

Abstract
It is accepted that confusion regarding the description of genetic variants occurs when researchers do not use standard nomenclature. The Human Genome Organization Gene Nomenclature Committee contacted a panel of consultants, all working on the KAL1 gene, to propose an update of the nomenclature of the gene, as there was a convention in the literature of using the ‘KAL1’ symbol, when referring to the gene, but using the name ‘anosmin-1’ when referring to the protein. The new name, ANOS1, reflects protein name and is more transferrable across species.
Keywords: : Kallmann, anosmin-1, KAL1, ANOS1, gene, nomenclature
Introduction
An analysis of research and review articles published in international journals has been carried out to briefly summarize both the well established and the most recent knowledge on the ANOS1 gene and anosmin-1 physiopathology in Kallmann’s and other diseases.
This article reports the most relevant information on ANOS1 and its product anosmin-1, from the identification to its new functions. It addresses a topic of importance to endocrinologists of reproduction, with indications for a better and correct classification of the genetic variants of Kallmann’s disease. The authors, together with the panel of consultants, hope that researchers and the scientific community, overall, will use the new nomenclature ANOS1 for the gene encoding for anosmin-1 and responsible for the X-linked form of Kallmann syndrome (KS), and ANOS2P for the pseudogene on the Y chromosome. The symbol ANOS1 will be propagated to orthologs in vertebrate species.
The application of a standard genetic nomenclature is fundamental to an accurate scientific communication and a correct classification of disease-related genes. In this regard, the Human Genome Organization (HUGO) Gene Nomenclature Committee (HGNC: www.genenames.org) [1] has recently proposed to the community to standardize the nomenclature of the KAL1 gene to ANOS1. The ANOS1 gene was first described as responsible for KS, a developmental genetic disease belonging to the wide family of congenital idiopathic hypogonadotropic hypogonadism.
KS was described as a genetic heterogeneous association between a lack of olfaction/smelling (anosmia) and hypogonadotropic hypogonadism, as the cause of sterility [2–4], with a prevalence estimated from around 1:8000 in men and 1:40 000 in women [5]. Adult patients typically show normal or low levels of circulating gonadotropins and low serum gonadal hormone levels because of an impairment of the function of gonadotropin-releasing hormone (GnRH) system, the master regulators of the hormonal reproductive axis. The lack of smell results from a developmental defect of the olfactory bulb (OB) [6].
The olfactory and reproductive dysfunctions may be combined with other defects or satellite symptoms, including renal agenesis, mirror movement (synkinesis), syndactyly, craniofacial abnormalities, coloboma and sensorineural deafness.
It took until 1989 to get the evidence that the biological defects observed in one 19 week-old KS-affected human fetus resided in the defective development of the olfactory axons and the arrest of hypothalamic GnRH neurons in their migration from the olfactory placode to the hypothalamus [7]; the results of this pioneering report have been confirmed more later in a 25 week-old KS-affected male fetus and in other fetuses affected by developmental arrhinencephalic disorders [8].
However, it was at the beginning of the 1990s decade that the assignment of a syndrome locus on the X chromosome was made [9], and the detection of a genetic lesion at Xp22.3 led to the definitive identification of the first KS candidate gene [10, 11]. A clinical severe form of hypogonadism is present in patients affected by the X-linked form of KS, which includes an absent or altered pulsatile gonadotropin secretion, low serum testosterone levels and a lack of puberty; however, the gonadotropin response to GnRH stimulation is preserved in these subjects [12]. X-linked KS phenotype is characterized by a severe anosmia, bimanual synkinesia, bilateral cryptorchidism and renal agenesis [13]. MRI analysis reveals aplastic or dysplastic OB and other brain changes associated with mirror movement [14].
The gene causative for the X-linked form of KS was named Kallmann syndrome 1 sequence (KAL1) [15]. The gene escapes X inactivation, has a homolog on the Y chromosome (originally named KALP) and shows an unusual pattern of conservation across species because it has not been so far identified in mouse and rat [15]. Other different modes of KS transmission (autosomal dominant and recessive) have since been described [8], among them the autosomal dominant form caused by mutation of the gene FGFR1, initially defined as KAL2 gene [16]. Interestingly, both KAL1 and FGFR1 have been recently proposed for noninvasive embryonic diagnosis of KS [17]. The contribution of FGFR1 to congenital hypopituitarism has been recently revisited [18]. Of interest, the first report on ANOS1 overexpression, because of a recessive X-chromosome microduplication, in humans reveals a phenotype characterized by hyperosmia, genital anomalies (testicular hydrocele and cryptorchidism), ectrodactyly and additional symptoms like mild intellectual disability, unilateral hearing loss, stocky build and facial dysmorphism, possibly related to a possible interference with the FGFR1 signal [19].
ANOS1 loss-of-function mutations are associated with the X-linked KS phenotype, but they have not so far been linked with other developmental defects (midline abnormality, cleft palate) found in other forms of KS [13].
Moreover, in recent years, the identification of a large proportion of oligogenic forms of the disease has changed the whole view of the genetic basis of KS [20].
The ANOS1 gene is largely conserved from invertebrates to primates including some rodents (Table 1). It is peculiar to note that although human ANOS1 gene product has been found to be functional in mice cells and tissues [21, 22], its immunoreactivity has been detected in rat tissue, using an antibody for the human protein, and human and mice share about 99% of genes; no KAL1 ortholog in mouse and rat has been so far identified [23], making a more complete genetic analysis of the role of ANOS1 in biological processes using classical rodent models not possible.
Table 1.
Orthologs of human KAL1 gene with updated nomenclature to ANOS1 and similarities in different species
| Organism | Gene name | Human gene similarity (%) |
|---|---|---|
| Chimpanzee | ANOS1 | 97 |
| Cow | ANOS1 | 79 |
| Dog | ANOS1 | 82 |
| Chicken | ANOS1 | 74 |
| Guinea Pig | ANOS1 | 84 |
| Rabbit | ANOS1 | 74 |
| Squirrel | Anos1 | 87 |
| Rat | No ANOS1 ortholog so far identified | – |
| Mouse | No ANOS1 ortholog so far identified | – |
| Zebrafish | anos1a, anos1b | 61 |
| C. elegans | kal-1 | 42 |
| Drosophila | Anos1 | 38 |
Anosmin-1
The ANOS1 gene contains 14 exons and encodes for the protein anosmin-1 whose lack or mutation is responsible for the main symptoms characterizing the X-linked form of this disease [24]. Anosmin-1 is a 680 amino acid glycoprotein of the extracellular matrix [25], with a high degree of sequence identity among species, that contains: (i) a cysteine-rich region (CR domain), (ii) a whey acidic protein (WAP)-like domain similar (like serine protease inhibitors), (iii) four consecutive fibronectin type III domains (FNIII, like many cell-adhesion molecules) and (iv) a C-terminal region rich in basic histidines and prolines [26] (Figure 1). The WAP domain is present in protease inhibitors and was found to play a role in axonogenesis and neuron migration. FnIII domains are present in proteins involved in cell adhesion, in tyrosine kinases and in phosphatases, implicated in neuronal migration and in axon guidance. In this sense, along this sequence, it is noteworthy that there are five potential heparan sulphate-binding sites as well as six sites for possible N-glycosylation.
Figure 1.
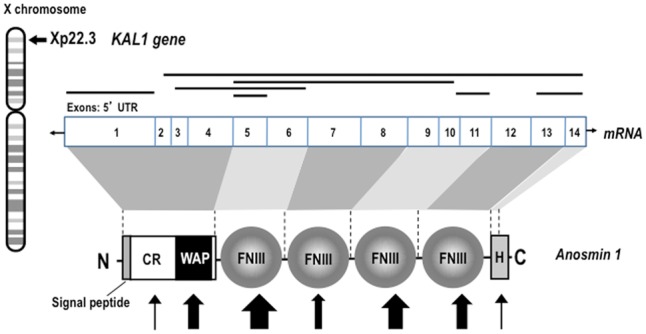
Structure of ANOS1 gene and of anosmin-1 protein. Schematic representation of the ANOS1 gene location on the short arm of X chromosome, of anosmin-1 mRNA and protein structures and mutations. The number on the mRNA structure indicates the exons covering the ORF of the protein anosmin-1; shaded area indicates the exons involved in the coding of the different protein domains. The position of the CR domain, the WAP-like domain, the four FNIII repeats and the histidine-rich domain of anosmin-1 are also indicated. Main exon deletions are shown with black lines, and point mutations are indicated by the arrows; the size of the arrows indicates approximately the frequency of the identified mutations (adapted from [31]).
The expression of ANOS1 messenger RNA (mRNA) and anosmin-1 protein extends from the embryonic development to adulthood, and it should be remarked that both mRNA and protein are largely more present in the different structures of the central nervous system (CNS, including cerebral cortex, OB and other components of the olfactory system, retina, cerebellum, spinal cord) than in other body organs and structures (inner ear, kidney, testis, skin and vascular endothelial cells), and overall explains the main and satellite symptoms in KS patients (for a comprehensive review on the subject, see [27]).
Although different putative interacting proteins for anosmin-1 have been described to date, the mechanism of action of this protein remains far from completely characterized. FGFR1, heparan sulphates, syndecan, glypicans, as well as different components of the extracellular matrix (uPA, fibronectin, laminin, integrin-beta, anosmin-1 itself—in many of these cases, the activity seems totally independent of FGF2–FGFR1 signaling) are able to interact with anosmin-1 either alone or, occasionally (this is the case of FGFR1 and heparan sulphate), together and with different results [23, 28–30]. Among them, FGFR1 is undoubtedly the most studied because mutations in FGFR1 gene (KAL2) are responsible for the autosomal dominant form of KS [16]. In total, >60 mutations of ANOS1 have been described, but clear hot spots have not been identified (Figure 1) [31]. Most of mutations are deletion, frameshift or nonsense; missense mutations in the FnIII domains of ANOS1 are among the most frequent in KS patients, while the real significance of the mutations in the WAP domain on the biological activity of anosmin-1 remains far from clear [21, 29, 32]. More details on the ANOS1 gene mutations underlying KS, including the suggested heterogeneity of effects related to the cell type, their extracellular environment or the combined interaction with different putative receptors, have been widely reported [23, 31]. More recently, a study carried out in Caenorhabditiselegans revealed that KAL-1/anosmin-1 may mediate neurite branching by acting as an autocrine cofactor of FGF signaling through a receptor complex consisting of the nematode orthologs of fibroblast growth factor receptor and cell adhesion molecule L1 [33].
Regarding its function in biology, in different in vitro models, anosmin-1 has been demonstrated to affect cell adhesion, neurite outgrowth and branching [34–36] and cell migration [21], whereas in in vivo models, it appears to regulate several aspects of neurogenesis [22], the motility/migration of different neural cell types during development and, both physiologically and in pathological scenarios in the adult [28, 30, 37], the outgrowth of axons and the genesis of axon collaterals [38, 39] as well as in the differentiation of oligodendrocytes and myelin formation [40]. Finally, the relevance of locally produced anosmin-1 on regulating three major morphogens like FGF8, BMP5 and WNT3a and therefore its crucial role in the formation of the neural crest has been also shown [41].
Other functions of Anosmin-1
Besides KS, anosmin-1 has been described as relevant in the pathogenesis of multiple sclerosis, the most frequent primary demyelinating disease and the most frequent neurological disease in young adults: the protein is upregulated in the core of chronic-active and chronic-inactive demyelinating lesions (in this zone of the lesions, there is no spontaneous remyelination), maybe produced by astrocytes, but not in the periplaque of the former type or in active lesions, the only sites where spontaneous remyelination by endogenous oligodendrocyte precursor cells occurs [37]. Also in samples from human cerebral cortices with multiple sclerosis, anosmin-1 is present in 13–14% of the nude axons crossing demyelinated lesions, but not in their periplaques or in the normal appearing white matter [37]. It is extremely interesting that endogenous oligodendrocyte precursor cells effectively recruited toward demyelinating lesions are mainly positive for FGFR1, which suggests that this lack of potential remyelinating cells would be because of anosmin-1 antagonizing the FGF2 motogenic effect [28, 37] (this scenario has been described more extensively by de Castro and coworkers [23, 42]). Indeed, the relationship between KS and demyelination/multiple sclerosis remains to be fully addressed after recent reports [43, 44, 45]. The real/direct contribution of ANOS1 gene to septo-optic dysplasia and deafness has been recently reevaluated together with the availability of new diagnostic tools [46–48].
Anosmin-1 protein also exerts functions outside the CNS: it modulates the response to immunoglobulin therapy in dermatomiositis [49] and regulates the density of nerve terminals in the epidermis affected by atopic dermatitis [50]. More recently, it has been shown that is concomitant SOX10 mutation the real cause of skin/hair/iris hypopigmentation [48].
Finally, ANOS1 has been involved in migration and metastasis of cancer cells; in particular, its expression was found modified in several tumors including brain, ovarian, colorectal, hepatocellular and oral squamous cancer [30, 51–55], where it may play a role as a possible modulator of the reactivation of developmental signal pathways, and therefore proposed as diagnostic or prognostic tool. In this regard, recently, it has been proposed that while the serum levels of anosmin-1 would be a good biomarker for gastric cancer prognosis and treatment stratification of patients [56], ANOS1 may be a tumor suppressor for hepatocellular cancer [55].
The change of nomenclature
The scientific community has previously used KAL1, KAL-1 and even ANOS-1 or ANOS1 (both, with or without hyphen, either in capital letters or not) to refer to the same gene, that encodes anosmin-1. In accordance with the suggestion of HGNC, who supported and encouraged us to write this position statement, we and the other researchers forming the group of consultants recruited by HGNC (see Acknowledgements) agree here to adopt the gene name anosmin1 (without hyphenation), encoding for anosmin-1 protein, with the approved symbol ANOS1 (Table 2).
Table 2.
Old and new nomenclatures for ANOS1
| Old nomenclature | New nomenclature | |
|---|---|---|
| Gene name | Kallmann syndrome 1 sequence | Anosmin 1 |
| Symbol aliases | KAL1, KAL-1, ADMLX, KAL, KALIG-1, KALIG1, HHA, KALM_HUMAN, KMS, WFDC19, | ANOS1 |
| Gene name aliases | Adhesion molecule-like X-linked anosmin-1, Kallmann syndrome interval gene 1, WAP four-disulfide core domain 19, Kallmann syndrome 1 protein, Kallmann syndrome protein | – |
The symbol ‘KAL’ was first approved in 1986 by the HGNC to represent the KS phenotype rather than a specific gene. This was later updated from KAL to KAL1 to distinguish it from the distinct form of KS known as KAL2. The causative gene for the X-linked form of KS was identified by two separate groups in 1991 as KALIG-1 for ‘Kallmann’s syndrome interval gene 1’ [10] and as ADMLX for ‘adhesion molecule-like from the X chromosome’ [11]. As neither symbol fitted with HGNC guidelines, the symbol KAL1 was retained for the cloned gene.
The first study on the protein encoded by KAL1 named this protein ‘anosmin-1’ to reflect the loss of smell, or anosmia, in patients with KS [24]. The name anosmin-1 grew in popularity over the years, and it became the convention to refer to the gene as KAL1 and the protein as anosmin-1. The protein name has been used for different species such as zebrafish [35], tammar wallaby [31] and Asian musk shrew [57].
In early 2015, the HGNC proposed updating the gene nomenclature to be in line with the protein name because this is more transferrable across species and avoids any confusion between ‘KAL1’ and other genes that cause forms of KS. Additionally, the name ‘anosmin’ is indicative of function since the lack of sense of smell in KS is because of the role of the protein in axonal outgrowth in the olfactory system. This nomenclature scheme may also be readily adapted to incorporate additional genes encoding other anosmins that would be eventually identified in the future. The symbol Anos1 [35] had already been published as an abbreviation for ‘anosmin-1’, and as this symbol is unique, the HGNC proposed the symbol ANOS1 and named anosmin1 for the gene. Hyphens were omitted to follow the conventions of gene nomenclature. The HGNC contacted researchers who had previously published on KAL1, and there was community support for the change.
The change in nomenclature of KAL1 resulted in a concordant change for the inactive homolog, which is present on the Y chromosome. This gene was first reported as a pseudogene because of frameshift and premature stop codons and named KALP [15]. Surrounding gene order and theories on Y chromosome evolution [58, 59] suggest that KALP has degraded in situ rather than being a duplication of ANOS1. According to annotation by the HAVANA group [60], this pseudogene is still transcribed. Therefore, KALP has been renamed as ANOS2P for ‘anosmin2, pseudogene’ to reflect its status as a degraded homolog of ANOS1 and not as a duplicated pseudogene of ANOS1 (which would have been given the symbol format ANOS1P1).
We hope that interested researchers and the scientific community, overall, will use the new nomenclature ANOS1 for the gene encoding for anosmin-1 and responsible for the X-linked form of KS, and ANOS2P for the pseudogene on the Y chromosome. The symbol ANOS1 will be propagated to orthologs in vertebrate species, and we request that researchers working on other species will also use the updated symbols.
Key Points
A different nomenclature is used to define Kallmann syndrome 1 gene (KAL1) and its proteic product (anosmin-1).
The most recent knowledge on the KAL1 gene and anosmin-1 confirms their involvement in the physiopathology of Kallmann’s disease but also in other diseases.
The HUGO Gene Nomenclature Committee (HGNC) with a panel of consultants propose an update of the nomenclature of the KAL1 gene in anosmin1 (ANOS1) and ANOS2P for the pseudogene present on the Y chromosome.
This nomenclature scheme may also be readily adapted to incorporate additional genes encoding other anosmins that would be eventually identified in the future.
Acknowledgements
The following researchers support the new nomenclature (in alphabetic order): Pierre-Marc Bouloux, Patricia Canto, Pedro F. Esteban, Soo-Hyun Kim, Lawrence C. Layman, Veronica Murcia-Belmonte, Christine Petit, Raghavan Raju, Nadia Sousssi-Yanicostas and Erika Trarbach.
The new nomenclature was also considered and unanimously approved by the European consortium on GnRH deficiency (COST Action BM1105, www.gnrhnetwork.eu).
The authors wish to thank the HUGO Gene Nomenclature Committee (HGNC) for proposing the update of the KAL1 gene nomenclature and for supporting the present publication, and all the scientists who have taken part in the discussions leading to the new nomenclature for this gene. The authors also thank Jean Pierre Hardelin for the fruitful discussion and the suggestions provided, and Carolina Melero-Jerez and Sonia Nocera for their help in the preparation of the list of references.
All members of the Group of HGNC consultants for KAL1 nomenclature gave the written permission for the use of his/her name.
Funding
This work was supported by grants from the Spanish Ministerio de Economia y Competitividad-MINECO (SAF2012-40023 and RD12-0032-12) and Consejo Superior de Investigaciones Científicas-CSIC (CSIC-2015201023) to F.dC., from National Human Genome Research Institute (U41HG003345) and Wellcome Trust (099129/Z/12/Z) to R.S. and from Fondazione Telethon (E.523) and Ministero dell’Istruzione Universitá e Ricerca (MIUR) to R.M.
Biographies
Fernando de Castro, MD, PhD, is Staff Scientist at the Spanish Research Council (Consejo Superior de Investigaciones Científicas-CSIC) and principal investigator at the Grupo de Neurobiología del Desarrollo-GNDe. Group leader of the Developmental Neurobiology Group (GNDe) at Instituto Cajal-CSIC, Madrid, Spain. The research activity of Dr F.dC. is focused on developmental neuroscience and on the genetic mechanisms controlling neuronal and glial migration and myelination and the correlated diseases. Author of 68 scientific papers and book chapters, with 2308 total number of citations.
Ruth Seal, PhD, is a Gene Nomenclature Advisor for the HUGO Gene Nomenclature Committee (HGNC), based at European Bioinformatics Institute (EBI-EBML), Hinxton, UK. She is working with the community to approve gene nomenclature, curating gene families, editing the HGNC newsletter, attending conferences and performing outreach for the HGNC.
Roberto Maggi, PhD, is a Professor of Physiology, Head and group leader of the Laboratory of Developmental Neuroendocrinology, Università degli Studi di Milano, Italy. The research activity of Prof. R.M. is focused on the isolation of hypothalamic stem cells and on the molecular mechanisms governing the development of the endocrine hypothalamus. He is a member of the Basic Science Working Group of COST action BM1105. Author of 111 papers with a sum of citations of 1986.
References
- 1. Gray KA, Yates B, Seal RL, et al. Genenames.org: the HGNC resources in 2015. Nucleic Acids Res 2015;43:D1079–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. de Morsier G. Studies in cranio-encephalic dysraphia. I. Agenesia of the olfactory lobe (lateral telencephaloschisis) and of the callous and anterior commissures (median telencephaloschisis); olfacto-genital dysplasia, Schweiz. Arch Neurol Psychiatr 1954;74:52. [PubMed] [Google Scholar]
- 3. Kallmann F, Schoenfeld W, Barrera S.. The genetic aspects of primary eunuchoidism. Am J Ment Defic 1944;48:33. [Google Scholar]
- 4. Maestre de San JA. Teratología: falta total de los nervios olfatorios con anosmia en un individuo en quien existía una atrofia congénita de los testículos y el miembro viril. El Siglo Médico Madrid 1856;131:10. [Google Scholar]
- 5. Dodé C, Hardelin JP.. Kallmann syndrome. Eur J Hum Genet 2009;17:139–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. MacColl G, Bouloux P, Quinton R.. Kallmann syndrome: adhesion, afferents, and anosmia. Neuron 2002;34:675–8. [DOI] [PubMed] [Google Scholar]
- 7. Schwanzel-Fukuda M, Bick D, Pfaff DW.. Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Brain Res Mol Brain Res 1989;6:311–26. [DOI] [PubMed] [Google Scholar]
- 8. Teixeira L, Guimiot F, Dode C, et al. Defective migration of neuroendocrine GnRH cells in human arrhinencephalic conditions. J Clin Invest 2010;120:3668–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meitinger T, Heye B, Petit C, et al. Definitive localization of X-linked Kallman syndrome (hypogonadotropic hypogonadism and anosmia) to Xp22.3: close linkage to the hypervariable repeat sequence CRI-S232. Am J Hum Genet 1990;47:664–9. [PMC free article] [PubMed] [Google Scholar]
- 10. Franco B, Guioli S, Pragliola A, et al. A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature 1991;353:529–36. [DOI] [PubMed] [Google Scholar]
- 11. Legouis R, Hardelin J, Levilliers J, et al. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell 1991;67:423–35. [DOI] [PubMed] [Google Scholar]
- 12. Pitteloud N, Hayes FJ, Dwyer A, et al. Predictors of outcome of long-term GnRH therapy in men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab 2002;87:4128–36. [DOI] [PubMed] [Google Scholar]
- 13. Quinton R, Duke VM, Robertson A, et al. Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization. Clin Endocrinol (Oxf) 2001;55:163–74. [DOI] [PubMed] [Google Scholar]
- 14. Manara R, Salvalaggio A, Citton V, et al. Brain anatomical substrates of mirror movements in Kallmann syndrome. Neuroimage 2015;104:52–8. [DOI] [PubMed] [Google Scholar]
- 15. del Castillo I, Cohen-Salmon M, Blanchard S, et al. Structure of the X-linked Kallmann syndrome gene and its homologous pseudogene on the Y chromosome. Nat Genet 1992;2:305–10. [DOI] [PubMed] [Google Scholar]
- 16. Dodé C, Levilliers J, Dupont JM, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet 2003;33:463–5. [DOI] [PubMed] [Google Scholar]
- 17. Sarfati J, Bouvattier C, Bry-Gauillard H, et al. Kallmann syndrome with FGFR1 and KAL1 mutations detected during fetal life. Orphanet J Rare Dis 2015;10:71.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Correa FA, Trarbach EB, Tusset C, et al. FGFR1 and PROKR2 rare variants found in patients with combined pituitary hormone deficiencies. Endocr Connect 2015;4:100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sowińska-Seidler A, Piwecka M, Olech E, et al. Hyperosmia, ectrodactyly, mild intellectual disability, and other defects in a male patient with an X-linked partial microduplication and overexpression of the KAL1 gene. J Appl Genet 2015;56:177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sykiotis GP, Plummer L, Hughes VA, et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci USA 2010;107:15140–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cariboni A, Pimpinelli F, Colamarino S, et al. The product of X-linked Kallmann's syndrome gene (KAL1) affects the migratory activity of gonadotropin-releasing hormone (GnRH)-producing neurons. Hum Mol Genet 2004;13:2781–91. [DOI] [PubMed] [Google Scholar]
- 22. García-González D, Murcia-Belmonte V, Esteban PF, et al. Anosmin-1 over-expression increases adult neurogenesis in the subventricular zone and neuroblast migration to the olfactory bulb. Brain Struct Funct 2016;221:239–60. [DOI] [PubMed] [Google Scholar]
- 23. de Castro F, Esteban PF, Bribián A, et al. The adhesion molecule anosmin-1 in neurology: Kallmann syndrome and beyond. Adv Neurobiol 2014;8:273–92. [DOI] [PubMed] [Google Scholar]
- 24. Soussi-Yanicostas N, Hardelin JP, Arroyo-Jimenez MM, et al. Initial characterization of anosmin-1, a putative extracellular matrix protein synthesized by definite neuronal cell populations in the central nervous system. J Cell Sci 1996;109:1749–57. [DOI] [PubMed] [Google Scholar]
- 25. Hardelin JP, Julliard AK, Moniot B, et al. Anosmin-1 is a regionally restricted component of basement membranes and interstitial matrices during organogenesis: implications for the developmental anomalies of X chromosome-linked Kallmann syndrome. Dev Dyn 1999;215:26–44. [DOI] [PubMed] [Google Scholar]
- 26. Legouis R, Lievre CA, Leibovici M, et al. Expression of the KAL gene in multiple neuronal sites during chicken development. Proc Natl Acad Sci USA 1993;90:2461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mitchell AL, Dwyer A, Pitteloud N, et al. Genetic basis and variable phenotypic expression of Kallmann syndrome: towards a unifying theory. Trends Endocrinol Metab 2011;22:249–58. [DOI] [PubMed] [Google Scholar]
- 28. Bribian A, Barallobre MJ, Soussi-Yanicostas N, et al. Anosmin-1 modulates the FGF-2-dependent migration of oligodendrocyte precursors in the developing optic nerve. Mol Cell Neurosci 2006;33:2–14. [DOI] [PubMed] [Google Scholar]
- 29. Hu Y, González-Martínez D, Kim SH, et al. Cross-talk of anosmin-1, the protein implicated in X-linked Kallmann's syndrome, with heparan sulphate and urokinase-type plasminogen activator. Biochem J 2004;384:495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Choy CT, Kim H, Lee JY, et al. Anosmin-1 contributes to brain tumor malignancy through integrin signal pathways. Endocr Relat Cancer 2014;21:85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hu Y, Bouloux PMX.. linked GnRH deficiency: role of KAL-1 mutations in GnRH deficiency. Mol Cell Endocrinol 2011;346:13–20. [DOI] [PubMed] [Google Scholar]
- 32. Murcia-Belmonte V, Esteban PF, García-González D, et al. Biochemical dissection of Anosmin-1 interaction with FGFR1 and components of the extracellular matrix. J Neurochem 2010;115:1256–65. [DOI] [PubMed] [Google Scholar]
- 33. Diaz-Balzac CA, Lazaro-Pena MI, Ramos-Ortiz GA, et al. The Adhesion molecule KAL-1/anosmin-1 regulates neurite branching through a SAX-7/L1CAM-EGL-15/FGFR receptor complex. Cell Rep 2015;11:1377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Soussi-Yanicostas N, Faivre-Sarrailh C, Hardelin JP, et al. Anosmin-1 underlying the X chromosome-linked Kallmann syndrome is an adhesion molecule that can modulate neurite growth in a cell-type specific manner. J Cell Sci 1998;111:2953–65. [DOI] [PubMed] [Google Scholar]
- 35. Bribián A, Esteban PF, Clemente D, et al. A novel role for anosmin-1 in the adhesion and migration of oligodendrocyte precursors. Dev Neurobiol 2008;68:1503–16. [DOI] [PubMed] [Google Scholar]
- 36. Soussi-Yanicostas N, de Castro F, Julliard AK, et al. Anosmin-1, defective in the X-linked form of Kallmann syndrome, promotes axonal branch formation from olfactory bulb output neurons. Cell 2002;109:217–28. [DOI] [PubMed] [Google Scholar]
- 37. Clemente D, Ortega MC, Arenzana FJ, et al. FGF-2 and Anosmin-1 are selectively expressed in different types of multiple sclerosis lesions. J Neurosci 2011;31:14899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gianola S, de Castro F, Rossi F.. Anosmin-1 stimulates outgrowth and branching of developing Purkinje axons. Neuroscience 2009;158:570–84. [DOI] [PubMed] [Google Scholar]
- 39. Di Schiavi E, Andrenacci D.. Invertebrate models of Kallmann syndrome: molecular pathogenesis and new disease genes. Curr Genomics 2013;14:2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murcia-Belmonte V, Esteban PF, Martínez-Hernández J, et al. Anosmin-1 over-expression regulates oligodendrocyte precursor cell proliferation, migration and myelin sheath thickness. Brain Struct Funct 2016;221:1365–85. [DOI] [PubMed] [Google Scholar]
- 41. Endo Y, Ishiwata-Endo H, Yamada KM.. Extracellular matrix protein anosmin promotes neural crest formation and regulates FGF, BMP, and WNT activities. Dev Cell 2012;23:305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. de Castro F, Bribian A, Ortega MC.. Regulation of oligodendrocyte precursor migration during development, in adulthood and in pathology. Cell Mol Life Sci 2013;70:4355–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Garcia-Gonzalez D, Murcia-Belmonte V, Clemente D, et al. Olfactory system and demyelination. Anat Rec (Hoboken) 2013;296:1424–34. [DOI] [PubMed] [Google Scholar]
- 44. DeLuca GC, Yates RL, Beale H, et al. Cognitive impairment in multiple sclerosis: clinical, radiologic and pathologic insights. Brain Pathol 2015;25:79–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Renukanthan A, Quinton R, Turner B, et al. Kallmann syndrome patient with gender dysphoria, multiple sclerosis, and thrombophilia. Endocrine 2015;50:496–503. [DOI] [PubMed] [Google Scholar]
- 46. Marlin S, Chantot-Bastaraud S, David A, et al. Discovery of a large deletion of KAL1 in 2 deaf brothers. Otol Neurotol 2013;34:1590–4. [DOI] [PubMed] [Google Scholar]
- 47. McCabe MJ, Hu Y, Gregory LC, et al. Novel application of luciferase assay for the in vitro functional assessment of KAL1 variants in three females with septo-optic dysplasia (SOD). Mol Cell Endocrinol 2015;417:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Suzuki E, Izumi Y, Chiba Y, et al. Loss-of-function SOX10 mutation in a patient with Kallmann syndrome, hearing loss, and iris hypopigmentation. Horm Res Paediatr 2015;84:212–6. [DOI] [PubMed] [Google Scholar]
- 49. Raju R, Dalakas MC.. Gene expression profile in the muscles of patients with inflammatory myopathies: effect of therapy with IVIg and biological validation of clinically relevant genes. Brain 2005;128:1887–96. [DOI] [PubMed] [Google Scholar]
- 50. Tengara S, Tominaga M, Kamo A, et al. Keratinocyte-derived anosmin-1, an extracellular glycoprotein encoded by the X-linked Kallmann syndrome gene, is involved in modulation of epidermal nerve density in atopic dermatitis. J Dermatol Sci 2010;58:64–71. [DOI] [PubMed] [Google Scholar]
- 51. Arikawa T, Kurokawa T, Ohwa Y, et al. Risk factors for surgical site infection after hepatectomy for hepatocellular carcinoma. Hepatogastroenterology 2011;58:143–6. [PubMed] [Google Scholar]
- 52. Kawamata H, Furihata T, Omotehara F, et al. Identification of genes differentially expressed in a newly isolated human metastasizing esophageal cancer cell line, T.Tn-AT1, by cDNA microarray. Cancer Sci 2003;94:699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jian B, Nagineni CN, Meleth S, et al. Anosmin-1 involved in neuronal cell migration is hypoxia inducible and cancer regulated. Cell Cycle 2009;8:3770–6. [DOI] [PubMed] [Google Scholar]
- 54. Liu J, Cao W, Chen W, et al. Decreased expression of Kallmann syndrome 1 sequence gene (KAL1) contributes to oral squamous cell carcinoma progression and significantly correlates with poorly differentiated grade. J Oral Pathol Med 2015;44:109–14. [DOI] [PubMed] [Google Scholar]
- 55. Tanaka Y, Kanda M, Sugimoto H, et al. Translational implication of Kallmann syndrome-1 gene expression in hepatocellular carcinoma. Int J Oncol 2015;46:2546–54. [DOI] [PubMed] [Google Scholar]
- 56. Kanda M, Shimizu D, Fujii T, et al. Function and diagnostic value of anosmin-1 in gastric cancer progression. Int J Cancer 2016;138:721–30. [DOI] [PubMed] [Google Scholar]
- 57. Dellovade TL, Hardelin JP, Soussi-Yanicostas N, et al. Anosmin-1 immunoreactivity during embryogenesis in a primitive eutherian mammal. Brain Res Dev Brain Res 2003;140:157–67. [DOI] [PubMed] [Google Scholar]
- 58. Incerti B, Guioli S, Pragliola A, et al. Kallmann syndrome gene on the X and Y chromosomes: implications for evolutionary divergence of human sex chromosomes. Nat Genet 1992;2:311–4. [DOI] [PubMed] [Google Scholar]
- 59. Iwase M, Satta Y, Hirai H, et al. Frequent gene conversion events between the X and Y homologous chromosomal regions in primates. BMC Evol Biol 2010;10:225.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Harrow JL, Steward CA, Frankish A, et al. The vertebrate genome annotation browser 10 years on. Nucleic Acids Res 2014;42:D771–9. [DOI] [PMC free article] [PubMed] [Google Scholar]