions regulate multiple genes cooperatively (Fig. 2F) and multiple TFBS-clustered regions regulate multiple gene cooperatively (Fig. 2G). We carried out Gene Ontology (GO) analysis for each group of genes regulated by these models. Genes regulated by the original model were related in the following functions: protein heterotetramerization, cell-cell adhesion, negative regulation of cell migration, telomere organization, nucleosome assembly and the sterol biosynthetic process. Genes regulated by the first new model (Fig. 2E) were related to the regulation of membrane potential, coronary vasculature development, protein transport, regulation of ion transmembrane transport, microtubule-based movement and the necroptotic process. Genes regulated by the second new model (Fig. 2F) were associated with erythrocyte development, negative regulation of transcription and the ceramide biosynthetic process. Finally, genes regulated by the third new model (Fig. 2G) contain the genes involving embryonic skeletal system morphogenesis and regulation of protein kinase C signalling.
We have shown that chromosomal conformation helps to reveal distinct models of TF regulation. TFs had been considered to regulate downstream genes because TFBSs have a clustering distribution at TSSs. Our presented regulatory models serve to explain the mechanisms that enable TFBS-clustered regions to regulate remote genes via spatial proximity in the context of the 3D chromosomal conformation.
4 Discussion
In this article, we combined TFBS-clustered regions and Hi-C data for the first time, revealing the SATs genome-wide. We proposed several new regulatory models in which SATs regulate gene expression in distinct manners, as a further upgrade of the previous model. These models help to explain how downstream TFs regulate gene expression, and the role that chromosomal conformation plays in gene regulation. The GO results showed that genes involved in different regulatory models and in different cell lines have different functions and are related to cell fate determination. Our future research will focus on the relationship between SATs/mSATs and cell fate.
As the resolution of Hi-C data and TFBS-clustered regions are at the kilobase-level, the chance that a TFBS-clustered region in SATs/mSATs would have missed the closest point of an interaction was minimized. This research and the associated data resources, especially for SATs, are expected to push forward the future research and facilitate a comprehensive understanding of the regulation mechanisms of familiar phenotypes. This model, however, is a presumed model based on currently available data and will require further experimental verification.
Supplementary Material
Acknowledgements
We wish to thank the ENCODE Project Consortium and Dr Bing Ren’s laboratory for making their data publicly available.
Funding
This work was supported by grants from the Major Research Plan of The National Natural Science Foundation of China (No. U1435222), the Program of International S&T Cooperation (No. 2014DFB30020), the National High Technology Research and Development Program of China (No.2015AA020108) and General Program [31401141, 81573251] of the Natural Science Foundation of China (www.nsfc.gov.cn).
Conflict of Interest: none declared.
References
- Boyer L.A. et al. (2005) Core transcriptional regulatory circuitry in human embryonic stem cells. Cell, 122, 947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. et al. (2015) An integrative analysis of TFBS-clustered regions reveals new transcriptional regulation models on the accessible chromatin landscape. Sci. Rep., 5, 8465.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J.R. et al. (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature, 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M.B. et al. (2010) Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science (New York, N.Y.), 330, 1775–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S. et al. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell, 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F. et al. (2013) A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature, 503, 290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne A. et al. (2014) 3D genome reconstruction from chromosomal contacts. Nat. Methods, 11, 1141–1143. [DOI] [PubMed] [Google Scholar]
- Li G. et al. (2012) Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell, 148, 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E. et al. (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (New York, N.Y.), 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre N. et al. (2011) A cis-regulatory map of the Drosophila genome. Nature, 471, 527–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S. et al. (2010) Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science (New York, N.Y.), 330, 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F. (2016) Gene regulation at a distance: from remote enhancers to 3D regulatory ensembles. Sem. Cell Dev. Biol., 57, 57–67. [DOI] [PubMed] [Google Scholar]
- Yan J. et al. (2013) Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell, 154, 801–813. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.