

Abstract
Motivation
Interpreting genetic variation in noncoding regions of the genome is an important challenge for personal genome analysis. One mechanism by which noncoding single nucleotide variants (SNVs) influence downstream phenotypes is through the regulation of gene expression. Methods to predict whether or not individual SNVs are likely to regulate gene expression would aid interpretation of variants of unknown significance identified in whole-genome sequencing studies.
Results
We developed FIRE (Functional Inference of Regulators of Expression), a tool to score both noncoding and coding SNVs based on their potential to regulate the expression levels of nearby genes. FIRE consists of 23 random forests trained to recognize SNVs in cis-expression quantitative trait loci (cis-eQTLs) using a set of 92 genomic annotations as predictive features. FIRE scores discriminate cis-eQTL SNVs from non-eQTL SNVs in the training set with a cross-validated area under the receiver operating characteristic curve (AUC) of 0.807, and discriminate cis-eQTL SNVs shared across six populations of different ancestry from non-eQTL SNVs with an AUC of 0.939. FIRE scores are also predictive of cis-eQTL SNVs across a variety of tissue types.
Availability and implementation
FIRE scores for genome-wide SNVs in hg19/GRCh37 are available for download at https://sites.google.com/site/fireregulatoryvariation/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Whole-genome sequencing technologies have enabled studies of genetic variation in increasingly large cohorts of healthy and diseased individuals. However, interpreting the biological and clinical significance of the variation identified in these studies remains a critical challenge, which has motivated the development of numerous computational tools to predict variant pathogenicity or deleteriousness (Adzhubei et al., 2010; Fu et al., 2014; Huang, et al., 2017; Ioannidis et al., 2016; Ionita-Laza et al., 2016; Jagadeesh et al., 2016; Kumar et al., 2009; Kircher et al., 2014; Li et al., 2009, 2016; Quang et al., 2015; Ritchie et al., 2014; Shihab et al., 2015). While most are specific to single nucleotide variants (SNVs) in protein coding regions, an increasing number of pathogenicity prediction tools also apply to SNVs in noncoding regions of the genome (Fu et al., 2014; Huang, et al., 2017; Ionita-Laza et al., 2016; Kircher et al., 2014; Li, et al., 2016; Quang et al., 2015; Ritchie et al., 2014; Shihab et al., 2015). As almost 90% of trait-associated SNVs identified in genome-wide association studies (GWAS) are either intergenic or intronic (Hindorff et al., 2009), noncoding regions are critical for clinical variant interpretation.
The potential functional effects of noncoding SNVs differ from those of coding SNVs, which often disrupt protein function by altering amino acid sequences. While most computational tools classify SNVs by predicting their overall pathogenicity or deleteriousness, a complementary approach is to develop predictors for specific downstream functional effects, which can then be combined to provide a more detailed mechanistic hypothesis about the potential role of any SNV in disease pathogenesis. A few recent tools for functional interpretation of noncoding SNVs predict specific effects such as altered DNase I sensitivity, transcription factor binding, or histone modification (Lee et al., 2015; Zhou and Troyanskaya, 2015), while unsupervised methods are argued to classify SNVs based on their overall functional importance (Ionita-Laza et al., 2016; Lu et al., 2015). As different functions are likely to be best predicted by different features of SNVs, prediction tools tailored to individual functional effects are needed to fully interpret the significance of any observed SNV.
Regulation of gene expression is a key functional mechanism by which noncoding SNVs may achieve clinical significance. RNA sequencing studies of gene expression across individuals have enabled the identification of SNVs associated with variation in expression levels of proximal genes in the genome, termed cis-expression quantitative trait loci (cis-eQTLs) (Battle et al., 2014; GTEx Consortium, 2015; Lappalainen et al., 2013). cis-eQTL SNVs partially overlap with SNVs associated with DNase I sensitivity (Degner et al., 2012) and are also enriched for GWAS trait associations (Nicolae et al., 2010). However, cis-eQTL studies have limited power to detect associations involving SNVs with low minor allele frequencies (MAFs) or small effects, and it is presently infeasible to experimentally catalog all human SNVs that regulate gene expression. Computational prediction of the potential of all genome-wide SNVs to alter gene expression levels would aid the functional and clinical interpretation of SNVs, particularly noncoding SNVs, discovered in genome sequencing studies. As previous studies have noted enrichment of cis-eQTL SNVs in various genomic annotations, here we explore the use of such annotations as predictive features to develop FIRE (Functional Inference of Regulators of Expression), a tool that scores all SNVs in the human genome based on their relative potential to regulate the expression level of one or more nearby genes.
2 Materials and methods
FIRE is a set of 23 random forests (Breiman, 2001; Hastie et al., 2009) trained using a chromosome exclusion approach, as described below, to prevent overfitting. The positive and negative SNVs in the FIRE training set, as well as their predictive features, are also described below. A random forest is a machine learning approach in which an ensemble of classification or regression trees is fit to bootstrapped samples from a labeled training set. Each classification tree in a FIRE random forest consists of a series of splits on individual feature values that best separate positive from negative SNVs in the bootstrapped sample, measured as a decrease in node impurity or Gini index (Breiman, 2001; Hastie et al. 2009). We fit random forests using the randomForest package (Liaw and Wiener, 2002) in R (R Core Team, 2014). After training, the score for any SNV is equal to the proportion of trees in the relevant random forest that classify the SNV as positive. Higher scores reflect stronger evidence that the SNV regulates the expression level of a nearby gene.
2.1 Training sets
2.1.1 Positive set
The positive training set consisted of 57 117 autosomal cis-eQTL SNVs identified by the Geuvadis Consortium (Lappalainen et al., 2013) in lymphoblastoid cell lines (LCLs) derived from 373 individuals of European descent (EUR). This set included only those SNVs in the Geuvadis EUR analysis that were associated at a nominal P-value < 10−10 with the expression level of at least one gene with transcription start site (TSS) within 50 kb of the SNV. Alternative definitions of the positive and negative training SNVs are discussed in Supplementary Text S.4.
2.1.2 Negative set
We first assembled a set of 537 291 non-eQTL autosomal SNVs meeting the following criteria: (1) located within 50 kb of the TSS of at least one gene included in the Geuvadis EUR analysis, and thus expressed in LCLs; (2) not associated (P > 0.1) in the Geuvadis EUR analysis with the expression level of any gene with TSS within one megabase of the SNV; and (3) not associated (false discovery rate, FDR > 5%) with the expression level of any tested gene with TSS within one megabase of the SNV in three other analyses: (i) the Geuvadis analysis of cis-eQTLs in Yoruban individuals (YRI) (Lappalainen et al., 2013), (ii) the GTEx Consortium V6p analysis of cis-eQTLs in 44 different tissue types (GTEx Consortium, 2015) and (iii) a separate analysis of prostate tissue cis-eQTLs (Larson et al., 2015). These eligible negative SNVs had a MAF distribution in the Geuvadis EUR dataset skewed towards lower MAFs than the positive set SNVs, due to reduced power to detect associations for low-MAF SNVs. Therefore, we extracted a subset of the eligible negative SNVs with the same MAF distribution as the positive SNVs by defining ten MAF bins of width 0.05 and randomly including each eligible negative SNV with a probability proportional to the ratio of the number of positive to eligible negative SNVs in the relevant bin, resulting in a set of 78 643 frequency-matched negative SNVs. Finally, we matched the number of positive and negative SNVs on each chromosome by taking a random subsample of whichever set, positive or negative, had the most SNVs on that chromosome. Thus the final frequency-matched training set consisted of 43 364 positive and 43 364 negative SNVs. We also explored performance on negative sets that were matched on TSS distance distribution or restricted to the same set of genes as the positive set SNVs (Supplementary Text S.4). However, the final FIRE training set was not matched on the exact distribution of TSS distances or on other features such as GC content, as these features are informative for the variant interpretation goal of predicting whether a particular SNV is likely to regulate gene expression.
2.2 SNV features
We assembled 92 features characterizing each SNV (Supplementary Table S1), including position relative to nearby gene boundaries, conservation scores, overlapping elements from ENCODE and Ensembl, and other features used in existing predictors of noncoding SNV pathogenicity and deleteriousness, CADD (Kircher et al., 2014) and GWAVA (Ritchie et al., 2014). We used median imputation within the training set to fill in missing feature annotations for quantitative features, and considered missing values as an additional category for categorical features. The importance of each feature was calculated as the total decrease in the Gini index measure of node impurity over all splits involving that feature within each tree, averaged over all trees in the full random forest (Breiman, 2001; Hastie et al., 2009) as implemented in the randomForest package in R.
2.3 Chromosome exclusion strategy for FIRE scores
A standard approach to cross-validation is to randomly exclude SNVs from training and then evaluate performance on these excluded SNVs. In this application, however, the standard approach could over-estimate performance due to correlation between proximally located SNVs in the genome. In particular, scores for excluded SNVs that are strongly correlated with nearby SNVs included in training may be biased. Scores for non-training set SNVs in future applications might also be biased by the presence of nearby correlated SNVs in the training set. To avoid this potential bias, we implemented a chromosome exclusion strategy in which we trained a total of 23 random forests: one was trained on the full training set described in Section 2.1, and 22 random forests were trained each excluding all SNVs on one of the 22 autosomes. We then obtained the FIRE score for any given SNV from the random forest that excluded its chromosome from training. Because the full training set in Section 2.1 did not include any sex chromosome SNVs, the random forest trained on the full training set was used to score SNVs on sex chromosomes. This chromosome exclusion strategy for computing FIRE scores ensured that there was no overlap between any test and training set SNVs, because the FIRE score for any given test set SNV was computed using the random forest that excluded not only the test SNV itself, but also all other SNVs on the same chromosome, from its training set. All genome-wide FIRE scores, including all scores used throughout the manuscript, were computed using this chromosome exclusion approach.
2.4 Evaluation of FIRE
We evaluated FIRE by examining its ability to distinguish cis-eQTL from non-eQTL SNVs in the Geuvadis dataset and in several other independent cis-eQTL datasets, including a uniform analysis of cis-eQTLs from 11 different gene expression studies across seven cell types (Brown et al., 2013), cis-eQTLs identified in six different ancestries (Stranger et al., 2012) with equal numbers of sampled individuals, and cis-eQTLs identified in 44 different tissue types (GTEx Consortium, 2015). We also compared FIRE to existing tools for scoring genome-wide SNVs based on pathogenicity, deleteriousness, or other predicted functional effects; in particular, CADD (Kircher et al., 2014), DANN (Quang et al., 2015), GWAVA (Ritchie et al., 2014), DeepSEA (Zhou and Troyanskaya, 2015), Eigen and Eigen-PC (Ionita-Laza et al., 2016), fathmm-MKL (Shihab et al., 2015), FunSeq2 (Fu et al., 2014), deltaSVM (Lee et al., 2015), GenoCanyon (Lu et al., 2015), PRVCS (Li et al., 2016), cepip (Li et al., 2017) and LINSIGHT (Huang et al., 2017). Receiver operating characteristic (ROC) curves and area under the curve (AUC) estimates with confidence intervals were computed using the pROC package (Robin et al., 2011) in R.
Additional methods details are available in Supplementary Text.
3 Results
3.1 FIRE performance on training set SNVs
We first assessed FIRE’s performance on the training set using the chromosome exclusion approach (Section 2.3) to compute the FIRE score for each SNV (Fig. 1A). FIRE discriminated between the positive and negative training SNVs with an AUC of 0.807 (Fig. 1B, Supplementary Table S2). For comparison, a standard cross-validation approach using random exclusion of SNVs from the training set estimated this AUC as 0.934, illustrating the importance of a cross-validation strategy such as chromosome exclusion to avoid bias arising from correlated training and testing SNVs located close to one another on the same chromosome. Other commonly used tools for genome-wide interpretation of SNVs were less well suited to this task of discriminating between expression-associated and unassociated SNVs (Figs 1B and C, Supplementary Table S2).
Fig. 1.
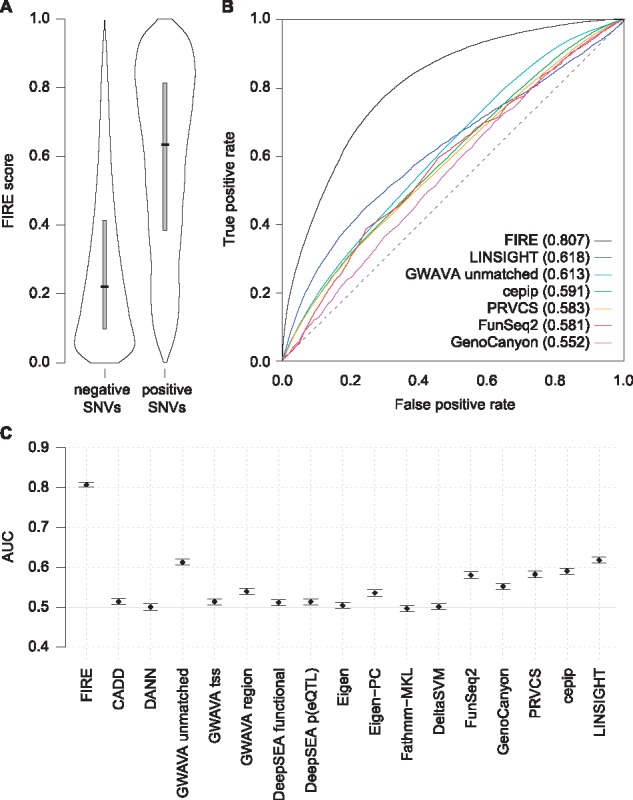
(A) FIRE score distributions for SNVs in the positive and negative training sets. FIRE scores throughout the manuscript were computed using the chromosome exclusion approach (Section 2.3). Gray boxes extend from the first to third quartiles, with a black horizontal line at the median. Means and quantiles of these distributions are also listed in Supplementary Table S3. (B) ROC curves for FIRE and six comparator tools with the highest AUCs (in parentheses) when discriminating between positive and negative SNVs in the FIRE training set. (C) AUC values for FIRE and all 16 tested comparator tools, plotted with error bars representing double the 95% confidence intervals. AUCs are also listed in Supplementary Table S2. The expected AUC for an uninformative null model is 0.5 (solid gray line)
SNVs in the negative set had a median chromosome exclusion FIRE score of 0.22 and a mean of 0.28, while the positive set SNVs had a median FIRE score of 0.63 and a mean of 0.59 (Fig. 1A, Supplementary Table S3). Higher FIRE scores reflect stronger evidence that a SNV regulates nearby gene expression levels. Score distributions were similar across most chromosomes (Supplementary Fig. S1) with several exceptions, including decreased separation of positive and negative SNVs on chromosome 6 and increased separation on chromosomes 16 and 17. We evaluated whether FIRE scores for the positive SNVs varied with the strengths of their gene expression associations in the Geuvadis EUR analysis and found a shift towards higher FIRE scores for SNVs with large effect sizes (Supplementary Fig. S2) and strong statistical significance (Supplementary Fig. S3). There was no clear dependence on Geuvadis MAF (Supplementary Figs S4 and S5), although the number of positive SNVs below 5% MAF in EUR was small due to reduced power to identify cis-eQTL associations for rare SNVs in the study population.
Random forest feature importances reflect both the predictive ability of each feature as well as correlation with other features. The most important SNV feature in the full FIRE random forest (Supplementary Fig. S6) was the B statistic, the inferred level of background selection at each position in the genome (McVicker et al., 2009), which reflects negative selection against deleterious variants and depends on local recombination rates, mutation rates and functional site density (Halligan et al., 2013; McVicker et al., 2009). Enrichment for high B values has been previously observed among trait-associated GWAS SNVs (Maher et al., 2012). Other important features included Segway chromatin element annotations (Hoffman et al., 2013) and distances to the nearest TSS, transcription end site and splice site (Supplementary Fig. S6), consistent with previous studies noting enrichment of such features among cis-eQTL SNVs (Battle et al., 2014; Brown et al., 2013; Lappalainen et al., 2013).
3.2 FIRE performance on an independent cis-eQTL dataset
We next tested the ability of FIRE to identify cis-eQTL SNVs from an independent cis-eQTL analysis of data from 11 different gene expression studies across seven cell types (Brown et al., 2013). That analysis used a stepwise regression approach to identify the most strongly associated cis-eQTL SNVs within linkage disequilibrium (LD) blocks (Supplementary Text S.1). We computed FIRE scores for these cis-eQTL SNVs identified at FDR < 1, 5, or 10% and found that FIRE could discriminate between them and non-eQTL SNVs with matched MAF distribution (drawn from the eligible non-eQTL set described in Section 2.1.2) with AUCs of 0.788, 0.770 and 0.755, respectively (Fig. 2, Supplementary Table S2). Other tools for genome-wide interpretation of SNVs all had AUC values less than 0.7 (Fig. 2, Supplementary Table S2). As described above, to ensure that none of these tested SNVs overlapped the FIRE training set, we used the chromosome exclusion approach in which not only each test SNV but also all SNVs on the same chromosome as the test SNV were excluded from the training set used to generate its FIRE score (Section 2.3).
Fig. 2.

Performance of FIRE and 16 comparator tools when discriminating between cis-eQTL SNVs (Brown et al., 2013) identified at FDR < 1% (black diamonds), <5% (gray squares), or < 10% (white circles) and MAF-matched non-eQTL SNVs. AUC values are plotted with error bars representing double the 95% confidence intervals and are also listed in Supplementary Table S2. All FIRE scores were computed using chromosome exclusion (Section 2.3) to eliminate overlap between training and test SNVs
3.3 FIRE scores for cis-eQTL SNVs in other ancestries
We evaluated the generalizability of FIRE to non-European ancestries by comparing FIRE scores for cis-eQTL SNVs identified in Geuvadis EUR and YRI individuals (Lappalainen et al., 2013). For this comparison, we considered all SNVs identified as cis-eQTLs in each ancestry at FDR < 5%, rather than the stricter P < 10−10 threshold used to train FIRE. We partitioned these SNVs into three subsets: those identified as cis-eQTLs in both ancestries, those identified in YRI but not EUR, and those identified in EUR but not YRI. FIRE score distributions were similar for all subsets, with a small shift towards higher scores for the YRI-only subset (Supplementary Fig. S7A, Supplementary Table S3). The smaller size of the YRI sample (89 YRI vs. 373 EUR individuals) could account for these higher FIRE scores in the YRI-only subset, since larger effect sizes are needed to meet statistical significance in smaller samples. To address this potential confounding factor, we also examined FIRE scores for cis-eQTL SNVs identified at FDR < 5% in LCLs from six different ancestries (Stranger et al., 2012) with equal sample sizes of 69 individuals each (Supplementary Text S.1). FIRE score distributions were similar for all six ancestries, with slightly higher scores in the CEU ancestry (Supplementary Fig. S8, Supplementary Table S3).
SNVs associated with gene expression levels in multiple ancestries are more likely to be causal expression regulators, rather than in LD with causal regulators, because of differing LD structures between ancestries. We found a consistent shift towards higher FIRE scores for cis-eQTL SNVs shared across greater numbers of ancestries (Fig. 3A, Supplementary Table S3). FIRE discriminates between cis-eQTL SNVs shared across all six ancestries, which we expect to be most enriched for causal regulators, and non-eQTL SNVs with matched MAF distribution with an AUC of 0.939 (Fig. 3B, Supplementary Table S2).
Fig. 3.

(A) FIRE score distributions for cis-eQTL SNVs shared in at least one, two, three, four, five, or all six ancestries in an analysis of equal numbers of CEU, CHB, GIH, JPT, LWK and YRI individuals (Stranger et al., 2012), compared to SNVs in the negative training set. Gray boxes extend from the first to third quartiles, with a black horizontal line at the median. Means and quantiles of these distributions are also listed in Supplementary Table S3. (B) Performance of FIRE and 16 comparator tools when discriminating between the cis-eQTL SNVs shared in all six ancestries and MAF-matched non-eQTL SNVs. AUC values are plotted with error bars representing 95% confidence intervals and are also listed in Supplementary Table S2. All FIRE scores were computed using chromosome exclusion (Section 2.3) to eliminate overlap between training and test SNVs
3.4 FIRE scores for cis-eQTL SNVs in other tissues
Since FIRE was trained on cis-eQTL SNVs identified in LCLs, we also evaluated its generalizability to other tissues by comparing FIRE score distributions for cis-eQTL SNVs identified in 44 different tissue types in the GTEx Consortium V6p analysis (GTEx Consortium, 2015). We found similar score distributions for cis-eQTL SNVs identified at FDR < 5% in each tissue (Supplementary Fig. S9, Supplementary Table S4). We also found enrichment for higher FIRE scores among cis-eQTL SNVs shared across multiple GTEx tissues when compared to tissue-specific cis-eQTL SNVs, as illustrated for example within the set of cis-eQTL SNVs identified in GTEx skeletal muscle (Fig. 4, Supplementary Table S3). Similar enrichment was observed when we compared FIRE scores for cis-eQTL SNVs identified in a separate prostate tissue analysis (Larson et al., 2015) to the Geuvadis EUR analysis in LCLs (Supplementary Text S.1). We partitioned the prostate and Geuvadis EUR LCL cis-eQTL SNVs into three subsets: those identified as cis-eQTLs in both tissues, those identified in LCLs but not prostate, and those identified in prostate but not LCLs. FIRE score distributions were similar for the LCL-only and prostate-only subsets, with a small shift towards higher scores for cis-eQTL SNVs identified in both tissues (Supplementary Fig. S7B, Supplementary Table S3).
Fig. 4.

FIRE score distributions for cis-eQTL SNVs identified in skeletal muscle tissue in the GTEx Consortium V6p analysis and for the subsets of these SNVs that were identified as cis-eQTLs only in skeletal muscle (tissue-specific) or shared as cis-eQTLs in other GTEx tissues (grouped by the total number of tissues, including skeletal muscle). Gray boxes extend from the first to third quartiles, with a black horizontal line at the median. Means and quantiles of these distributions are also listed in Supplementary Table S3. All FIRE scores were computed using chromosome exclusion (Section 2.3)
3.5 FIRE scores for clinically relevant cis-eQTL SNVs
Finally, we tested whether SNVs identified as cis-eQTLs at FDR < 5% in the Geuvadis EUR analysis were enriched for higher FIRE scores if they also overlapped with either the GWAS Catalog of trait-associated SNVs (MacArthur et al., 2017; Welter et al., 2014) or the Human Gene Mutation Database (HGMD) of disease-relevant SNVs (Stenson et al., 2014). We found moderate enrichment for higher FIRE scores among cis-eQTL SNVs in HGMD, but only minor enrichment among those in the GWAS Catalog (Fig. 5A, Supplementary Table S3), suggesting that fewer GWAS SNVs than HGMD SNVs are causal expression regulators. This observation is consistent with the current consensus that a majority of GWAS risk SNVs are not causal but are in LD with a causal variant (MacArthur et al., 2017). We compared the ability of FIRE and existing genome-wide variant interpretation tools to distinguish these two subsets of clinically relevant cis-eQTL SNVs from non-eQTL SNVs with matched MAF distributions. FIRE had the highest performance (AUC = 0.798) on the GWAS-overlapping cis-eQTL SNVs and the second highest performance (AUC = 0.880) on the HGMD-overlapping cis-eQTL SNVs (Fig. 5B, Supplementary Table S2), although the AUC of the top performer, GWAVA unmatched, might be overestimated on this dataset because GWAVA was trained on SNVs from HGMD.
Fig. 5.

(A) FIRE score distributions for cis-eQTL SNVs identified at FDR < 5% in the Geuvadis EUR analysis and for subsets of these SNVs overlapping either the GWAS Catalog or HGMD. Gray boxes extend from the first to third quartiles, with a black horizontal line at the median. Means and quantiles of these distributions are also listed in Supplementary Table S3. (B) Performance of FIRE and 16 comparator tools when discriminating between these Geuvadis cis-eQTL SNVs overlapping the GWAS Catalog (top panel) or HGMD (bottom panel) and MAF-matched non-eQTL SNVs. AUC values are plotted with error bars representing 95% confidence intervals and are also listed in Supplementary Table S2. All FIRE scores were computed using chromosome exclusion (Section 2.3) to eliminate overlap between training and test SNVs
4 Discussion
FIRE is a genome-wide variant annotation tool that assigns higher scores to SNVs that are more likely to alter the expression levels of nearby genes. Since FIRE is specific to gene expression regulation, FIRE scores do not directly correspond to pathogenicity or deleteriousness. In addition, FIRE is specific to expression regulation at the mRNA level and does not apply to SNVs that alter protein expression independently of mRNA expression. We have shown that FIRE significantly outperforms existing genome-wide variant annotation tools in the task of discriminating cis-eQTL SNVs from non-eQTL SNVs across multiple independent cis-eQTL datasets. Our results also demonstrate the importance of computing genome-wide scores for FIRE and other similar tools using an approach such as chromosome exclusion that avoids biased scores from nearby correlated SNVs in the training set.
Of the comparator tools that we tested, the best performers for identifying cis-eQTL SNVs included LINSIGHT (Huang et al., 2017), GWAVA (Ritchie et al., 2014), cepip (Li et al., 2017), PRVCS (Li et al., 2016) and FunSeq2 (Fu et al., 2014). Many of the comparator tools were designed for other purposes, such as predicting pathogenic or deleterious SNVs, and have advantages over FIRE for such applications. Tools such as cepip can also predict tissue-specific regulatory potentials of SNVs, which is of interest for many biological applications. We did not test tools that infer the most likely causal regulatory SNV within sets of candidate cis-eQTL SNVs near individual genes but that do not provide genome-wide scores (Battle et al., 2014; Gaffney et al., 2012; Hormozdiari et al., 2016).
We found that the performance of FIRE was robust across different ancestries and tissues. Although FIRE was trained on cis-eQTL SNVs identified in LCLs from individuals of European descent, it was effective at scoring cis-eQTL SNVs identified in six tested ancestries and 44 tested tissue types, indicating an absence of substantial ancestry or tissue specificity in the features of regulatory SNVs among those in our feature set. One reason that FIRE may perform well in diverse tissue types is that we removed cis-eQTL SNVs identified in these other tissue types from the negative training set to reduce the number of false negatives. Future studies will explore the development of tissue-specific FIRE scores by incorporating tissue-specific genomic annotations and training data.
The approach used here to develop FIRE has limitations due to its dependence on association testing for constructing the training sets. Specificity suffers when non-causal SNVs are associated with gene expression due to LD with the underlying causal variant, and sensitivity suffers when causal SNVs are not significantly associated with expression due to low MAF, low effect size, or tissue specificity. Therefore, we expect some overlap between FIRE score distributions for SNVs in the positive and negative training sets. Nevertheless, enrichment of causal eQTLs within the positive set enables the FIRE random forest to identify genomic features that are most predictive of these regulatory SNVs.
Importantly, FIRE scores for subsets of cis-eQTL SNVs that are more likely to be causal expression regulators were higher than for other cis-eQTL SNVs. SNVs associated with gene expression in six different ancestries with different LD structures had much higher FIRE scores than SNVs associated in one ancestry. FIRE scores for cis-eQTL SNVs in HGMD, which requires evidence of involvement in disease or functional effects, were also higher than for other cis-eQTL SNVs. FIRE outperformed existing variant interpretation tools when discriminating non-eQTL SNVs from clinically relevant subsets of cis-eQTL SNVs overlapping either HGMD or the GWAS Catalog in almost all cases. However, the fact that many existing tools had higher performance on these clinically relevant subsets than they did on cis-eQTL SNVs as a whole suggests that these cis-eQTL subsets contain SNVs that are clinically relevant for other reasons, not recognized by FIRE, in addition to SNVs that are causal expression regulators.
In future studies, FIRE scores can be combined with tools tailored to other functional effects to obtain a more complete prediction of the significance of any observed SNV and to develop a mechanistic hypothesis about its biological function or role in disease. FIRE scores can be used to prioritize genome-wide SNVs of unknown significance for follow up studies or to weight them in statistical association tests combining many SNVs. Pre-computed genome-wide FIRE scores for all possible alternative SNV alleles at every position in hg19/GRCh37 can be downloaded from https://sites.google.com/site/fireregulatoryvariation/.
Supplementary Material
Acknowledgements
We thank Tuuli Lappalainen, Joseph Rothstein, Sumit Middha, Saurabh Baheti, Daniel Schaid, Xin Li and Sharon Plon for helpful discussions.
Funding
This work was supported by the National Institutes of Health [F32HG008330 to N.M.I., U01HG007436 to C.D.B., R01HG008150 to S.B.M., C.D.B. and A.J.B., U01CA089600 to S.N.T. and A.S.W., R01CA151254 to S.N.T.]; and by a Stanford Center for Computational, Evolutionary and Human Genomics postdoctoral fellowship to N.M.I. and a Lucille P. Markey Biomedical Research Fellowship to J.R.D.
Conflict of Interest: none declared.
References
- Adzhubei I.A. et al. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle A. et al. (2014) Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res., 24, 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. (2001) Random forests. Mach. Learn., 45, 5–32. [Google Scholar]
- Brown C.D. et al. (2013) Integrative modeling of eQTLs and cis-regulatory elements suggests mechanisms underlying cell type specificity of eQTLs. PLoS Genet., 9, e1003649.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner J.F. et al. (2012) DNase I sensitivity QTLs are a major determinant of human expression variation. Nature, 482, 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y. et al. (2014) FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol., 15, 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney D.J. et al. (2012) Dissecting the regulatory architecture of gene expression QTLs. Genome Biol., 13, R7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium (2015) Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science, 348, 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halligan D.L. et al. (2013) Contributions of protein-coding and regulatory change to adaptive molecular evolution in murid rodents. PLoS Genet., 9, e1003995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie T. et al. (2009) The Elements of Statistical Learning: data Mining, Inference, and Prediction. Springer, New York. [Google Scholar]
- Hindorff L.A. et al. (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA, 106, 9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman M.M. et al. (2013) Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res., 41, 827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F. et al. (2016) Colocalization of GWAS and eQTL signals detects target genes. Am. J. Hum. Genet., 99, 1245–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.F. et al. (2017) Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat. Genet., 49, 618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis N.M. et al. (2016) REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet., 99, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionita-Laza I. et al. (2016) A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet., 48, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagadeesh K.A. et al. (2016) M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet., 48, 1581–1586. [DOI] [PubMed] [Google Scholar]
- Kircher M. et al. (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet., 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P. et al. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc., 4, 1073–1081. [DOI] [PubMed] [Google Scholar]
- Lappalainen T. et al. (2013) Transcriptome and genome sequencing uncovers functional variation in humans. Nature, 501, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson N.B. et al. (2015) Comprehensively evaluating cis-regulatory variation in the human prostate transcriptome by using gene-level allele-specific expression. Am. J. Hum. Genet., 96, 869–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D. et al. (2015) A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet., 47, 955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B. et al. (2009) Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics, 25, 2744–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.J. et al. (2016) Predicting regulatory variants with composite statistic. Bioinformatics, 32, 2729–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.J. et al. (2017) cepip: context-dependent epigenomic weighting for prioritization of regulatory variants and disease-associated genes. Genome Biol., 18, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw A., Wiener M. (2002) Classification and regression by randomForest. R News, 2, 18–22. [Google Scholar]
- Lu Q. et al. (2015) A statistical framework to predict functional non-coding regions in the human genome through integrated analysis of annotation data. Sci. Rep., 5, 10576.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur J. et al. (2017) The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res., 45, D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher M.C. et al. (2012) Population genetics of rare variants and complex diseases. Hum. Hered., 74, 118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVicker G. et al. (2009) Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet., 5, e1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolae D.L. et al. (2010) Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet., 6, e1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang D. et al. (2015) DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics, 31, 761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. (2014) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Ritchie G.R. et al. (2014) Functional annotation of noncoding sequence variants. Nat. Methods, 11, 294–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X. et al. (2011) pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics, 12, 77.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihab H.A. et al. (2015) An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics, 31, 1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson P.D. et al. (2014) The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet., 133, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger B.E. et al. (2012) Patterns of cis regulatory variation in diverse human populations. PLoS Genet., 8, e1002639.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter D. et al. (2014) The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res., 42, D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Troyanskaya O.G. (2015) Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods, 12, 931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.