

Abstract
Motivation
LncRNAs play important roles in various biological processes. Although more than 58 000 human lncRNA genes have been discovered, most known lncRNAs are still poorly characterized. One approach to understanding the functions of lncRNAs is the detection of the interacting RNA target of each lncRNA. Because experimental detections of comprehensive lncRNA–RNA interactions are difficult, computational prediction of lncRNA–RNA interactions is an indispensable technique. However, the high computational costs of existing RNA–RNA interaction prediction tools prevent their application to large-scale lncRNA datasets.
Results
Here, we present ‘RIblast’, an ultrafast RNA–RNA interaction prediction method based on the seed-and-extension approach. RIblast discovers seed regions using suffix arrays and subsequently extends seed regions based on an RNA secondary structure energy model. Computational experiments indicate that RIblast achieves a level of prediction accuracy similar to those of existing programs, but at speeds over 64 times faster than existing programs.
Availability and implementation
The source code of RIblast is freely available at https://github.com/fukunagatsu/RIblast.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Long non-coding RNAs (lncRNAs) play integral roles in diverse biological processes including transcriptional regulation (Kino et al., 2010) and subnuclear structure formation (Naganuma and Hirose, 2013). The dysfunctions of many lncRNAs are associated with severe diseases such as diabetes and various cancers (Wapinski and Chang, 2011), and thus elucidating lncRNA functions is an important research topic. Although large-scale transcriptome analysis has revealed that more than 58 000 lncRNA genes are encoded by the human genome (Iyer et al., 2015), most of these lncRNAs are still poorly characterized (de Hoon et al., 2015).
Sequence similarity search and RNA secondary structure similarity search have achieved substantial success in characterizing the function of protein-coding genes and small RNAs (sRNAs), respectively (Altschul et al., 1990; Nawrocki and Eddy, 2013). However, these strategies are unsuitable for inferring the function of lncRNAs because lncRNAs frequently lack sequence and structure conservation (Cabili et al., 2011). In contrast, the identification of interaction partners for each lncRNA should be a powerful approach for inferring the function of lncRNAs because lncRNAs function by being assembled with other proteins or RNAs (Hirose et al., 2014). Several lncRNAs have been experimentally confirmed to regulate biological processes through their interactions with target RNAs. For example, Abdelmohsen et al. determined that lncRNA 7SL reduces p53 protein translation levels by binding TP53 mRNA (Abdelmohsen et al., 2014). Gong and Maquat also discovered that lncRNA 1/2-sbsRNAs inhibit the translation of the interaction target RNA through a Staufen1-mediated mRNA decay process (Gong and Maquat, 2011). These examples show that the identification of lncRNA–RNA interactions is an important step in characterizing lncRNA functions.
Several sequencing-based technologies have been developed for the experimental discovery of RNA–RNA interactions. RIA-seq (Kretz et al., 2013) and RAP-RNA (Engreitz et al., 2014) can identify target RNAs attached to an anchored RNA. Although these methods are outstanding technologies to exhaustively detect interaction targets of a specific lncRNA, repeating these experiments across many lncRNAs is labour intensive. In contrast, PARIS (Lu et al., 2016), SPLASH (Aw et al., 2016), LIGR-seq (Sharma et al., 2016) and MARIO (Nguyen et al., 2016) can comprehensively identify RNA–RNA interactions in vivo. However, the majority of the detected interactions have been related to ribosomal RNAs or small RNAs, and the number of identified lncRNA–RNA interactions has been limited. In addition, because most of the lncRNAs show tissue-specific expression patterns (Cabili et al., 2011; Iyer et al., 2015), these experiments on various tissues are necessary but they require quite hard work. Since the detection of genome-wide lncRNA–RNA interactions through experiments is difficult, computational prediction of lncRNA–RNA interactions is an indispensable technique.
Szcześniak and Makalowska predicted entire lncRNA–RNA interactions across the human transcriptome using sequence similarity search without consideration of RNA secondary structure (Szcześniak and Makałowska, 2016). However, benchmarking results of RNA–RNA interaction predictions showed that omitting RNA secondary structure information decreases prediction accuracy (Lai and Meyer, 2016). To date, many RNA–RNA interaction prediction tools that consider RNA secondary structure have been proposed, e.g. IntaRNA (Busch et al., 2008), RNAplex (Tafer and Hofacker, 2008; Tafer et al., 2011) and RactIP (Kato et al., 2010), and can detect sRNA interactions with high accuracy. However, as these programs were designed for detecting sRNA interactions, the computational costs are too high to predict lncRNA interactions comprehensively. To predict a comprehensive lncRNA interactome with consideration of RNA secondary structure, Terai et al. (2016) firstly screened interaction candidates based on only sequence complementarity and then exhaustively predicted interactions using IntaRNA. Although their approach effectively narrowed down interaction candidates, it still required extensive computational resources to utilize IntaRNA. Therefore, a much faster RNA–RNA interaction prediction program that considers RNA secondary structure is required for further progress in lncRNA function estimation.
In the present study, we developed an ultrafast RNA–RNA interaction prediction algorithm for comprehensive lncRNA interaction analysis. The key idea of our algorithm is the utilization of seed-and-extension approach, which is widely adopted in sequence homology search tools including BLAST (Altschul et al., 1990). We implemented this high-speed algorithm as a program named RIblast, which detects seed regions using suffix arrays, and subsequently extends both ends of seed regions based on an RNA secondary structure energy model. While the prediction accuracies of RIblast were comparable to those of existing programs, RIblast was more than 64 times faster than existing tools.
2 Materials and methods
2.1 Concept of the RIblast algorithm
RIblast enumerates potentially interacting segments between a query RNA x and a target RNA y. RIblast uses two energies as the evaluation criteria to determine whether two segments, (xs and ys) in sequences x and y, intermolecularly interact: accessible energy and hybridization energy. Accessible energy is the energy required to prevent the segments from forming intramolecular base pairs and can be calculated by utilizing a partition function algorithm (McCaskill, 1990). Briefly, a segment with high accessible energies tends to not form intermolecular base pairs because the segment forms intramolecular base pairs (Fig. 1A). Hybridization energy is the free energy derived from intermolecular base pairs between two segments and can be calculated as the sum of stacking energies and loop energies in the formed base-paired structure based on a nearest-neighbour energy model (Fig. 1B). When calculating hybridization energies, intra-molecular base pairs are not taken into consideration. Here, we defined the interaction energy between two segments xs and ys as the sum of the accessible energy of xs, accessible energy of ys and hybridization energy between xs and ys. RIblast outputs two segments with a particularly low interaction energy as a detected RNA–RNA interaction. Note that RNAup (Mückstein et al., 2006), IntaRNA (Busch et al., 2008) and RNAplex-a (Tafer et al., 2011) also predict RNA–RNA interactions based on this combination of hybridization energy and accessible energy, and each showed high prediction accuracies in a previous benchmarking test (Lai and Meyer, 2016).
Fig. 1.

(A) A schematic illustration of the effect of accessible energies. While a segment with low accessible energy tends to form inter-molecular base pairs, a segment with high accessible energy tends not to form inter-molecular base pairs because such a segment tends to form intra-molecular base pairs. (B) Example of hybridization energy calculation. Hybridization energy can be calculated as the sum of stacking energies and loop energies in the formed base-paired structure. Generally, stacking energies stabilize RNA–RNA interactions but loop energies destabilize interactions. This calculation is based on Turner’s parameter. (C) Overview of the RIblast algorithm
For all-to-all interaction predictions of lncRNAs, the calculation time of accessible energies scales linearly with the number of sequences. This is because accessible energies of an RNA sequence can be calculated independently of the other RNA sequences. On the other hand, the calculation time of hybridization energies is quadratic with the number of sequences when an all-to-all interaction prediction is conducted. This calculation is the obstacle to comprehensive lncRNA–RNA interaction prediction. The calculation of hybridization energy between two RNA segments is similar to the calculation of a local alignment score between two sequences (Tjaden et al., 2006). Therefore, hybridization energy can be calculated based on a Smith-Waterman algorithm-like method. In the subject of local sequence alignment, many researches have been conducted to speed up the calculation of alignments. Seed-and-extension heuristic is one of the most successful approaches and has been adopted by many sequence alignment tools, such as BLAST (Altschul et al., 1990) and LAST (Kiełbasa et al., 2011). This method first finds short matching regions, which are called seeds, between a query and target sequence and subsequently extends alignments from both end points of the detected seeds. We recognized that the application of this approach to the calculation of hybridization energy should accelerate the computation speed considerably.
2.2 Methodology overview
RIblast implements two major steps: database construction and an RNA interaction search. Figure 1C shows the flowchart of the RIblast algorithm. In the database construction step, RIblast first calculates the accessible energy of each segment in the target RNA dataset. To speed up calculation, RIblast calculates approximated accessible energies. Second, target RNA sequences are reversed and concatenated with delimiter symbols inserted between the two sequences. Third, a suffix array of the concatenated sequence is constructed. The suffix array is an efficient text-indexing data structure that comprises a table of the starting indices of all suffixes of the string in alphabetical order. It can be constructed in linear-time relative to sequence length (Nong et al., 2011; Shrestha et al., 2014). Fourth, in order to speed-up the RNA interaction search, search results of short strings are exhaustively pre-calculated. Then, the approximated accessible energies, concatenated sequences, suffix array and search results of short strings are stored in a database.
In the RNA interaction search step, RIblast first calculates approximated accessible energies and constructs a suffix array for a query sequence. Second, RIblast finds seed regions whose hybridization energy is less than a threshold energy T1 based on two suffix arrays of the query and the database. Third, the interaction energies of the detected seed regions are calculated by summation of hybridization energy and two accessible energies. In this step, RIblast removes seed regions whose interaction energies exceed 0 kcal/mol. Fourth, RIblast extends interactions from seed regions without a gap. RIblast terminates the extension when the duplexes is no longer formed in the extension. Fifth, the interactions that fully overlap with other interactions are removed. In addition, those interactions with interaction energies exceeding the threshold energy T2 are also excluded. Note that no interactions are removed if T2 is set to 0 kcal/mol. Finally, RIblast extends interactions from seed regions with a gap. RIblast terminates the extension when the duplexes is no longer formed in the extension.
2.3 The method for calculating accessible energy
We defined as a segment from position i to position j in an RNA sequence x. Here, we defined the accessible energy that is required to make the segment form a single-stranded structure as , and the accessible probability that the segment is single-stranded as . For a fixed segment length, Raccess can calculate accessible energies and accessible probabilities of all segments with using dynamic programming (Kiryu et al., 2011). Here, N is the sequence length and W is the constraint of maximal distance between the bases that may form base pairs.
However, RIblast requires the accessible energies of segments with arbitrary length, and the exhaustive calculation is computationally expensive. Therefore, RIblast uses approximated accessible energies instead of . This method was proposed in RNAplex-a (Tafer et al., 2011). was defined as follows:
R represents the gas constant and T represents the absolute temperature (we used T = 310.15 K in this study). By this approximation, we only have to calculate the accessible energies of segments with length δ and . In addition, by restricting the minimum length of seeds to δ, we need not calculate accessible energies of segments whose length is less than δ.
2.4 Seed search
BLAST searches seeds with a fixed length, but this method is unsuitable for RNA–RNA interaction search from the viewpoint of RNA energy model. For example, in Andronescu’s energy model, the hybridization energy of a 6-mer seed consisting of only G-C base pairs is about -10 kcal/mol, but that consisting of only G-U base pairs is about -1 kcal/mol. The large difference in hybridization energies between seeds of the same length may depress the performance of tools. Therefore, RIblast adopts score-based seeds, as proposed in GHOSTX (Suzuki et al., 2014). Our score-based seeds were defined as the perfect base-pairing region whose hybridization energy is less than the threshold energy T1 and length is at least δ.
In RIblast, the enumeration of duplexes satisfying the seed criteria and the search of the seeds in the database and the query are conducted simultaneously based on the suffix array and the depth-first search. Supplementary Figure S1 shows the schematic illustration of the seed search. First, RIblast searches for a single inter-molecular base pair such as G-C. If this pair is found in the query and the database, then RIblast extends the base pair by one base pair as GG-CC, GC-CG, …, GU-CG and then checks whether these extended strings are found in the query and the database. If the extended strings are detected and meet the conditions for score-based seeds, then RIblast stores the string pair as a seed. If extended strings are detected but do not meet the conditions for score-based seeds, then RIblast extends the strings by one base pair again and repeats this step. If extended strings are not detected, then the extension is stopped. The search of extended duplexes in the database and the query is efficiently calculated based on the suffix array (Shrestha et al., 2014). To avoid overly long seeds, we restricted the max seed length lengthmax. Supplementary Figure S2 shows the pseudo-code of the RIblast seed search algorithm. Here, Sq and Sdb are the query sequence and the reversed and concatenated database sequence, respectively. SAq and SAdb are the suffix arrays of Sq and Sdb, respectively. seedq and seeddb represent the temporary seeds for the query and database, respectively. spq, epq, spdb and epdb are the indices of SAq and SAdb. The SASearchNextString function returns the indices of the new extended string in a suffix array. If the string exists in the query and the database, then the returned sp() is smaller than the returned ep().
In order to accelerate this seed search step, we pre-calculate the indices of the strings whose length is shorter than l for a database sequence in the database construction step. The results of the short sequence search are used on the database sequences. Therefore, this binary search of the suffix array is needed only for the search of query sequences or long strings in the database sequence.
2.5 Extension
After the seeds are found in the query and the database, RIblast tries to extend interactions from both end points of these seed regions. The gapless extension is first conducted, and then the gapped extension is performed in a similar way to BLAST, LAST and GHOSTX.
RIblast first extends interactions without a gap from seed regions. If extended interactions have lower interaction energies than the present minimum interaction energy in this extension step, then RIblast updates the minimum interaction energy. Otherwise, extensions are repeated. If RIblast extends Y nucleotides from the length that requires the minimum interaction energy in this extension but the energy has not been updated, then RIblast terminates the gapless extension. In this step, we assume that the possible complementary bases always interacts with each other. After the gapless extension step, if two interactions and satisfy the conditions and , then we exclude the later interaction. In addition, if the interaction energy of an interaction exceeds threshold T2, then we remove the interaction.
Next, RIblast tries to extend interactions with a gap. Like the gapless extension step, if the interaction energy of extended interactions is lower than the present minimum interaction energy in this extension step, then the minimum interaction energy is updated. If RIblast extends X nucleotides from the length that requires the minimum interaction energy in this extension but the energy has not been updated, then RIblast terminates the gapless extension. The calculation of the interaction energy of extended interactions is as follows (Supplementary Fig. S3 shows the schematic illustration). Here, we regard as an interaction after gapless extension. In the extension towards the end of the query sequence (and end of the database sequence), RIblast calculates , which is the minimum interaction energy for sequences and , as the following equation.
 |
where indicates the free energy of the loop consisting of base pairs (a, b) and (c, d) and n is the sequence length of the database sequence. Here, and are satisfied. In addition, the internal loop size is restricted to within X. This formula is the same as the calculation formula of interaction energy in RNAup and IntaRNA. The extension in the opposite direction is calculated in the same manner. Dangling energies are added only after gapped extensions are finished.
2.6 Evaluation method
We assessed the performance of RIblast using three evaluation methods: base pair prediction performance, bacterial sRNA target prediction performance, and lncRNA TINCR target prediction performance.
2.6.1 Method for evaluating base pair prediction performance
To evaluate the base pair prediction performance, we used 109 validated bacterial sRNA-mRNA pairs and 52 validated fungal snoRNA-rRNA pairs as datasets, which were constructed by Lai and Meyer (Lai and Meyer, 2016). The bacterial sRNA-mRNA interaction dataset was composed of 64 E.coli and 45 Salmonella enterica interactions as well as 18 query sRNAs and 82 target mRNAs. Following the benchmark research of Lai and Meyer (Lai and Meyer, 2016), we used the sequences between 150 bp upstream and 150 bp downstream of each start codon as the target sequences. All fungal snoRNA-rRNA interactions in the dataset were S. cerevisiae C/D box interactions, and these interactions were between 43 snoRNAs and 2 rRNAs. For target rRNAs, full rRNA sequences were used. We selected IntaRNA and RNAplex-a as the software to compare with RIblast although there are many programs to predict RNA–RNA interactions. This is because both these programs consider accessible energies as with RIblast and show the best prediction performance in current tools (Lai and Meyer, 2016). The command line options used for IntaRNA and RNAplex-a were the same as those used by previous benchmark research (Lai and Meyer, 2016). As the energy parameter characterizing RNA secondary structures, we used two energy parameters, Turner’s 2004 parameter (Mathews et al., 2004) and Andronescu’s BL* parameter (Andronescu et al., 2010). Because IntaRNA did not have an option to change the energy parameter, we used only the default Turner’s 1999 parameter (Mathews et al., 1999) in the IntaRNA evaluation. TPR, PPV and MCC were calculated for each RNA–RNA interaction, and the averaged scores were evaluated. The definitions of these three scores are as follows: TPR = TP/(TP + FN), PPV = TP/(TP + FP), and
We only evaluated the minimum energy interactions.
2.6.2 Method for evaluating sRNA target prediction performance
We evaluated the sRNA target prediction performance by predicting all-to-all interactions between 18 sRNAs and all 4319 E.coli mRNAs. This evaluation method was originally proposed by Richter and Backofen (2012). As target mRNA sequences, we used sequences between 150 base-pairs (bp) upstream and 50 bp downstream from each start codon. This sequence length setting is the same as that used by Terai et al. (2016). The sequence data were downloaded from NCBI (http://www.ncbi.nlm.nih.gov/nuccore/NC_000913). We used 64 experimentally validated interactions as positive data, which were also used to evaluate base pair prediction performance. As negative data, we used all non-positive interactions in all-to-all interactions between 18 sRNAs and all 4319 E.coli mRNAs.
2.6.3 Method for evaluating lncRNA target prediction accuracy
To evaluate the human lncRNA target prediction performance, we used an RIA-seq-based TINCR interaction dataset (Kretz et al., 2013). We used the same dataset and evaluation method as Terai et al. (2016). The dataset was composed of 5195 target RNAs and 1062 RNAs among them that interact with TINCR at one or more interacting segments. The target RNAs that have more interacting segments are more likely to be TINCR-interacting RNAs. As positive data, we used RNAs that at least had a threshold number of the interacting segments. When this threshold was set to 1, 2, 3, 4 and 5 interactions, the numbers of positive data were 1062, 434, 191, 104 and 65, respectively. Instead of comparing RIblast to IntaRNA or RNAplex-a, we compared the performance of RIblast with those of the Terai et al. pipeline (Terai et al., 2016) and LAST (Kiełbasa et al., 2011), a fast local alignment tool. This is because lncRNA target predictions by IntaRNA and RNAplex-a have heavy computational costs. As LAST alignment parameter, we used the parameter settings used by Szcześniak and Makalowska (2016). We regarded the alignment score × (–1) as the interaction energy. The simple repeat regions were masked by TANTAN (Frith, 2010) with the default options. In order to match our study with previous research by Terai et al., we excluded masked regions in the seed search step but considered them in the RIblast extension step. The short summary of the Terai et al. pipeline is as follows. First, accessible energies were calculated by Raccess, and inaccessible regions were removed. Second, pairs of complementary gapless subsequences were detected as interaction regions by LAST. Finally, the interaction energies of the interaction regions were calculated by IntaRNA.
2.7 Assesment of parameter settings
RIblast uses multiple parameters in the algorithm, and the appropriate parameters have to be set in order to predict RNA–RNA interaction accurately and efficiently. Because the influence of these parameters on the performance is not independent, the simultaneous optimization of all parameters is desirable. However, there are too many parameters in RIblast, it is difficult to optimize all parameters simultaneously. Therefore, we determined parameter settings as follows. First, we fixed lengthmax, which is the parameter of a maximum seed length, to 20 because this parameter does not influence software performance when the value is sufficiently large. Second, l, which is the parameter for pre-search of seed in the database construction step, was fixed to 8. This is because this parameter influences only the calculation speed of seed search step and the calculation time of this step is very short compared to the total calculation time. Third, we investigated the influence of parameter T1 and X on the base pair prediction performance of the bacterial sRNA-mRNA dataset with the other parameters fixed. T1 is a threshold energy for seed detection, and X is a threshold length for extension termination. We adopted the parameter combination that yielded the highest MCC score. If there were several parameter combinations with the best performance, we adopted the smallest X and largest parameter combination in order to accelerate the computation. In this step, W, δ, Y and T2 is set to 70, 5, 5, and 0, respectively. The parameter setting of W is the same as that of a benchmark research for RNA–RNA interaction prediction tools (Lai and Meyer, 2016). Fourth, we assessed the effect of parameter W, δ and Y on the base pair prediction performance of the bacterial sRNA-mRNA dataset with determined X and T1 values. Fifth, to determine values for the parameter T2, we examined the dependence of lncRNA target prediction accuracy decreases from area under the receiver operating characteristic curve (AUROC) scores of SUMENERGY on T2. We used AUC scores of -16 kcal/mol and -8 kcal/mol as interaction energy thresholds for SUMENERGY when the energy parameters were Turner and Andronescu parameters, respectively.
3 Results
3.1 Evaluation of basepair prediction performance
We investigated base pair prediction performance by evaluating whether programs predict correct base pairs between two RNAs with experimental interaction evidence.
We firstly determined the values of the parameter T1 and X in RIblast based on the base pair prediction performance of the bacterial sRNA-mRNA dataset. The performances of various T1 and X values were investigated, and the parameter set that yielded the best performance was adopted (Supplementary Tables S1 and S2). These determined values of T1 and X were used in the following analyses. T2 was set to 0 kcal/mol in this evaluation. As a result, we set X and T1 to 18 and -10.0, respectively, when we used Turner’s model, and we set X and T1 to 16 and -6.0, respectively, when we used Andronescu’s model. Next, we assessed the effect of parameter W, δ, and Y on the base pair prediction performance with determined X and T1 values. (Supplementary Tables S3–S5). In consequence, we set W, δ and Y to 70, 5 and 5, respectively.
Tables 1 and 2 show the evaluation results of base pair prediction performance. For the bacterial sRNA-mRNA dataset, RIblast with Andronescu’s parameter achieved the best PPV (0.73) and MCC (0.67) performance. The best TPR score was obtained by IntaRNA (0.66). For the fungal snoRNA-rRNA dataset, RNAplex-a with Andronescu’s parameter was the best performing tool according to all three accuracy measures (TPR, 0.74; PPV, 0.69; MCC, 0.71), and was followed by RIblast using Andronescu’s parameter (TPR, 0.66; PPV, 0.60; MCC, 0.62). In both datasets, tools using Andronescu’s parameter showed superior performance to the same tool with Turner’s parameter.
Table 1.
The base pair prediction performance on the bacterial sRNA dataset
| Program | TPR | PPV | MCC |
|---|---|---|---|
| IntaRNA | 0.66 | 0.61 | 0.62 |
| RNAplex-a (Turner) | 0.63 | 0.56 | 0.58 |
| RNAplex-a (Andronescu) | 0.60 | 0.68 | 0.63 |
| RIblast (Turner) | 0.58 | 0.66 | 0.61 |
| RIblast (Andronescu) | 0.63 | 0.73 | 0.67 |
Note: The columns correspond to the three evaluation criteria. The rows indicate the performance of each program. The bold values are the highest scores in each column.
Table 2.
The base pair prediction performance on the fungal snoRNA dataset
| Program | TPR | PPV | MCC |
|---|---|---|---|
| IntaRNA | 0.61 | 0.53 | 0.56 |
| RNAplex-a (Turner) | 0.56 | 0.49 | 0.52 |
| RNAplex-a (Andronescu) | 0.74 | 0.69 | 0.71 |
| RIblast (Turner) | 0.57 | 0.49 | 0.53 |
| RIblast (Andronescu) | 0.66 | 0.60 | 0.62 |
Note: The columns correspond to the three evaluation criteria. The rows indicate the performance of each program. The bold values are the highest scores in each column.
3.2 Evaluation of sRNA target prediction accuracy
Although the information of interacted base pair is useful for designing experiments for further functional analyses of the RNAs, what most of the users would like to know may be whether an RNA is a target or not. Therefore, we evaluated bacterial sRNA target prediction performance by validating whether the predicted interaction energies of positive sRNA-mRNA interactions are lower than those of negative sRNA-mRNA interactions. After all-to-all interaction between mRNAs and sRNAs were predicted by each software, we sorted mRNAs for each sRNA by the minimum interaction energy. Then, we plotted ROC-like curves whose x- and y-axes were the number of true positive predictions and the total number of target predictions per sRNA, respectively. The parameter T2 was also set to 0 kcal/mol in this evaluation.
Figure 2 shows the bacterial sRNA target prediction performance. The best performing tool was RNAplex-a with Andronescu’s parameter. The prediction performance of RIblast with Turner’s parameter was slightly lower, but RIblast with Andronescu’s parameter showed similar performance to the other programs.
Fig. 2.
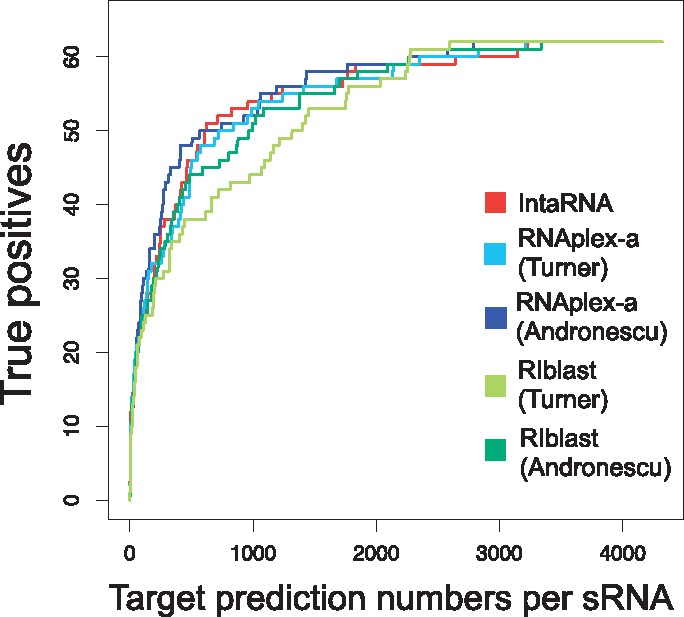
The performance of bacterial sRNA target prediction. The x- and y-axes represent target prediction numbers per sRNA and true positives, respectively.
3.3 Evaluation of lncRNA TINCR target prediction accuracy
Third, we validated human lncRNA target prediction performance by comparing predicted interactions of human lncRNA TINCR with interactions experimentally validated by RIA-seq (Kretz et al., 2013). After all interactions between TINCR and target RNAs were calculated, we sorted target RNAs based on the minimum interaction energy among all predicted interactions in the target RNA (denoted by MINENERGY) or the sum of the interaction energies that are lower than some threshold value in the target RNA (denoted by SUMENERGY). Then, we calculated AUROC scores using the pROC R package (Robin et al., 2011).
Supplementary Table S6 shows AUROC results for MINENERGY sorting. LAST, Terai et al. pipeline and RIblast exhibited performances that were similar to each other in this case. On the other hand, Figure 3 and Supplementary Tables S7–S9 show AUROC scores for SUMENERGY sorting. This result illustrates that SUMENERGY sorting performs better than MINENERGY sorting among all methods. This result is consistent with at least one previous study (Terai et al., 2016). In addition, for SUMENERGY sorting, RIblast achieved higher AUROC scores than the other methods for any threshold number of interacting segments. Unlike the evaluation of base pair prediction or sRNA target prediction performance, there was no difference in performance between Turner’s and Andronescu’s parameters. Finally, to obtain the appropriate parameter T2, we investigated the influence of T2 on TINCR target prediction accuracy (Supplementary Tables S10–S11). Although lower T2 values cause faster computation speed with lower prediction accuracy, the accuracy was robust to the T2 parameter setting. We set T2 to -6 and -4 when the energy models were Turner’s and Andronescu’s parameters, respectively.
Fig. 3.

The performance of human lncRNA TINCR target prediction. The x-axis represents the threshold number of interacting segments in the positive data. The y-axis represents the AUROC score.
3.4 Evaluation of running time
We finally evaluated the computational speed of RIblast by comparing its run time with the times required for IntaRNA, RNAplex-a and Terai et al. pipeline. We excluded joint secondary structure prediction using RactIP (Kato et al., 2010) in the Terai et al. pipeline (Terai et al., 2016) because this step does not affect prediction accuracy. The calculation time for RNAplex-a included the run time of accessibility calculation by RNAplfold (Bernhart et al., 2006). The query and target sequences were randomly selected from human lncRNAs and mRNAs in Gencode version 24, respectively (Harrow et al., 2012). Then, all-to-all interaction predictions between query and target sequences were conducted. The computation was performed on an Intel(R) Xeon E5 2670 2.6GHz CPU with 4 GB of memory. Table 3 and Figure 4 show the computational times depended on the dataset size for each software tool. In all cases, RIblast was much faster than the other programs. As the dataset size increased, the speed advantage over the other programs became quite large. In particular, when the dataset consisted of 500 lnRNAs and mRNAs, RIblast was 64-fold and 73-fold and faster than the Terai et al. pipeline and RNAplex-a, respectively.
Table 3.
The results of the run time evaluation on partial human lncRNA and mRNA datasets
| The number of lncRNAs and mRNAs | |||||
|---|---|---|---|---|---|
| Program | 5 × 5 | 10 × 10 | 50 × 50 | 100 × 100 | 500 × 500 |
| IntaRNA | 59 m 04 s (126.6) | 3 h 30 m 17 s (252.3) | − (-) | − (-) | − (-) |
| RNAplex-a | 2 m 34 s (5.5) | 10 m 37 s (12.8) | 4 h 20 m 42 s (47.0) | 17 h 56 m (63.4) | 19 d 20 h 53 m (74.1) |
| Terai et al. pipeline | 1 m 02 s (2.2) | 3 m 08 s (3.8) | 2 h 26 m 43 s (26.4) | 14 h 50 m (52.4) | 17 d 09 h 44 m (64.9) |
| RIblast | 28 s (1.0) | 50 s (1.0) | 5 m 33 s (1.0) | 17 m (1.0) | 6 h 26 m (1.0) |
Note: The columns correspond to the number of lncRNAs assessed in the dataset. The rows indicate the run times and the run time ratio of each program to RIblast. The symbol ‘−’ indicates that we did not investigate the computational speed for a particular combination of dataset size and program because the calculation time was prohibitively long.
Fig. 4.

The results of the run time evaluation on partial human lncRNA and mRNA datasets. The x-axis represents the number of lncRNAs and mRNAs. The y-axis represents the runtime ratio of each program to RIblast. The dotted line, the dashed line and the solid line represent the performances of IntaRNA, RNAplex-a and RIblast, respectively
4 Discussion
In this study, we developed RIblast, which is an RNA–RNA interaction prediction algorithm based on the seed-and-extension approach. RIblast is the fastest software that can be applied to large-sclae lncRNA datasets.
We used an interaction energy cutoff to exclude likely incorrect predictions, but this method may be highly arbitrary. As such, we should determine the reliability of the predicted interactions based on a statistical score like the e-values generated by BLAST. Rehmsmeier et al. developed a calculation method for the statistical significance of predicted RNA–RNA interactions (Rehmsmeier et al., 2004). However, their calculation method cannot be applied to our software directly because the method did not consider the effect of accessible energies. Therefore, we need to develop an e-value calculation method for RIblast’s predicted interactions.
While the previous benchmarking paper of bacterial sRNA target prediction found that IntaRNA shows the superior performance to RNAplex (Pain et al., 2015), these two programs show almost the same performances in our evaluation. This difference of evaluation may be caused by the difference of some experimental conditions such as the benchmark dataset or the version of software. Specifically, the difference of energy parameter can cause large difference of evaluation results. The previous paper uses Turner’s 1999 parameter for the energy parameter in RNAplex, but we use Turner’s 2004 parameter for that. Therefore, RNAplex in our evaluation should show high performance than RNAplex in the previous paper.
Although Hajiaghayi et al. reported that the accuracy of RNA secondary structure prediction with Andronescu’s parameter outperforms those that use other energy parameters (Hajiaghayi et al., 2012), we provide the first report that Andronescu’s parameter also delivers superior performances compared with Turner’s parameter in small RNA–RNA interaction predictions. Currently, major miRNA or snoRNA target prediction tools (Agarwal et al., 2015; Betel et al., 2010; Tafer et al., 2010) utilize Turner’s parameter, and thus the application of Andronescu’s parameter to these programs may improve the accuracies.
RIblast efficiently predicts RNA–RNA interaction, but further acceleration is an essential task. Considering that the seed-and-extension approach contributes to the acceleration of RNA–RNA interaction predictions, other acceleration techniques in sequence homology search may be effective for RNA–RNA interaction predictions. Specifically, parallelization is a promising technique. At present, many parallelization methods (Rognes, 2011; Suzuki et al., 2012, 2016) have been proposed for sequence homology search and have successfully speed up calculation.
While typical mRNAs tend to be localized in the cytoplasm, typical lncRNAs tend to be localized in the nucleus (Ulitsky and Bartel, 2013). This tendency may suggest that lncRNAs exert their functions by interacting with pre-mRNAs (Engreitz et al., 2014). Thus, interaction prediction between lncRNAs and pre-mRNAs is a fascinating research topic, but the current RIblast cannot be applied to this task. This is because the accessible energy calculation of pre-mRNAs by the Raccess algorithm is computationally difficult for too long RNA sequences. For this purpose, we will integrate the ParasoR (Kawaguchi and Kiryu, 2016), which can calculate accessible energies for quite long RNAs, with RIblast.
The evolution of lncRNA is a hot topic (Ulitsky, 2016). Although the majority of lncRNAs are lineage-specific, a thousand human lncRNAs have homologs (Hezroni et al., 2015). In addition, Ngueyn et al. revealed that experimentally validated RNA–RNA interaction sites are evolutionarily conserved (Nguyen et al., 2016). These results suggest that the interaction relationships between lncRNA and RNA are widely conserved among species. We aim to validate this hypothesis by comparing RIblast-based lncRNA interactome networks between species.
Supplementary Material
Acknowledgements
We thank Dr Junichi Iwakiri and Ms Risa Kawaguchi for critically reading the manuscript, and Dr Kun Qu and Dr Paul A. Khavari for providing the TINCR RIA-seq dataset.
Funding
This work was supported by the Japan Society for the Promotion of Science [grant numbers JP16J00129 to T.F. and JP16H05879 to M.H.]
Conflict of Interest: none declared.
References
- Abdelmohsen K. et al. (2014) 7SL RNA represses p53 translation by competing with HuR. Nucleic Acids Res., 42, 10099–10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal V. et al. (2015) Predicting effective microRNA target sites in mammalian mRNAs. Elife, 4, e05005.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F. et al. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Andronescu M. et al. (2010) Computational approaches for RNA energy parameter estimation. RNA, 16, 2304–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aw J.G.A. et al. (2016) In vivo mapping of eukaryotic RNA interactomes reveals principles of higher-order organization and regulation. Mol. Cell, 62, 603–617. [DOI] [PubMed] [Google Scholar]
- Bernhart S.H. et al. (2006) Local RNA base pairing probabilities in large sequences. Bioinformatics, 22, 614–615. [DOI] [PubMed] [Google Scholar]
- Betel D. et al. (2010) Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol., 11, R90.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A. et al. (2008) IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics, 24, 2849–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili M.N. et al. (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes. Dev., 25, 1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon M. et al. (2015) Paradigm shifts in genomics through the FANTOM projects. Mamm. Genome, 26, 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz J.M. et al. (2014) RNA–RNA interactions enable specific targeting of noncoding RNAs to nascent pre-mRNAs and chromatin sites. Cell, 159, 188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith M.C. (2010) A new repeat-masking method enables specific detection of homologous sequences. Nucleic Acids Res., 39, e23.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C., Maquat L.E. (2011) lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with UTRs via Alu elements. Nature, 470, 284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajiaghayi M. et al. (2012) Analysis of energy-based algorithms for RNA secondary structure prediction. BMC Bioinformatics, 13, 22.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J. et al. (2012) GENCODE: the reference human genome annotation for the ENCODE project. Genome Res., 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hezroni H. et al. (2015) Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep., 11, 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose T. et al. (2014) Elements and machinery of non-coding RNAs: toward their taxonomy. EMBO Rep., 15, 489–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer M.K. et al. (2015) The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet., 47, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y. et al. (2010) RactIP: fast and accurate prediction of RNA–RNA interaction using integer programming. Bioinformatics, 26, i460–i466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi R., Kiryu H. (2016) Parallel computation of genome-scale RNA secondary structure to detect structural constraints on human genome. BMC Bioinformatics, 17, 203.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiełbasa S.M. et al. (2011) Adaptive seeds tame genomic sequence comparison. Genome Res., 21, 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino T. et al. (2010) Noncoding RNA Gas5 is a growth arrest and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal., 3, ra8.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu H. et al. (2011) A detailed investigation of accessibilities around target sites of siRNAs and miRNAs. Bioinformatics, 27, 1788–1797. [DOI] [PubMed] [Google Scholar]
- Kretz M. et al. (2013) Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature, 493, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai D., Meyer I.M. (2016) A comprehensive comparison of general RNA–RNA interaction prediction methods. Nucleic Acids Res., 44, e61.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z. et al. (2016) RNA duplex map in living cells reveals higher-order transcriptome structure. Cell, 165, 1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews D.H. et al. (1999) Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol., 288, 911–940. [DOI] [PubMed] [Google Scholar]
- Mathews D.H. et al. (2004) Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. U. S. A., 101, 7287–7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaskill J.S. (1990) The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers, 29, 1105–1119. [DOI] [PubMed] [Google Scholar]
- Mückstein U. et al. (2006) Thermodynamics of RNA–RNA binding. Bioinformatics, 22, 1177–1182. [DOI] [PubMed] [Google Scholar]
- Naganuma T., Hirose T. (2013) Paraspeckle formation during the biogenesis of long non-coding RNAs. RNA Biol., 10, 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki E.P., Eddy S.R. (2013) Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics, 29, 2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T.C. et al. (2016) Mapping RNA–RNA interactome and RNA structure in vivo by MARIO. Nat. Commun., 7, 12023.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nong G. et al. (2011) Two efficient algorithms for linear time suffix array construction. IEEE Trans. Comput., 60, 1471–1484. [Google Scholar]
- Pain A. et al. (2015) An assessment of bacterial small RNA target prediction programs. RNA Biol., 12, 509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehmsmeier M. et al. (2004) Fast and effective prediction of microRNA/target duplexes. RNA, 10, 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter A., Backofen R. (2012) Accessibility and conservation: general features of bacterial small RNA–mRNA interactions? RNA Biol., 9, 954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X. et al. (2011) pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics, 12, 77.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognes T. (2011) Faster Smith-Waterman database searches with inter-sequence SIMD parallelisation. BMC Bioinformatics, 12, 221.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma E. et al. (2016) Global mapping of human RNA–RNA interactions. Mol. Cell, 62, 618–626. [DOI] [PubMed] [Google Scholar]
- Shrestha A.M.S. et al. (2014) A bioinformaticianś guide to the forefront of suffix array construction algorithms. Brief. Bioinf., 15, 138–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S. et al. (2012) GHOSTM: a GPU-accelerated homology search tool for metagenomics. PloS One, 7, e36060.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S. et al. (2014) GHOSTX: an improved sequence homology search algorithm using a query suffix array and a database suffix array. PloS One, 9, e103833.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S. et al. (2016) GPU-acceleration of sequence homology searches with database subsequence clustering. PLoS One, 11, e0157338.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szcześniak M.W., Makałowska I. (2016) lncRNA–RNA interactions across the human transcriptome. PloS One, 11, e0150353.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafer H. et al. (2010) RNAsnoop: efficient target prediction for H/ACA snoRNAs. Bioinformatics, 26, 610–616. [DOI] [PubMed] [Google Scholar]
- Tafer H. et al. (2011) Fast accessibility-based prediction of RNA–RNA interactions. Bioinformatics, 27, 1934–1940. [DOI] [PubMed] [Google Scholar]
- Tafer H., Hofacker I.L. (2008) RNAplex: a fast tool for RNA–RNA interaction search. Bioinformatics, 24, 2657–2663. [DOI] [PubMed] [Google Scholar]
- Terai G. et al. (2016) Comprehensive prediction of lncRNA–RNA interactions in human transcriptome. BMC Genomics, 17, (1–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjaden B. et al. (2006) Target prediction for small, noncoding RNAs in bacteria. Nucleic Acids Res., 34, 2791–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I. (2016) Evolution to the rescue: using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet., 17, 601–615. [DOI] [PubMed] [Google Scholar]
- Ulitsky I., Bartel D.P. (2013) lincRNAs: genomics, evolution, and mechanisms. Cell, 154, 26–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski O., Chang H.Y. (2011) Long noncoding RNAs and human disease. Trends Cell Biol., 21, 354–361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.