

Abstract
Motivation
Computational methods for phosphorylation site prediction play important roles in protein function studies and experimental design. Most existing methods are based on feature extraction, which may result in incomplete or biased features. Deep learning as the cutting-edge machine learning method has the ability to automatically discover complex representations of phosphorylation patterns from the raw sequences, and hence it provides a powerful tool for improvement of phosphorylation site prediction.
Results
We present MusiteDeep, the first deep-learning framework for predicting general and kinase-specific phosphorylation sites. MusiteDeep takes raw sequence data as input and uses convolutional neural networks with a novel two-dimensional attention mechanism. It achieves over a 50% relative improvement in the area under the precision-recall curve in general phosphorylation site prediction and obtains competitive results in kinase-specific prediction compared to other well-known tools on the benchmark data.
Availability and implementation
MusiteDeep is provided as an open-source tool available at https://github.com/duolinwang/MusiteDeep.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Post-translational modification (PTM) generally refers to the addition of a functional group covalently to a protein as in phosphorylation, acetylation, methylation or ubiquitination. It is a key mechanism to increase proteomic diversity (Prabakaran et al., 2012). The most studied PTM is phosphorylation on serine and threonine. It has been estimated that one-third of mammalian proteins may be phosphorylated (Sefton and Shenolikar, 2001). This modification plays a vital role in intracellular signal transduction, and is involved in regulating cell cycle progression, differentiation, transformation, development, peptide hormone response and adaptation (Cohen, 2002; Hubbard and Cohen, 1993; Pawson and Scott, 1997). Therefore, identifying and understanding phosphorylation are critical in cell biology and diseases. In contrast to conventional experimental methods, which are expensive and time-consuming, computational identification of phosphorylation provides an alternative strategy for proteome-wide annotation and large-scale experimental design with advantages of fast speed and low cost.
There are two categories of phosphorylation prediction tools: tools for general phosphorylation site prediction and tools for kinase-specific phosphorylation site prediction. Given protein sequences, the first category of tools predicts sites that can be phosphorylated, such as the DISPHOS (Iakoucheva et al., 2004), ModPred (Pejaver et al., 2014) and AMS 4.0 (Plewczynski et al., 2012), while the second category of tools predicts sites that can be phosphorylated by a specific kinase, such as KinasePhos 2.0 (Wong et al., 2007), the series of GPS methods (Liu et al., 2011; Xue et al., 2011, 2008; Zhao et al., 2014) and NetPhosK 1.0 (Blom et al., 2004). Most of the existing methods share a common strategy that can be summarized as two main steps: (1) to extract features from the original sequence or other domain knowledge, which is known as ‘feature engineering’ in machine learning; (2) to choose a machine-learning algorithm for training and prediction by using the extracted features. Other than different machine-learning algorithms, such as support vector machine (SVM) and random forest, the success of the prediction largely depends on the effective feature extraction and protein representation. Different features are extracted from protein sequences or domain knowledge: such as the amino acid substitution matrix used in the series of GPS methods (Liu et al., 2011; Xue et al., 2010, 2008; Zhao et al., 2014), the three sets of features (k nearest neighbor scores, disorder scores and amino acid frequencies) used in the Musite (Gao et al., 2010; Yao et al., 2012), and the physicochemical properties used in (Plewczynski et al., 2012; Yao et al., 2015). Although these features helped many existing methods achieve good performance on phosphorylation sites predictions, there is limitation of feature engineering, which requires human design that may result in incomplete or biased features.
One promising and attractive solution for such a problem is the deep-learning approach. Compared with conventional machine-learning techniques, deep-learning methods allow their computational models to be fed with raw data and automatically discover the complex representations needed for classification. There has been a growing interest in applying deep-learning methods for biological sequence analysis. For example, convolutional neural network (CNN) (LeCun et al., 2010) was used in DeepBind for predicting sequence specificities of DNA- and RNA-binding proteins (Alipanahi et al., 2015); a hybrid of CNN and bidirectional long short-term memory network (BLSTM) (Graves and Schmidhuber, 2005) was used in DanQ for predicting properties and functions of DNA sequences (Quang and Xie, 2016). These approaches by using only the raw sequence have achieved significantly better performance than previous machine learning methods. However, there is no deep-learning framework for PTM prediction and it is highly nontrivial to apply a deep-learning framework for a new biology problem, especially to address the kinase-specific prediction problem by deep learning using small-sample data. Currently there are only ∼6000 phosphorylation sites with known catalytic enzyme information in the public databases (http://phospho.elm.eu.org/).
Here we present MusiteDeep, a novel deep-learning framework for general and kinase-specific phosphorylation site prediction. MusiteDeep is a update of our previous tool Musite (Gao et al., 2010) with a novel deep-learning method. Different from existing phosphorylation site prediction methods, MusiteDeep predicts directly from the raw protein sequence avoiding feature engineering. To address the small-sample problem in kinase-specific site prediction, MusiteDeep utilizes the concept of transfer learning to fine-tune the kinase-specific models from the pre-trained general phosphorylation model. By augmenting the convolutional network with an attention mechanism on both sequence dimension and feature map dimension, a biologically interpretable representation of protein sequence is obtained, by which protein fragments can be clustered into biologically meaningful groups. To our best knowledge, MusiteDeep is the first deep-learning framework for general and kinase-specific phosphorylation site prediction. MusiteDeep is provided as an open source tool and implemented in Python at https://github.com/duolinwang/MusiteDeep. At present, MusiteDeep only provides predictions of human phosphorylation sites; however, MusiteDeep also provides customized model training that enables users to train other PTM prediction models of any species by using their own training datasets.
2 Materials and methods
2.1 Benchmark dataset
For general phosphorylation site prediction, phosphorylation data for Homo sapiens were collected from UniProt/Swiss-Prot (Bairoch et al., 2005). Phosphorylation sites on serine (S), threonine (T) or tyrosine (Y) annotated by UniProt/Swiss-Prot were used as positive data, while the same amino acid excluding annotated phosphorylation sites from the same proteins were regarded as the negative data. For kinase-specific phosphorylation site prediction, the protein sequences were also collected from UniProt/Swiss-Prot, while the annotations of human kinases were extracted from RegPhos (Lee et al., 2011), which contains information of kinase-specific phosphorylation sites from six phosphorylation-associated resources such as Phospho.ELM (Dinkel et al., 2011), PhosphoSitePlus (Hornbeck et al., 2012), PHOSIDA (Gnad et al., 2011), SysPTM (Li et al., 2009), HPRD (http://www.hprd.org/) and UniProtKB/Swiss-Prot. We extracted kinase family data from RegPhos according to the categorization used in (Xue et al., 2008). For each kinase family, we trained a specific prediction model and only the sites annotated by the specific kinase family were used as positive data, whereas all other residues of the same types (serine, threonine or tyrosine) in the same substrates were used as negative data.
To compare with different deep-learning architectures and other existing phosphorylation site prediction methods, we used independent sets for training and testing. To avoid any overlap of the testing set with any training processes of other tools, we used the recently created data as the testing set. In particular, the annotation entries that were created after the year 2008 were used as the testing set and the remaining annotation was used as the training set. We constructed non-redundant training and testing set, and removed any protein sequences in the training set having high similarities with the testing set by using Blastp (2.2.25) (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/) with a sequence identity threshold of 50%. Table 1 summarizes the collected phosphorylation data used in this study. Because a serine/threonine-specific kinase typically can phosphorylate both serine and threonine residues (Shi, 2009), we combined phosphoserine and phosphothreonine sites in the data collection and trained one model for both serine and threonine sites.
Table 1.
Phosphorylation data collected in this study
| General phosphorylation sites | ||||
| Homo sapiens | Data source | Residue type | # of positive sites | # of negative sites |
| Training | Swiss-Prot | S/T | 34 401 | 677 157 |
| Y | 1883 | 128 007 | ||
| Testing | Swiss-Prot | S/T | 2074 | 60 880 |
| Y | 47 | 9174 | ||
| Kinase-specific phosphorylation sites | ||||
| Kinase family | Data source | Residue type | # of positive sites | # of negative sites |
| CDK | Swiss-Prot | S/T | 315 | 15 878 |
| RegPhos | ||||
| PKA | Swiss-Prot | S/T | 354 | 20 321 |
| RegPhos | ||||
| CK2 | Swiss-Prot | S/T | 303 | 9687 |
| RegPhos | ||||
| MAPK | Swiss-Prot | S/T | 399 | 16 572 |
| RegPhos | ||||
| PKC | Swiss-Prot | S/T | 456 | 19 779 |
| RegPhos | ||||
2.2 Methods
Figure 1 summarizes our deep-learning framework for both general and kinase-specific phosphorylation site prediction.
Fig. 1.
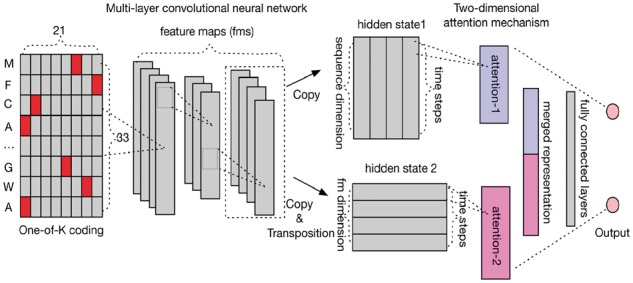
Deep-learning architecture of MusiteDeep. The input layer is the one-of-K coding of a 33-residue protein fragment centered at the prediction site. Multi-layer CNN is used as the feature extractor but no pooling layers are used. The last hidden state of multi-layer CNN is copied twice, where one directly inputs into the attention mechanism (attention-1) and the other first trans-positioned and then inputs into another attention mechanism (attention-2). The output of the two attention mechanisms is combined and input into the fully connected neural network layers. The final layer is a single neural network layer with the softmax output
2.2.1 General phosphorylation site prediction
Given protein sequences, the general phosphorylation site prediction predicts sites that can be phosphorylated by serine/threonine or tyrosine. It can be formulated as a binary classification problem, namely, each potential phosphorylation site can be classified as either a phosphorylation site or a non-phosphorylation site. In contrast to other traditional phosphorylation prediction tools, our method takes the raw sequence as input exclusively. Given a protein sequence, a peptide of 33 residues centered at the potential phosphorylation site is extracted. We choose the window size of 33 (with the potential phosphorylation site and 16 residues at each side), since it is long enough compared with other tools: Musite uses a window size of 27, NetPhos3.1 uses up to 33 and GPS2.0 uses 15. The protein fragments were coded by one-of-K coding, i.e. a K-dimensional vector with value 1 at the index corresponding to the amino acid in the protein sequence, and with 0 at all other positions. For unknown or non-standard amino acids, for example amino acids with abbreviation ‘X’, 0.05 was assigned to all positions. Since there are 20 common amino acids, K is set to 20. However, when the left part or right part for a potential phosphorylation site is not as long as the window size, a dash (‘-’) was given and treated at one additional amino acid. Thus, we actually used the one-of-21 coding.
In the deep-learning architecture of MusiteDeep, the multi-layer CNN encodes an input protein sequence into a fixed two-dimensional hidden state (as shown in Fig. 1). Then, one copy of the two-dimensional hidden state (hidden state 1) is input into attention-1, and another copy of the hidden state (hidden state 2) is trans-positioned and input into attention-2. The implementation of the attention-based decoder is inspired by (Bahdanau et al., 2014), which extended a basic RNN encoder-decoder architecture by introducing an attention mechanism to neural machine translation. By augmenting with the attention mechanism, it allows their model to automatically search for important positions to learn a soft transformation between the input and output sequences. We modified their approach by (i) replacing the RNN encoder with a multi-layer CNN; (ii) providing a two-dimensional attention mechanism on both sequence dimension and feature map dimension; and (iii) changing the RNN decoder into a feedforward neural network to generate a single representation vector. The two independent attention mechanisms built on top of the multi-layer CNN were designed to quantitatively estimate the contributions of each element on both sequence and feature map dimensions and finally to obtain a merged soft-weighted representation of protein sequence. These two attention mechanisms work in the same way. Taking the attention-2 as an example, the graphical illustration of the attention-based decoder is shown in Figure 2. The output H' is a weighted sum of the hidden states:
| (1) |
where ht is a hidden state (hidden state 2) from the multi-layer CNN, t = 1, 2,…T (T = 200 for attention-2). is the softmax weight of each hidden state ht, which is formulated by:
| (2) |
| (3) |
where et is generated from the hidden state ht by a feedforward neural network function (3), W is an attention hidden matrix, U is an attention hidden vector and represents the linear activation function. The attention-based decoder decodes the two copies of the hidden state (ht) into a merged representation (H'), which is used as input to the following fully connected neural network layers and the softmax output layer (Fig. 1).
Fig. 2.

Graphical illustration of the attention-based decoder on the feature map dimension. It decodes the feature maps (h1, h2…, hT) from the last hidden state of multi-layer CNN into a single target representation (H'). All the parameters within each layer are scaled between 0 and 1. The grey scale is shown according to the values of parameters
Different deep-learning architectures were explored in this work to compare them with the proposed architecture, including one-layer CNN architecture as in DeepBind (Alipanahi et al., 2015), the hybrid CNN and LSTM as in DanQ (Quang and Xie, 2016) and a multi-layer RNN (multi-layer LSTM) with a deep neural network. The exact same hyperparameters in their models will not be applicable to our problem since they were designed for different applications. Hence, we need to choose optimal hyperparameters for these architectures to fit our task. The selection of hyperparameters in deep learning could be a tedious task. After the main architecture of each model has been decided, we applied one of Bayesian optimization methods (Snoek et al., 2012) to guide the selection of the hyper-parameters for these architectures; specifically, the Spearmint package (https://github.com/HIPS/Spearmint) was used. Bayesian optimization is an automatic tuning approach for optimizing the performance of a given learning algorithm by modeling the algorithm’s generalization performance through sampling the hyperparameters from a Gaussian process. For each model, a small subset of training data was used to train different models with different hyperparameters suggested iteratively by the Bayesian optimization. After enough iterations, the best performance will not improve and the optimal hyperparameters which get the best performance were used in the final model. All the details about the architectures and parameters for these models can be found in Section 1 in Supplementary Material.
2.2.2 Kinase-specific phosphorylation site prediction
For kinase-specific phosphorylation prediction, the challenge is how to train a highly accurate generalized model with small sample of kinase-specific training data. According to the RegPhos database, only kinase families, CDK, PKA, CK2, MAPK and PKC have more than 100 known phosphorylation sites. To compare with other kinase-specific prediction tools, we focused on these five big kinase families. Because the size of the training data is much smaller than that of the parameters of the model, it is very easy to overfit the training data. As shown in (Yosinski et al., 2015), the multi-layer CNN in the general model can be regarded as feature extraction layers and easily generalized to other datasets for transfer learning (Caruana, 1995). Instead of frozen all the transferred layers, fine-tuning the transferred layers improves generalization (Yosinski et al., 2014). Therefore, to solve the small-sample problem of kinase-specific phosphorylation site prediction, we trained a base network on the general phosphorylation data and then transferred the whole layers except for the last output layer of the base network to kinase-specific models. At last we fine-tuned the whole network using the kinase-specific data. In this way, the kinase-specific models learn from the general feature representations and the overfitting problem is relieved. This approach has successfully been applied to a number of image classification problems and demonstrated good classification performance by using small-sample data (Esteva et al., 2017; Zeiler and Fergus, 2014).
2.2.3 Bootstrapping
The sizes of positive and negative data in this study were highly imbalanced. The size of negative data was more than two orders of magnitude larger than the positive data as shown in Table 1. To address this issue, we extended our deep-learning framework with a bootstrapping method. The realization of the bootstrapping is similar to (Yan et al., 2015), but was implemented in a different way. Given the training samples from positive and negative datasets, the bootstrap procedure is as follows. Let n and p be the number of negative and positive samples in the imbalanced training dataset with n ≫ p. For each bootstrap iteration, the same number (Sp) of samples of positive data and negative data were selected and one model was trained on this balanced dataset. To go through all the negative data, the n negative samples were divided into N bins according to Sp; therefore, . Totally, N times of bootstrap iterations will be trained to generate one classifier. This procedure will be repeated for m times (m = 5 by default) and m classifiers will be generated. When predicting for a query site, the average output calculated by the m classifiers was taken as the final prediction. For each bootstrap iteration i, early stop strategy (Yao et al., 2007) was used to control the number of epochs for each bootstrap training (patience = 20 by default).
3 Results
Although MusiteDeep is realized based on deep learning, a method well-known for being time-consuming in training, the prediction time is actually less than other feature-extraction based tools due to saving of feature calculations. For example, the running time was less than 5 minutes for predicting general phosphorylation sites on 1000 protein sequences using a 8 GB GeForce GTX 1080 machine, although it took nearly 24 h to train the model. It is worth noting that the training process is only needed once for a prediction model.
3.1 Evaluating the performance of MusiteDeep for general phosphorylation site prediction
To evaluate the performance of MusiteDeep against Musite and other deep-learning architectures described in Section 2.2.1, a five-fold cross-validation was performed. For all the methods, the same training sets and same testing sets were used. The average ROC (receiver operating characteristic) and precision-recall curves of the five tests were plotted in Figure 3. It shows that all the deep-learning architectures outperformed the feature-extraction based tool Musite. The performance of MusiteDeep was better than other deep-learning architectures. By using the L1 regularization and dropout (Srivastava et al., 2014), MusiteDeep relieved the overfitting by showing very similar performance in the training and validation processes for long epochs, as shown in Supplementary Figure S1 in Supplementary Material. Furthermore, the early stop mechanism monitored on the cost of validation set broke off the training process when the cost of validation set did not decrease for some epochs.
Fig. 3.
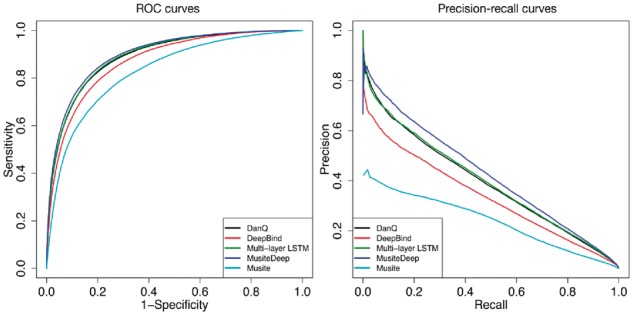
ROC and precision-recall curves comparing MusiteDeep with Musite and other deep-learning architectures by five-fold cross-validation
We compared MusiteDeep with several well-known and publicly available tools for general phosphorylation site prediction, such as the original Musite, NetPhos 3.1 (http://www.cbs.dtu.dk/services/NetPhos-3.1/), ModPred (Pejaver et al., 2014) and one recently published tool PhosPred-RF (Wei et al., 2017). We used one training set to train our model and predicted on one independent testing set, as described in Table 1. Because these tools did not provide customized model training, we used their tools and their pre-trained model as is to do prediction for the testing set. We also trained MusiteDeep on one strict training set in which sequences have no more than 10% identity with the testing set (removed 1590 additional proteins from the training set). By taking different thresholds according to the scores provided by each method, the ROC curves and the precision-recall curves were plotted in Figure 4 and used for calculating the AUC (area under the ROC curves) and the mean precision (area under the precision-recall curves). Figure 4 shows that MusiteDeep had much better performance than other methods in both ROC and precision-recall estimators. The performance using the training set of 10% identity threshold had a very similar performance to that of the original training set, which demonstrates that the redundancy of sequences in the training set had little effect on its performance of the testing set in this case.
Fig. 4.
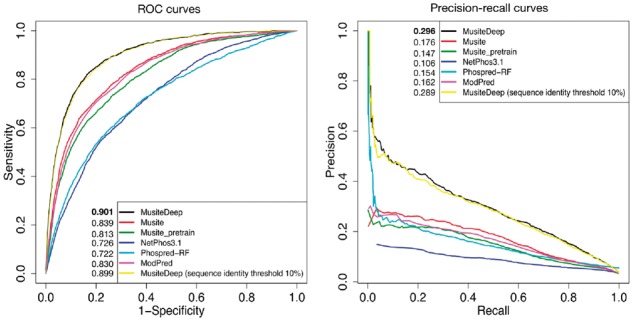
ROC and precision-recall curves comparing MusiteDeep with other well-known general phosphorylation site prediction tools on the testing set
We evaluated the contribution of different strategies that affect the performance of MusiteDeep by five-fold cross-validation. We compared the performance of MusiteDeep with and without different attentions, including using a two-dimensional attention, no attention and two single-dimensional attentions by training on the same balanced training set. Supplementary Figure S2 shows that the two-dimensional attention obtained the best performance (average AUC = 0.886, average mean precision = 0.372), while no attention got the second best results (average AUC = 0.884, average mean precision = 0.363). Interestingly, no attention performed better than the single-dimensional attention. This result is similar to (Sønderby et al., 2015), in which the regular LSTM performed better than the LSTM with an attention mechanism on the sequence dimension. This result may be because that all the information of the previous hidden state was kept for the next layers with no attention, while the information of either dimension would be weaken through the weighted sum operation (Equation (2)) by the single dimensional attention mechanism. The two-dimensional attention obtained the best performance probably because it got more information back through attention mechanisms on both dimensions. We also compared the performance of MusiteDeep with and without bootstrapping. The bootstrapping version contained 10-ensemble classifiers. It was compared with two versions of non-bootstrapping. One was trained directly on the unbalanced datasets (unbalanced training). The other was trained on the balanced datasets (balanced training), which selected 1: 1 positive and negative training data randomly for one cross-validation run. Supplementary Figure S3 shows that the bootstrapping strategy with 10-ensemble classifiers improved MusiteDeep from 0.886 (and 0.372) of the balanced training and 0.888 (and 0.388) of the unbalanced training without bootstrapping to 0.897 (and 0.404) with bootstrapping in terms of AUC (and mean precision).
3.2 Comparison with other kinase-specific phosphorylation site prediction tools
We compared MusiteDeep with several well-known tools for kinase-specific prediction, including Musite, NetPhos3.1, GPS 2.0 (Xue et al., 2008) and GPS 3.0 (http://gps.biocuckoo.org/). The five big kinase families, CDK, PKA, CK2, MAPK and PKC were used for the comparison. Since known kinase-specific sites were limited, we could not just separate one training dataset from one testing dataset according to their creation dates as what we did for the general prediction. Thus, to evaluate the performance for each of the five kinase families, a five-fold cross-validation test was performed. Each time, the four-fifths of the data were used to train MusiteDeep, and the remaining one-fifth of the data were used as the testing set for MusiteDeep, and other tools by using their pre-trained models to do the prediction. During the training process of MusiteDeep, a separate validation set was extracted from the four-fifth of the data. Furthermore, to make sure there was no overlap between the testing set and the data used during any training procedure, we used separate pre-trained general models which were trained from data without including any proteins of the five kinase annotations. Some of the testing proteins might have been trained in other tools, and thus the performance could be biased in favor of them. In the practical application, we can use all the available sites for each kinase family to train the model and the performance is expected to be improved. The ROC curves and the precision-recall curves were plotted for kinase families CDK and PKA in Figure 5, and for CK2, MAPK and PKC in Supplementary Figure S4. Figure 5 and Supplementary Figure S4 show that MusiteDeep has a comparable sensitivity with other tools under certain specificity, and has superior precision than other tools in most cases.
Fig. 5.
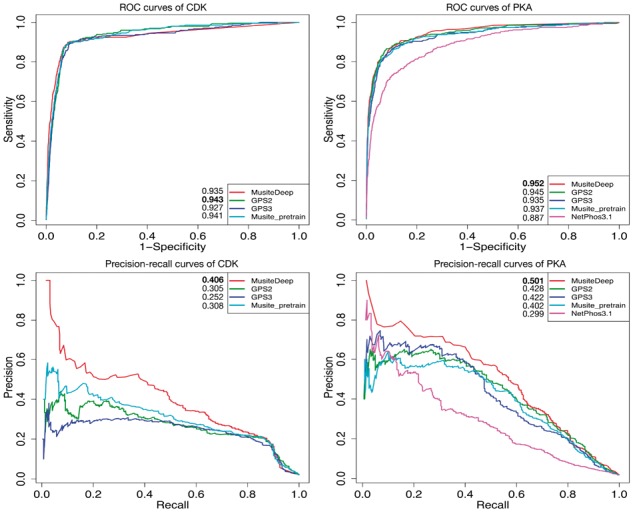
ROC and precision-recall curves comparing MusiteDeep with other well-known kinase-specific phosphorylation site prediction tools by five-fold cross-validation of CDK (left) and PKA (right)
We present all the AUCs and the mean precisions for all these five kinase families in five-fold cross-validation (Supplementary Fig. S5) to show the robustness of MusiteDeep comparing with GPS 3.0 and MusiteDeep without the transfer learning. The average AUCs, average mean precisions and the ranges (difference between the lowest and highest values) were labeled in the plots. Supplementary Figure S5 shows that MusiteDeep had comparable ranges with GPS 3.0 under the five-fold cross-validation, which means MusiteDeep had comparable robustness with traditional machine-learning method on small sample of training data. On the other hand, the MusiteDeep without transfer learning could not achieve good generalization in most cases compared with the other two, which demonstrated the important role of the transfer learning in small-sample learning. A set of novel kinase-specific phosphorylation sites with high confidence is reported in Supplementary Table S1 in Supplementary Material for future experimental verification.
3.3 Effect of transfer learning on small-sample data learning
From the concept of transfer learning, the base network trained using the general phosphorylation data is expected to generate general feature representations. Meanwhile, the attention mechanism built on top of the multi-layer CNN is designed to generate a comprehensive representation for the peptide of 33 residues by soft weights on both sequence and feature map dimension. In this section, we will show how the general feature representations from the base network can be transferred and helps the small-sample data learning for the kinase-specific phosphorylation site prediction. Using t-SNE (Maaten and Hinton, 2008), the merged representations of two dimensions (H' in Equation (1)) for the five kinase families calculated from the same general model are plotted in Figure 6a. The original one-of-K representations are presented in Figure 6b. A more detailed representation of Figure 6 with each kinase family highlighted in one plot is shown in Supplementary Figure S6. Through t-SNE, these two representations were projected to their corresponding two-dimensional space. The between/within class scatter ratio (Johnson and Wichern, 2002) is labeled in each figure. From Figure 6 and Supplementary Figure S6, it is apparent that the protein fragments from the same kinase family generally grouped together by the merged representation learned from the base network, while the original one-of-K representation could hardly distinguish these kinase families apart. Here, PKA and PKC are from the AGC group and CDK and MAPK are from the CMGC group (Xue et al., 2008); therefore, it is not surprising that they tend to overlap within each other. Based on the base networks, after we fine-tuned the network using the kinase-specific data, the specific kinase family stood out further from other kinase families, as shown in Supplementary Figure S7. Through visualizing the representations generated by the two-dimensional attention mechanism, we show that the raw protein fragments can be transformed into a biologically meaningful representation; in particular, even without the kinase labels, it could classify unknown kinase families to some extent.
Fig. 6.
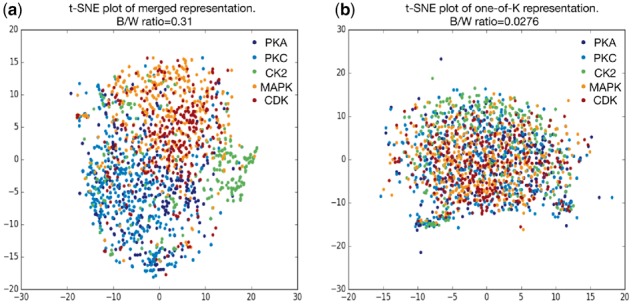
t-SNE plot of the merged representation and the original one-of-K representation
4 Discussion
In this paper, we present MusiteDeep as a novel method for both general and kinase-specific phosphorylation site prediction. MusiteDeep takes raw sequence data as input without using other tools to generate features. MusiteDeep has been demonstrated to significantly outperform some other well-known tools on benchmark datasets. Especially in the general phosphorylation site prediction, MusiteDeep achieves over a 50% relative improvement in the area under the precision-recall curve. After justifying the performance of MusiteDeep by running it with different strategies separately, we believe the superior performance is mainly due to the following three aspects: (1) our deep-learning architecture captured the underlying sequence patterns related to phosphorylation better than feature engineering-based methods; (2) the bootstrapping integrated deep-learning method utilized all the negative data in an unbiased way; and (3) the dual-attention mechanism further improved the performance. To our best knowledge, this is the first application of any deep-learning method in general or kinase-specific phosphorylation site prediction. Besides the human phosphorylation site prediction in the pre-trained model, MusiteDeep also provides customized model training that enables advanced users to train other PTM prediction models by their own data.
In this work, we have explored different deep-learning architectures, including the one-layer CNN architecture as in DeepBind, the hybrid of CNN and LSTM as in DanQ and a multi-layer RNN architecture. In both DeepBind and DanQ, the one-layer CNN is used as motif detectors, and the LSTM in DanQ is used to capture the global features of a sequence. Notice that DeepBind and DanQ were designed for different applications both taking longer DNA sequences as input, while in our study the input is just the 33-residue protein fragments, and hence these architectures are not suitable for our application, which was also demonstrated by Figure 3. Although in general, the CNNs are ideal for images that contain spatial invariant features while RNNs are ideal for text that contains sequential features, there are some successful examples of pure CNNs that obtain start-of-the-art performance when applied to sequential data (Gehring et al., 2017; Sainath et al., 2013). Comparing with the RNNs, CNNs are easier to interpret and faster to train. In addition, the multi-layer CNN has shown very powerful in extracting complex features. Therefore, in this study, the multi-layer CNN architecture was used and better performance was obtained compared with the multi-layer RNN architecture.
In most deep-learning frameworks, the attention mechanism is typically introduced in an RNN model and applied to only the input dimension; for example, only the sequence dimension can be regarded as the time steps instead of the feature dimension. However, the feature maps of a CNN model are regarded as independent feature detectors which can capture features from different aspects. By applying an independent attention mechanism on the feature map dimension, we can assign soft weight to each feature map for a specific input sequence. The two-dimensional attention mechanism built on top of the same CNN hidden state provides a more comprehensive way for representing protein sequence and also obtains superior performance than single-dimensional attention mechanisms and without attention, as shown in Supplementary Figure S2.
By visualizing the representations generated by the two-dimensional attention mechanism (Fig. 6 and Supplementary Fig. S6), the base network shows some ability to extract biological interpretable representations that tend to distinguish kinase families even without the kinase labels. Since the pre-trained base network is effective in general phosphorylation feature representations, the transfer learning strategy is powerful in transferring the base network to achieve a robust kinase-specific phosphorylation site prediction model by fine-tuning on the significantly smaller kinase-specific data (Supplementary Figs S5 and S7). We expect this architecture and framework of deep learning to be useful for other PTM predictions and even some other biological sequence analyses.
Although deep-learning method has improved the performance of classification and become a promising approach, there are still significant challenges for its applications in biological sequence analyses, especially its interpretability and biologically meaningful discoveries. In the future work, we will collaborate closely with biologists and continue to modify the architecture to make the models more interpretable and realizable to reveal the underlying identification mechanism between a kinase and its substrates.
Funding
This work was partially supported by National Institutes of Health grant R01-GM100701. The high-performance computing infrastructure is supported by the National Science Foundation under grant number CNS-1429294.
Conflict of Interest: none declared.
Supplementary Material
References
- Alipanahi B. et al. (2015) Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol., 33, 831–838. [DOI] [PubMed] [Google Scholar]
- Bahdanau D. et al. (2014) Neural machine translation by jointly learning to align and translate, arXiv preprint arXiv: 1409.0473.
- Bairoch A. et al. (2005) The Universal Protein Resource (UniProt). Nucleic Acids Res., 33, D154–D159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom N. et al. (2004) Prediction of post‐translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics, 4, 1633–1649. [DOI] [PubMed] [Google Scholar]
- Caruana R. (1995) Learning many related tasks at the same time with backpropagation. In: Advances in Neural Information Processing Systems, 7, pp. 657–664. [Google Scholar]
- Cohen P.T. (2002) Protein phosphatase 1-targeted in many directions. J. Cell Sci., 115, 241–256. [DOI] [PubMed] [Google Scholar]
- Dinkel H. et al. (2011) Phospho.ELM: a database of phosphorylation sites–update 2011. Nucleic Acids Res., 39, D261–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteva A. et al. (2017) Dermatologist-level classification of skin cancer with deep neural networks. Nature, 542, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J. et al. (2010) Musite, a tool for global prediction of general and kinase-specific phosphorylation sites. Mol. Cell. Proteomics MCP, 9, 2586–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring J. et al. (2017) Convolutional Sequence to Sequence Learning, arXiv preprint arXiv: 1705.03122.
- Gnad F. et al. (2011) PHOSIDA 2011: the posttranslational modification database. Nucleic Acids Res., 39, D253–D260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves A., Schmidhuber J. (2005) Framewise phoneme classification with bidirectional LSTM and other neural network architectures. Neural Netw. Off. J. Int. Neural Netw. Soc., 18, 602–610. [DOI] [PubMed] [Google Scholar]
- Hornbeck P.V. et al. (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res., 40, D261–D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard M.J., Cohen P. (1993) On target with a new mechanism for the regulation of protein phosphorylation. Trends Biochem. Sci., 18, 172–177. [DOI] [PubMed] [Google Scholar]
- Iakoucheva L.M. et al. (2004) The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res., 32, 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R.A., Wichern D.W. (2002) Applied Multivariate Statistical Analysis. Prentice hall Upper Saddle River, NJ. [Google Scholar]
- LeCun Y. et al. (2010) Convolutional networks and applications in vision. In: Proceedings of 2010 IEEE International Symposium on Circuits and Systems (ISCAS). IEEE, 2010, pp. 253–256.
- Lee T.Y. et al. (2011) RegPhos: a system to explore the protein kinase-substrate phosphorylation network in humans. Nucleic Acids Res., 39, D777–D787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2009) SysPTM: a systematic resource for proteomic research on post-translational modifications. Mol. Cell. Proteomics MCP, 8, 1839–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. et al. (2011) GPS-YNO2: computational prediction of tyrosine nitration sites in proteins. Mol. bioSystems, 7, 1197–1204. [DOI] [PubMed] [Google Scholar]
- Maaten L.vd., Hinton G. (2008) Visualizing data using t-SNE. J. Mach. Learn. Res., 9, 2579–2605. [Google Scholar]
- Pawson T., Scott J.D. (1997) Signaling through scaffold, anchoring, and adaptor proteins. Science (New York, N.Y.), 278, 2075–2080. [DOI] [PubMed] [Google Scholar]
- Pejaver V. et al. (2014) The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci. Publ. Protein Soc., 23, 1077–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plewczynski D. et al. (2012) AMS 4.0: consensus prediction of post-translational modifications in protein sequences. Amino Acids, 43, 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabakaran S. et al. (2012) Post-translational modification: nature's escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscipl. Rev. Syst. Biol. Med., 4, 565–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang D., Xie X. (2016) DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res., 44, e107.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainath T.N. et al. (2013) Deep convolutional neural networks for LVCSR. In: 2013 IEEE International Conference on Acoustics, Speech and Signal Processing IEEE, pp. 8614–8618.
- Sefton B.M., Shenolikar S. (2001) Overview of protein phosphorylation. Curr. Protoc. Protein Sci., Chapter 13, Unit13 11. [DOI] [PubMed]
- Shi Y. (2009) Serine/threonine phosphatases: mechanism through structure. Cell, 139, 468–484. [DOI] [PubMed] [Google Scholar]
- Snoek J. et al. (2012) Practical bayesian optimization of machine learning algorithms. In: Advances in Neural Information Processing Systems 25, pp. 2960–2968. [Google Scholar]
- Sønderby S.K. et al. (2015) Convolutional LSTM networks for subcellular localization of proteins. In: International Conference on Algorithms for Computational Biology Springer, pp. 68–80.
- Srivastava N. et al. (2014) Dropout: a simple way to prevent neural networks from overfitting. J. Mach. Learn. Res., 15, 1929–1958. [Google Scholar]
- Wei L. et al. (2017) PhosPred-RF: a novel sequence-based predictor for phosphorylation sites using sequential information only, In: IEEE Transactions on Nanobioscience. [DOI] [PubMed]
- Wong Y.H. et al. (2007) KinasePhos 2.0: a web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic Acids Res., 35, W588–W594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y. et al. (2011) GPS 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng. Des. Select. PEDS, 24, 255–260. [DOI] [PubMed] [Google Scholar]
- Xue Y. et al. (2008) GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics, 7, 1598–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y. et al. (2008) GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics MCP, 7, 1598–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y. et al. (2015) Deep learning for imbalanced multimedia data classification. In: IEEE International Symposium on Multimedia (ISM) IEEE, pp. 483–488.
- Yao Q. et al. (2012) Predicting and analyzing protein phosphorylation sites in plants using musite. Front. Plant Sci., 3, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Q. et al. (2015) Phosphorylation site prediction in plants. Methods Mol. Biol. (Clifton, N.J.), 1306, 217–228. [DOI] [PubMed] [Google Scholar]
- Yao Y. et al. (2007) On early stopping in gradient descent learning. Construct. Appr., 26, 289–315. p [Google Scholar]
- Yosinski J. et al. (2014) How transferable are features in deep neural networks? Adv. Neural Inf. Process. Syst.27, 3320–3328.
- Yosinski J. et al. (2015) Understanding neural networks through deep visualization, arXiv preprint arXiv: 1506.06579.
- Zeiler M.D., Fergus R. (2014) Visualizing and understanding convolutional networks. In: ECCV volume 8689 of Lecture Notes in Computer Science, pp. 818–833.
- Zhao Q. et al. (2014) GPS-SUMO: a tool for the prediction of sumoylation sites and SUMO-interaction motifs. Nucleic Acids Res., 42, W325–W330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.