

Abstract
Summary
Identifying genomic regions with higher than expected mutation count is useful for cancer driver detection. Previous parametric approaches require numerous cell-type-matched covariates for accurate background mutation rate (BMR) estimation, which is not practical for many situations. Non-parametric, permutation-based approaches avoid this issue but usually suffer from considerable compute-time cost. Hence, we introduce Mutations Overburdening Annotations Tool (MOAT), a non-parametric scheme that makes no assumptions about mutation process except requiring that the BMR changes smoothly with genomic features. MOAT randomly permutes single-nucleotide variants, or target regions, on a relatively large scale to provide robust burden analysis. Furthermore, we show how we can do permutations in an efficient manner using graphics processing unit acceleration, speeding up the calculation by a factor of ∼250.
Availability and implementation
MOAT is available at moat.gersteinlab.org.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
A common analysis strategy in cancer driver detection is to look for genomic elements with high variant accumulation across patients. However, the background mutation rate (BMR) is highly heterogeneous across the genome due to numerous influences. Inaccurate modeling of BMRs could in turn introduce false positives into cancer driver detection. Our Mutations Overburdening Annotations Tool (MOAT) differs from parametric schemes and does not make any assumption except that the BMR remains constant within a local context.
MOAT offers an annotation-centric algorithm (MOAT-a), a variant-centric algorithm (MOAT-v) and a somatic variant simulator (MOAT-s) built on MOAT-v’s variant placement algorithm. Moreover, we can use MOAT to gauge the functional impact burden of annotations relative to the surrounding genome. MOAT is useful for comparing observed and permuted variant impact scores. Here, we provide an example using FunSeq2 scores (Fu et al., 2014). In the following sections, we describe MOAT’s implementation and recall of known non-coding cancer drivers.
2 Materials and methods
Several covariates jointly affect the BMR in a complicated and dynamic manner, making variant burden analysis very challenging (Lawrence et al., 2013). The length of the test region usually varies from hundreds to thousands of bases, while external features such as replication timing can work at up to a megabase resolution. To address these challenges, MOAT circumvents the need for parametric models by explicitly permuting the variants or annotations within a region where the levels of all the covariates are essentially constant. One important issue with these permutation algorithms is that their running times do not scale well to whole-genome annotation sets. We addressed this issue by taking advantage of large-scale graphics processing unit (GPU) parallelization.
2.1 MOAT-a: annotation-centric permutation
MOAT requires two input files: an annotation file (afile) and a variant file (vfile). MOAT-a uses NVIDIA’s compute unified device architecture language (Nickolls et al., 2008) for general-purpose GPU acceleration (Fig. 1a). MOAT-a iterates through each annotation, computing the intersecting variant count. It defines a genomic block with user-defined boundaries for permuting the annotation n times. MOAT-a then finds the variant counts of the n random bins and compares them to the annotation’s observed variant count to provide empirical P-values. When MOAT-a is used with a variant impact signal file, it generates observed and permuted annotation impact scores by summing the intersecting variants’ impact scores to calculate P-values.
Fig. 1.
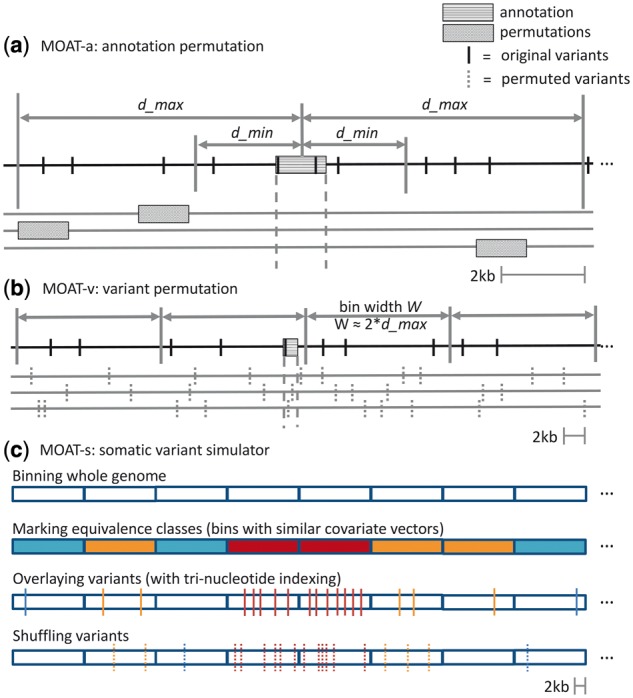
(a) MOAT-a shuffles each annotation to a new location within the local genome context bounded by user-defined parameters d_min and d_max, producing n permutations. (b) In MOAT-v, the whole genome is divided into bins of user-defined width W, within which variants are moved to new coordinates, thereby preserving the local mutation context. As with MOAT-a, MOAT-v produces n permutations. (c) MOAT-s bins the entire genome, whereupon it calculates the covariate values for each bin. The program then clusters bins with similar covariate values, represented here as bins with the same color (we refer to these clusters as equivalence classes). The input variants that fall within each cluster are then permuted to new locations chosen from the bins within the same cluster, honoring trinucleotide context preservation if requested
We can adjust the boundaries of the intervals for choosing permuted annotations—d_min and d_max—to scale the surrounding genome context with respect to the size of the original annotation. Ideally, the permutation intervals will provide enough range to enable non-overlapping sampling. As a rule of thumb, the choice of d_min should be large enough to avoid potential mutation burden signal from ‘bleeding’ into the permutation intervals. Simultaneously, the selected d_max must be small enough that the BMR covariates remain approximately constant within the permutation intervals. For example, in our analysis of transcription start site(s) (TSS) mutation burdens, where TSS are roughly 100 bp in length, we used a d_min of 2 kb and a d_max of 50 kb.
2.2 MOAT-v: variant-centric permutation
MOAT-v creates permuted datasets by assigning new coordinates to each variant within a local genomic region to account for the covariate effects from known genomic features (Fig. 1). MOAT-v and MOAT-s offer the option to preserve the trinucleotide context of the original variant when choosing a new variant location (see Supplementary Material). This constraint reflects the differential mutation probabilities of different trinucleotides while preserving the mutational signatures. MOAT-v generates a permuted dataset by subdividing the genome into blocks of a user-defined size within which variants are permuted, thus generating n permutations (Fig. 1b). We can determine the empirical P-value for each annotation based on the fraction of permutations with variants equal to or greater than the observed variant count. Unlike MOAT-a, we designed MOAT-v to parallelize its workflow across multiple central processing unit (CPU) cores using the OpenMPI framework (Gabriel et al., 2004), due to the more memory intensive nature of the trinucleotide context preservation.
The ability to adjust the width of the whole-genome bins in MOAT-v enables users to select a width that represents regions in which the BMR covariates are expected to be approximately constant. Hence, the permutations that MOAT-v creates will honor the expected density of regional mutations due to these covariates. Our analyses of a few of the most significant covariates, such as DNA replication timing, histone marks and guanine–cytosine content, indicate that a suitable bin size range is 50–100 kb (see Supplementary Material).
2.3 MOAT-s: simulated somatic variant datasets
In addition to the main MOAT programs, we developed a variant simulator, MOAT-s, which reflects the levels of whole-genome covariates that directly influence the BMR. MOAT-s evaluates covariate signals over a set of whole-genome bins. The simulator then clusters these bins based on their covariate signal profiles and allows variants to be permuted not just within their local genome context, but across all bins that share the same covariate signal profile (i.e. across bins in the same equivalence class). Specifically, MOAT-s clusters the whole-genome bins using k means, which use the distances between the bins’ covariate signal profiles to group them into a pre-defined number of clusters (see Supplementary Material).
3 Results
3.1 MOAT-a
We demonstrated the parallel speedup by running MOAT-a on datasets of various sizes. Using a dataset of ∼8 million cancer variants from Alexandrov et al. (2013) and Wang et al. (2014), we used three different annotation sets to demonstrate the scalability of MOAT-a (Harrow et al., 2012; Thurman et al., 2012; Yip et al., 2012). We demonstrate that the GPU version of MOAT-a scales very well with respect to the number of annotations (e.g. ∼9-fold speedup on ∼3 million annotations), and with respect to the number of permutations (e.g. ∼256-fold speedup on 100 000 permutations), resulting in dramatically improved running times (Supplementary Table S1).
Due to the lack of a gold standard, assessing MOAT’s predictions is challenging. Nevertheless, we used the aforementioned cancer variant dataset to demonstrate how MOAT-a can find elevated mutation burdens in genomic elements by identifying highly mutated GENCODE elements. TERT, which has well-documented cancer-associated promoter mutations, carried a significant mutation burden. Other well-known cancer-associated TSS, such as TP53, LMO3 and AGAP5, also had significant mutation burdens.
3.2 MOAT-v and MOAT-s
Using the same set of cancer variants as in the MOAT-a tests, we evaluated MOAT-v’s running time. The running time scales close to linear with the number of CPUs, indicating an even division of labor between each CPU core. MOAT-s’s running time exhibited similar characteristics (data not shown).
We then applied MOAT-v on the same variant and annotation sets to find elevated cancer mutation burdens. MOAT-v produced comparable results as MOAT-a, flagging the same known cancer-associated TSS as significant.
4 Discussion
Here, we introduce MOAT, a new software tool to facilitate identification of high mutation burden. We demonstrate the usefulness of this tool for flagging putative non-coding cancer drivers and provide parallelized versions that dramatically improve running time. Given the demand for efficient and meaningful analysis of genome sequence data, which scientists are producing at very high rates, we believe that MOAT’s provision of such analysis for genetic disease drivers is timely.
Funding
This work was supported by the National Institutes of Health [grant number 5U41HG007000-04].
Conflict of Interest: none declared.
Supplementary Material
References
- Alexandrov L.B. et al. (2013) Signatures of mutational processes in human cancer. Nature, 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y. et al. (2014) FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol., 15, 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel E. et al. (2004) Open MPI: goals, concept, and design of a next generation MPI implementation. Springer, 97–104. [Google Scholar]
- Harrow J. et al. (2012) GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res., 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M.S. et al. (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature, 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickolls J. et al. (2008) Scalable parallel programming w/CUDA. Queue, 6, 40–53. [Google Scholar]
- Thurman R.E. et al. (2012) The accessible chromatin landscape of the human genome. Nature, 489, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. et al. (2014) Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet., 46, 573–582. [DOI] [PubMed] [Google Scholar]
- Yip K.Y. et al. (2012) Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol., 13, R48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.