

Abstract
Summary
With the development of new high-throughput DNA sequencing technologies and decreasing costs, large gene expression datasets are being generated at an accelerating rate, but can be complex to visualize. New, more interactive and intuitive tools are needed to visualize the spatiotemporal context of expression data and help elucidate gene function. Using tomato fruit as a model, we have developed the Tomato Expression Atlas to facilitate effective data analysis, allowing the simultaneous visualization of groups of genes at a cell/tissue level of resolution within an organ, enhancing hypothesis development and testing in addition to candidate gene identification. This atlas can be adapted to different types of expression data from diverse multicellular species.
Availability and Implementation
The Tomato Expression Atlas is available at http://tea.solgenomics.net/. Source code is available at https://github.com/solgenomics/Tea.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Large-scale transcriptome profiling has become widely adopted as a means to characterize the status and diversity of biological samples, and as a platform for functional genomic studies. Such analyses typically target whole organisms or specific organs; however, there is an increasing interest in using cell- or tissue-related transcriptome profiling to provide enhanced spatiotemporal understanding of gene function (Martin et al., 2016). The quantity and resolution of such gene expression information necessitates the development of new data visualization tools that are more interactive, intuitive and accessible.
Tools such as the eFP browser (Winter et al., 2007) or MapMan (Thimm et al., 2004) are available to visualize RNA-seq expression and metabolomics data in intuitive graphical formats. At the Sol Genomics Network (SGN, https://solgenomics.net/, Fernandez-Pozo et al., 2015) we have developed the Tomato Expression Atlas (TEA), a web tool to store and display RNA-Seq data derived from complex organs/organisms down to the cell-type level of resolution, with the versatility to show different stages of development, genotypes, treatments, or other variables. TEA is currently based on expression data from tomato (Solanum spp.) fruit, but could be adapted for any multicellular organ/organism (See supplementary materials for more information).
2 Materials and methods
2.1 The code underlying the expression atlas
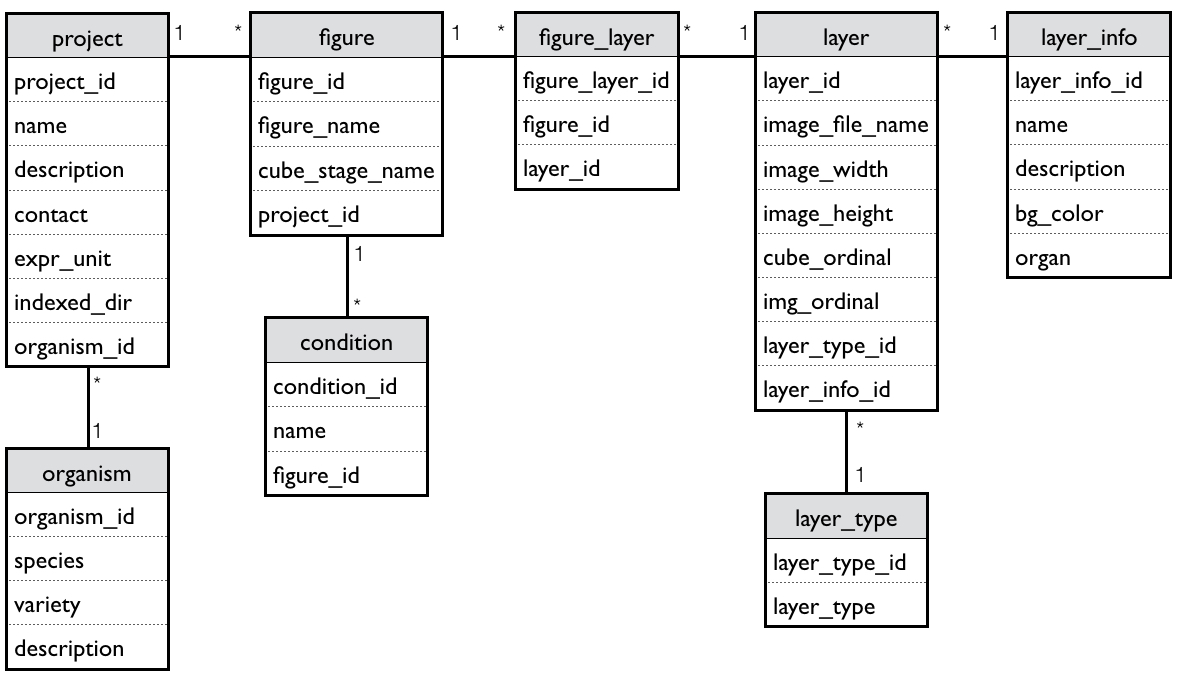
The TEA is developed on a Catalyst framework, using Perl as a programming language for the controllers, Mason as the templating toolkit, and Perl DBIx classes as an object-relational mapping tool to query a PostgreSQL database. The database (Supplementary Fig. S1) contains tables to store the samples metadata needed to construct the TEA cube and expression images (Fig. 1), and to apply the stage, organ, tissue and condition input filters. This design allows for flexibility with expression images, which can represent different levels, from one single tissue to multi-organ figures.
Fig. 1.

The Tomato Expression Atlas cube (a) and expression images (b). Colors represent expression values in Reads Per Million (RPM)
Gene expression and correlation values, as well as the gene descriptions are stored in indexed files for fast access and easy maintenance. These indices are created and queried using a Perl module wrapper for Lucy Apache (Lucy::Simple) (See supplemental methods for more information about the expression and correlation data and the front-end code).
3 Results
The TEA was designed to be highly interactive and to produce publication quality graphics (Fig. 1; Supplementary Figs S2 and S3). Three different gene input options are provided: (i) the gene ID to retrieve the expression values for this gene and genes with similar expression profiles; (ii) amino acid or nucleotide sequence to find homologous genes using BLAST (Altschul et al., 1990); and (iii) a custom list of genes.
3.1 Gene expression visualization
The main output of the TEA is the ‘Expression Cube’, which allows users to visualize and compare expression profiles of multiple genes simultaneously, and gives direct access to detailed information for each gene with bar plots and downloadable data (Fig. 1a). The expression cube displays the expression profile of the query gene, together with the genes with the most highly correlated expression profiles (Fig. 1a). The horizontal slices of the cube represent the genes. On each slice, from left to right, are the developmental stages, sorted chronologically, and from front to back are the tissues/cell types.
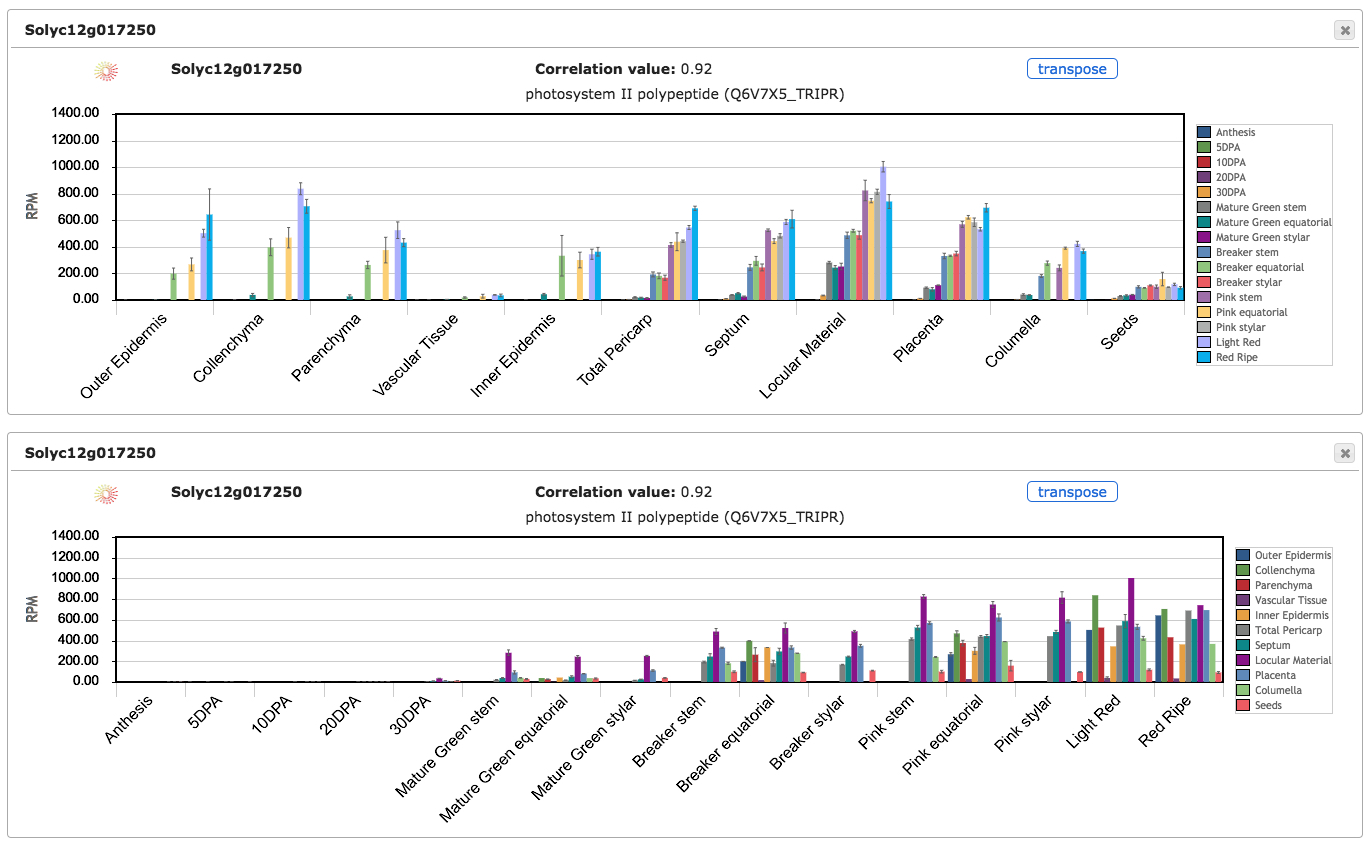
The cube is designed to be highly interactive. When the cursor is placed over the gene name, the description and the correlation value of expression profiles between that gene and the query gene are displayed (Fig. 1a). The cube can be split into multiple stacked layers by clicking on the gene names (Fig. 1a). Clicking again on the same gene name will restore the cube. When the cursor is over the tiles of the cube, the conditions and expression values are displayed. Clicking on the tiles will display a bar plot with gene expression values for the respective gene (Supplementary Fig. S2). Multiple bar plot overlays can be displayed simultaneously to facilitate comparisons between genes.
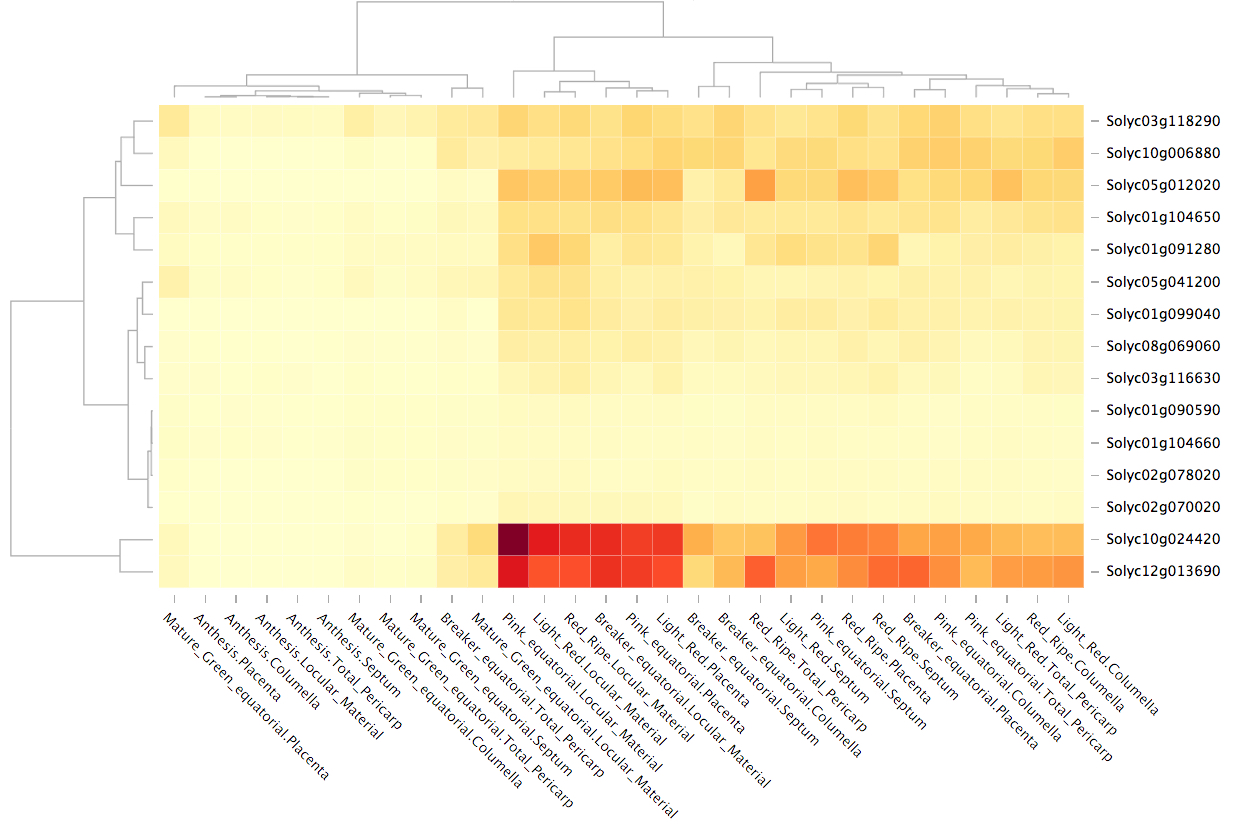
A second option to visualize the results is ‘Expression images’, which displays images representing organs, tissues or cellular types for each one of the stages (Fig. 1b). These images show colors representing the expression values, corresponding to the same ranked colors on the expression cube. The third TEA output tab shows an interactive hierarchical heatmap, which clusters genes and samples using dendrograms, allowing the simultaneous comparison of multiple genes (Supplementary Fig. S3).
Funding
This work has been supported by a grant from the US National Science Foundation (Plant Genome Research Program; IOS-1339287) and a Grant-in-Aid for JSPS Fellows from the Japan Society for the Promotion of Science (16J00582).
Conflict of Interest: none declared.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
References
- Altschul S.F. et al. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Fernandez-Pozo N. et al. (2015) The Sol Genomics Network (SGN)—from genotype to phenotype to breeding. Nucleic Acids Res., 43, D1036–D1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L.B.B. et al. (2016) Laser microdissection of tomato fruit cell and tissue types for transcriptome profiling. Nat. Protoc., 11, 2376–2388. [DOI] [PubMed] [Google Scholar]
- Thimm O. et al. (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J., 37, 914–939. [DOI] [PubMed] [Google Scholar]
- Winter D. et al. (2007) An “Electronic Fluorescent Pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS One, 2, e718.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.