

Abstract
Motivation
Bioinformatic analyses are becoming formidably more complex due to the increasing number of steps required to process the data, as well as the proliferation of methods that can be used in each step. To alleviate this difficulty, pipelines are commonly employed. However, pipelines are typically implemented to automate a specific analysis, and thus are difficult to use for exploratory analyses requiring systematic changes to the software or parameters used.
Results
To automate the development of pipelines, we have investigated expert systems. We created the Bioinformatics ExperT SYstem (BETSY) that includes a knowledge base where the capabilities of bioinformatics software is explicitly and formally encoded. BETSY is a backwards-chaining rule-based expert system comprised of a data model that can capture the richness of biological data, and an inference engine that reasons on the knowledge base to produce workflows. Currently, the knowledge base is populated with rules to analyze microarray and next generation sequencing data. We evaluated BETSY and found that it could generate workflows that reproduce and go beyond previously published bioinformatics results. Finally, a meta-investigation of the workflows generated from the knowledge base produced a quantitative measure of the technical burden imposed by each step of bioinformatics analyses, revealing the large number of steps devoted to the pre-processing of data. In sum, an expert system approach can facilitate exploratory bioinformatic analysis by automating the development of workflows, a task that requires significant domain expertise.
Availability and Implementation
1 Introduction
Due to the increasing breadth and complexity of bioinformatics analyses, even routine pre-processing of data can be technically challenging. As one example, calling polymorphisms from next-generation sequencing data involves many steps (quality control, read trimming, alignment, recalibration, variant calling, annotation, filtering, etc.) that can be carried out by a range of possible software programs, each with technical idiosyncrasies and distinct semantic requirements (DePristo et al., 2011; Kroigard et al., 2016; McKenna et al., 2010; Wang et al., 2013). A great deal of expertise is required even before the domain-specific analysis and interpretation of the data.
To facilitate analyses, a common strategy is to develop computational pipelines that can link together series of tools in a pre-defined manner. These range in complexity from ad-hoc scripts to more polished software made publicly available. In addition, formal systems to develop pipelines are also used, including GenePattern, Taverna or Galaxy (Goecks et al., 2010; Oinn et al., 2004; Reich et al., 2006). Regardless of the underlying technology, pipelines have been widely adopted because they reduce effort, minimize technical errors, implicitly capture the expertise of the developer, and increase the reproducibility of the results (Hoon et al., 2003).
The main drawback of pipelines as currently implemented is that they are typically developed for a single purpose, within a narrow domain, and are not designed for any substantial extensibility. In practice, the choice of algorithms used in pipelines are relatively limited, and changing them may require re-coding one or more steps in the pipeline. Thus, they are difficult to use for exploratory analysis, which is critical for bioinformatics as frequently multiple approaches need to be attempted. Further, as more methods are being developed, the number of potential pipelines grows exponentially. To manage these challenges, a new framework must be developed that can accommodate the flexibility that is demanded in an exploratory bioinformatic analysis.
To facilitate pipeline development, we have investigated using expert systems. Their capabilities closely match the needs of bioinformatics as they provide the ability to advise (which programs to run), plan (the sequence of programs to run), document (what was run and how), as well as execute (run the programs). More specifically, for a typical bioinformatic analysis, we surmise that the desired goal (e.g. a set of variant calls) is known a priori, and help is needed to determine the sequence of steps required to produce the output correctly. This can be provided by a backwards-chaining expert system.
An expert system consists of a knowledge base of rules and an inference engine that operates on the rules. Each rule describes formally how a set of inputs (the antecedents) is converted to an output (the consequent), and typically corresponds to a software program. The inference engine then reasons on the rules to produce a workflow that generates the target of interest. In this manuscript, we use workflow to mean the set of computations, including all valid algorithms and parameters, that can generate a desired output. A workflow can be represented as a graph. In contrast, a pipeline is a linear path through a workflow representing a series of computations with a specific set of parameters.
Inference engines have previously been used in bioinformatics, including makefiles, Snakemake or Ruffus (Feldman, 1979; Goodstadt, 2010; Koster and Rahmann, 2012). These systems link together a set of computational steps according to user defined constraints. Another project, Wings takes a different approach by applying previously developed templates for common workflows, and then leveraging an expert system to help users instantiate the templates with specific algorithms and datasets by validating the constraints imposed by their choices (Gil et al., 2011). Workflows for genomic analysis are being developed in Wings, including those that perform association tests, copy number variant detection, and variant calling (Gil et al., 2013). However, these systems cannot develop workflows autonomously.
One challenge in applying an expert system to bioinformatics stems from the richness of the biological data. For example, one commonly used data object is a matrix of gene expression values. The matrix can be described by many orthogonal attributes (e.g. has log transformation been applied, are there missing values, how are the genes sorted, etc.), and the number of possible states grows exponentially with the number of attributes. Therefore, an expert system for bioinformatics must have the ability to model the richness of the data, and possess strategies to manage the exponential complexity of the reasoning.
Outside of bioinformatics, backwards chaining inference engines, such as Jess or Prolog, can represent the data objects in a more structured format (Colmerauer and Roussel, 1996; Friedman-Hill, 2003). However, it is difficult to manage the exponential search in these systems. Second, they also lack an ability to integrate with external tools that can perform introspection to ascertain the state of the input data, which can dynamically alter the execution of the pipeline. As an example for gene expression analysis, this introspection would allow the system to check for potential batch effects (Leek et al., 2010), and if necessary, apply the tools to remove them.
To determine the feasibility of in silico generation of workflows, we developed an expert system, BETSY (Bioinformatics ExperT SYstem), that combines a rich data model with an inference engine tuned to reduce the exponential expansion of the search (Fig. 1). Users interact with the system by specifying the desired target (e.g. a set of differentially expressed genes), allowing BETSY to propose a solution, and then entering a dialog with the system to refine the proposed pipeline (e.g. indicating preferences for algorithms or parameters). Finally, BETSY is able to run the pipeline and generate the result. Currently, the BETSY knowledge base is populated with modules focused on the analysis of microarray and next generation sequencing data, enabling it to develop workflows to perform analyses that include differential expression analysis, clustering, classification, variant calling, peak calling, and visualization. Importantly, the expert system provides the ability to develop, refine and execute these complex analyses in a flexible and reproducible manner.
Fig. 1.

BETSY Architecture. (A) The BETSY knowledge base system consists of a Rule Base that describes (1) the types of data encountered in bioinformatics, and (2) the ability of bioinformatics software to transform data from one type to another. Given these components, the Inference Engine uses a backwards-chaining algorithm to produce a pipeline that describes how to generate a target object of interest, given the inputs available. Then, the Processing Engine can run the pipeline to produce the output requested. (B) This shows the structure of an example rule from the knowledge base. This rule describes a module that aligns next generation sequencing reads using the Bowtie2 algorithm. It requires a set of antecedents including a folder of the FASTQ format files to be aligned, a mapping of the files to samples, a description of the orientation of the reads, as well as the reference genome. Given these inputs, the module produces a folder of BAM files. The Constraints set the conditions on the antecedents that must exist before this rule can be applied. For instance, the FASTQ files cannot be compressed. However, the adapters may or may not be trimmed. The Consequences describe the changes that are imposed on the consequent after this module is run. Here, the aligner attribute for the BAM files is set to bowtie2
2 Materials and methods
2.1 BETSY knowledge base
The BETSY knowledge base is built upon data and modules that represent computations to be applied to the data. Data objects (e.g. a database identifier, a file containing a gene expression matrix, or a folder of BAM files) typically correspond to files or directories. Each data object is associated with a set of key-value attributes (e.g. format of gene expression file, whether data is logged, or the algorithm used to preprocess) that further parameterize it for inference.
Modules convert one or more input data objects into exactly one output data object. They are generally implemented as software programs, and the semantics of what they do are described by rules. Rules are structured descriptions of how inputs are converted to an output, and consist of (1) one or more input data objects (antecedents), (2) exactly one output object (consequent), (3) a set of zero or more constraints that describe restrictions on the attributes of the input data objects (e.g. this rule only accepts gene expression matrices that have not been logged) and (4) a set of zero of more consequences, that describe the state of the attributes of the output data object (e.g. this rule produces a gene expression matrix that is logged).
2.2 Inference engine
Given a desired target data object (with specific attributes), the BETSY inference engine applies the rules to generate a data flow graph. To do this, it leverages a FOL-BC-Ask strategy that is modified to handle the attributes associated with the data objects (Russell and Norvig, 2009). To minimize the exponential expansion, it uses a heuristic to merge data objects when possible (e.g. if the downstream algorithms are indifferent to the log states of the gene expression matrix, then no distinction need be made between matrices with different states). Secondly, when searching for pipelines, it aggressively prunes the search in the branches where no solutions are possible (e.g. if no BAM files are provided or can be generated, then it does not search for any solutions requiring BAM files).
The inference engine generates a bipartite directed acyclic graph, where one class of nodes represent data, and the other modules. To describe the graph in a way that facilitates comparison with those of other workflow systems, we follow a framework previously used to review Taverna and other systems (Curcin and Ghanem, 2008). The workflow graph generated by BETSY allows no loop structures and no logical (e.g. AND, OR, NOT) branching constructs. IF statements are handled and enable the system to perform introspection. They are implemented as module nodes that produce more than one possible alternative output data nodes. The specific one is determined at run time. For example, a gene expression record downloaded from the GEO database may contain Affymetrix CEL files or Illumina IDAT files, which require different methods for preprocessing. In this situation, BETSY will generate a workflow that downloads the data, and then executes a module (identify_type_of_expression_files) that identifies the type of data, allowing control to be directed toward the pipeline downstream that is capable of processing the data downloaded.
In the BETSY data model, there is no explicit handling of collections of data objects. The BETSY inferencing engine can only compute on individual data objects. However, the data objects themselves can be associated with either files or directories that are handled by the modules. As an example, there is a data type called a FastqFolder that represents a folder of FASTQ formatted files. Although this appears as an individual unit to the inference engine, the modules can perform operations over this entire collection of files.
Finally, the execution of the workflow passes discretely from one module node to the next. Each module must be completed before the next one begins. Streaming, in which execution passes from a partially completed module to the next, is not supported.
While the knowledge base is specific to bioinformatics, the inference engine contains no domain-specific functionality, and can, in principle, be applied to other domains.
2.3 Processing engine
BETSY also contains a processing engine. Given a data flow graph and set of inputs, it can execute the graph, running the appropriate programs to generate the target output. To facilitate exploratory analysis, where the same analysis may be re-run with few attributes changed, it uses a caching strategy so that only the steps affected by the changes will be re-run. Further, the processing engine can be run concurrently. Before running a program, BETSY can detect if another instance is already running the same program on the same inputs, and will wait for the first instance to finish rather than replicating the computation or overwriting the result. Finally, to ensure reproducibility, the processing engine documents the software, version (when available), parameters and other salient properties.
2.4 Implementation
BETSY is implemented in Python 2 and calls data analysis software implemented on a range of platforms, including R, Matlab, LINUX binaries and via the internet. The knowledge base, and thus the capabilities of BETSY, is extensible by adding rules (implemented as Python objects) that execute new analysis software.
BETSY is currently usable via a command line interface. In a typical interaction with the system, the user begins by browsing through the knowledge base to identify the desired output data type. Then, they ask BETSY to develop an initial workflow that can produce this data. The user examines this workflow, which can be rendered visually as a graph or as text. Typically, the workflow will require further iterative refinement, which is accomplished by specifying attributes of appropriate data objects. For example, when creating a workflow to generate a heat map of a gene expression data set, the user can specify the algorithm used to preprocess the data, the approach to normalize the data, or the number of genes to plot by setting the desired attribute on the SignalFile object, Similarly, the color scheme or size of the heatmap can be configured by setting attributes on the Heatmap object. Once the user is satisfied with the workflow, they may then elect to execute it to produce the desired output.
The BETSY modules are implemented to take advantage of multi-processor machines and, when possible, use as many cores as the user allocates to the BETSY process. We typically run jobs distributed over 40 cores on a 96 core machine. It has been applied to medium-sized next generation sequencing data sets comprised of over 100 mammalian RNA-Seq or Whole Exome Sequencing samples.
3 Results
3.1 Meta-analysis of the bioinformatics landscape
Within the BETSY framework, we have developed a knowledge base focused on common bioinformatics analyses encountered by the UTHealth Bioinformatics Service Center (Chang, 2015). The rules are focused on microarray pre-processing (preprocessing, batch effect removal), next generation sequencing analysis (alignment, gene expression estimation, variant calling), as well as common downstream processing (pathway analysis, clustering, classification, visualization). Currently, the knowledge base consists of 233 rules covering 119 types of data and 339 attributes on those data.
To describe the BETSY knowledge base, we generated a network that reveals the scope of bioinformatics that it can perform. Since there is not a single target that can utilize the entire knowledge base, we approximated the extent of the knowledge by enumerating a broad set of targets and asked BETSY to generate graphs that can produce those targets. Then, we merged the resultant graphs into a single network (Fig. 2A). This network summarizes the common tasks that we have encountered in bioinformatics and encoded into BETSY. To understand the nature of these analyses, we categorized the steps into different types of activities, including Acquisition (downloading data from databases), Data Wrangling (non-statistical conversion of data, e.g. standardizing the columns of a data file), Preprocessing (statistical conversion of the form of the data, e.g. RMA preprocessing of microarray data), Normalization (adjusting the values of the data for consistency or to conform with assumptions), Analysis (application of bioinformatic algorithms to derive information from the data), Visualization, Summarization (organizing the data for human interpretation) and Introspection (eliciting properties of the data for processing). While Analysis (e.g. clustering microarray data, predicting disease outcomes, etc.) is the key step of the pipeline, and what is typically considered bioinformatics, preparing the data for analysis comprises the bulk of the network. Indeed, the graph resembles an iceberg where the mass of data preparation is hidden under the desired bioinformatic analysis.
Fig. 2.

Summarization of the BETSY knowledge base. (A) This directed acyclic graph shows the common outputs that can be generated by BETSY. The square nodes represent modules, and the intervening small circles represent the data that is computed upon. The colors of the module nodes indicate the type of work done. Each path through this graph constitutes a pipeline that can be executed. The potential inputs to the network are shown on the bottom, and the output data types are placed at the top; the pipelines flow from the bottom upwards. At the bottom, the figures is labeled according to the data that is handled. The text labels show the analyses being performed. The dashed line roughly separates the Analysis nodes (on top), those that represent the algorithmic computation that produces the desired outcome, from the steps to prepare the data for analysis (on bottom). (B) (Left panel) This table shows the total number of module nodes for each type of work (Total) column, as well as the number of transitions that originate from each type of module (rows) to the next type of module (columns). (Right panel) This graph is a visualization the transitions shown in the table. Each circle represents a type of module, and are colored the same as panel A. The area of the circles are relative to the number of modules. The arrows indicate transitions from one type to the next, and their relative sizes correspond to the number of transitions
By quantifying the type of modules included in the BETSY network, we revealed that 42% of the steps are comprised of data wrangling, roughly comparable with previous estimates of 50–80% (Endel, 2015; Lohr, 2014). While we previously found that the actual time spent on non-analysis activities was in the minority (Chang et al., in review), this nevertheless demonstrates quantitatively the large technical burden imposed by data wrangling (Fig. 2B). Further, a comparable number of steps were devoted to introspection, signifying the abundance of incremental decisions that need to be made based on the state of the data. Taken together, this illustrates that the majority of the steps in performing bioinformatics do not require creative intellectual input, but instead, expertise is required in the construction and selection of the proper pipelines, and the specification of the target goal.
3.2 The knowledge base can recreate prior analyses
To determine whether BETSY has the capacity to perform useful bioinformatics, we tested whether the system can reproduce the results of previously published analyses. We chose to reproduce a landmark cancer classification result (Golub et al., 1999), as well as two others selected from a study that identified reproducible microarray results (Ioannidis et al., 2009).
For the first case, the original study reported on a machine learning classifier to distinguish two leukemia subtypes, ALL and AML, based on gene expression patterns found in microarray data. To reproduce this, we used BETSY to generate a network that will produce classification predictions, using the same algorithm (weighted voting) and leave-one-out cross validation (Fig. 3A). Although BETSY opted to use a support vector machine (Noble, 2006), an algorithm that is commonly used and shown to perform well across a range of applications, we changed this to be consistent with the previous analysis by specifying that the classification results should be generated with the weighted voting classifier, causing BETSY to revise the workflow. Then, based on the inputs we provided, along with the gene expression data and class labels that were made available with the original manuscript, BETSY selected a pipeline that included steps for preprocessing, gene filtering and thresholding, introspection for missing values and log normalization, and then classification with cross validation.
Fig. 3.
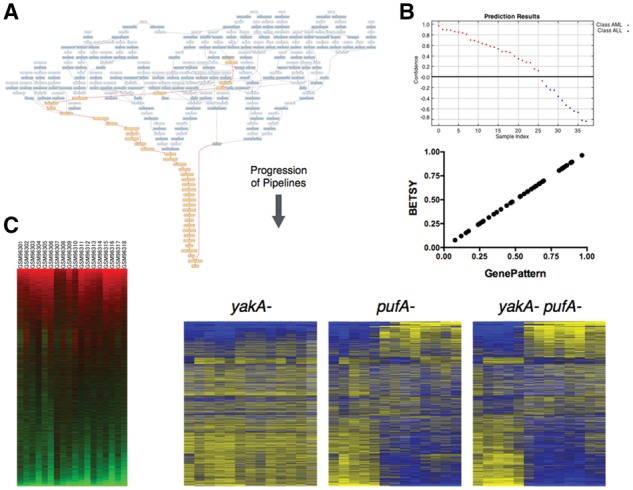
Reproduction of prior analyses. (A) This graph shows the workflow that can reproduce the cancer classification problem reported in (Golub et al., 1999). In contrast to Figure 2A, the inputs here are shown at the top of this network, and the pipelines flow downward to the output at the bottom. The dark blue nodes represent modules, and the light blue ones indicate data. The highlighted yellow nodes show the pipeline given the input files (green). (B) (Top) The predictions for the classes based on the pipeline published in the original manuscript. (Bottom) This scatter plot compares the scores predicted from the GenePattern (x-axis) pipeline following the original manuscript, with the scores from the pipeline produced by BETSY (y-axis). (C) These heatmaps (but not the labels) were generated using pipelines from BETSY. The one in the left panel is a reproduction of Figure 5 in Loh et al. (2006), and right panel Figure 1C in Van Driessche et al. (2005)
Running the pipeline resulted in predicted scores for each of the samples in the dataset (Fig. 3B). We compared the scores generated against the published scores and found that they were identical. This demonstrates that BETSY had the ability to create a pipeline that could accurately generate useful bioinformatic results.
To test whether BETSY can reproduce the results of other manuscripts, we applied it to reproduce a result from two other manuscripts (Loh et al., 2006; Van Driessche et al., 2005). With the data from these manuscripts, we used BETSY to re-create their published heatmaps. In contrast to the classification example, these pipelines required BETSY to acquire data, filter genes for missing values, zero fill missing values, reorder genes, and finally create a heatmap given specifications for color and size. The resultant heatmaps reproduced the ones published in Figure 5 (Loh et al., 2006) and Figure 1C (Van Driessche et al., 2005) in the original manuscripts (Fig. 3C).
3.3 Expert systems facilitate exploratory analyses
Since the ALL/AML manuscript described above was published, there have been further advances in the development of machine learning algorithms. Therefore, to test the flexibility by which BETSY can alter workflows, we repeated the classification task, except replacing the weighted voting classifier with a more modern random forests classifier (Breiman, 2001). We requested a change in the classification algorithm, resulting in a new workflow. After running it, we found that the random forests classifier could perform more accurately than weighted voting (both with leave-one-out cross validation) (Fig. 4). On the training set, random forests made no classification errors, while weighted voting made one. On the test, random forests had one error, while weighted voting had two. Nevertheless, comparing the accuracy of the methods is not the goal of this analysis, but instead, this result demonstrates that a knowledge based system facilitates the application of new algorithms by dynamically generating new workflows.
Fig. 4.
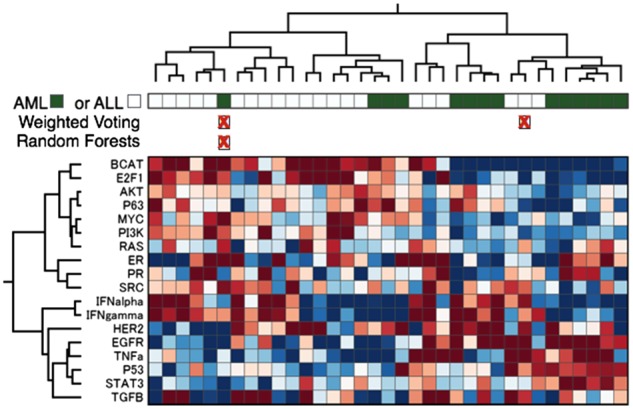
Exploratory analyses. This figure shows the results of a supervised machine learning classification (top) and pathway analysis (bottom heatmap) of the ALL/AML test set. Each column contains an AML or ALL cancer sample. In the bar directly below the top dendrogram, AML samples are colored with a dark green box, and ALL white. The following rows indicate the samples, marked with a red X, that Weighted Voting or Random Forests classifiers predicted incorrectly. The heatmap shows the results of a pathway prediction. Red indicates high activation of pathways (rows), and blue low
We next applied BETSY to perform a pathway analysis on the same dataset with the SIGNATURE toolkit (Chang et al., 2011; Gatza et al., 2010). Because SIGNATURE was described in its knowledge base, BETSY could infer and execute a new workflow that generates pathway predictions rather than cancer type predictions. As a result, while the original paper demonstrated that AML and ALL shared distinct gene expression patterns, this new analysis revealed that they also differ in terms of predicted pathway activities. The majority of the ALL samples cluster on the left, and exhibited relatively higher E2F, β-Catenin, Myc and PI3K activation, while the AML samples on the right have relatively higher activation of HER2, EGFR and TNF-α. Thus, with very little work from the user, BETSY could apply the same dataset in a different workflow that provides further insight into the biology of the data.
4 Discussion
It has been argued that spreadsheets were a boon to finance, because they allowed users to consider ‘what if’ questions. Given the diversity seen across bioinformatics projects (Chang, 2015), an analogous ability to try a range of approaches is needed. Here, we have demonstrated that an expert system can provide the flexibility to support exploratory analyses.
One consequence of switching from hand crafted scripts to an expert system to run analysis is that the maintenance of the system is converted from a software maintenance task to knowledge base curation. Although BETSY can document what was done in an analysis, creating the workflow itself depends on the knowledge base, which may be frequently updated to add new rules, potentially impacting existing capabilities. While there is a substantial literature on the development and curation of knowledge bases, arguably the analogs in software are currently better understood. Nevertheless, we argue that the higher level of abstraction provided by a knowledge base makes it easier to maintain, but this is not yet evident given the more limited experience with this approach.
While much attention has been devoted to developing biologist friendly software (Chang et al., 2011), we do not believe that it is appropriate to use BETSY in this way. BETSY could in principle be used as a black box without an understanding of the workflow developed. In most cases, this would generate correct results. However, biology is complex, and there will always be situations that BETSY does not understand. For example, we recently used BETSY to call variants from RNA-Seq data. Because it had no knowledge of RNA, it produced a workflow to call variants from DNA-Seq data, ignorant of the splicing that occurs in the RNA transcripts. This error was detected by manually reviewing the workflow. To handle this situation correctly, we had to expand the BETSY knowledge base by adding attributes to distinguish RNA and DNA data transcripts so the right alignment strategy would be selected. This both illustrates that BETSY possesses an incomplete (yet ever expanding) knowledge of biology, and also highlights the value of having a theoretical framework that enables to us to explicitly encode and build upon expert knowledge.
Acknowledgements
We thank members of the Chang lab and the UTHealth Bioinformatics Service Center for testing the software. We thank Micheal Hewett for helpful discussions.
Funding
This work was supported by grants R1006 and UL1 TR000371 from the Cancer Prevention and Research Institute of Texas and TL1TR000371 from the National Institutes of Health.
Conflict of Interest: none declared.
References
- Breiman L. (2001) Random forests. J. Mach. Learn., 45, 5–32. [Google Scholar]
- Chang J. (2015) Core services: reward bioinformaticians. Nature, 520, 151–152. [DOI] [PubMed] [Google Scholar]
- Chang J.T. et al. (2011) SIGNATURE: a workbench for gene expression signature analysis. BMC Bioinformatics, 12, 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J.T. et al. Fulfilling the need for bioinformatics in biomedical research. in review.
- Colmerauer A., Roussel P. (1996) The Birth of Prolog In: Bergin T.J.J., Gibson R.G.J. (eds) History of Programming Languages–II. ACM, New York, NY, USA, p. 331–367. [Google Scholar]
- Curcin V., Ghanem M. (2008) Scientific workflow systems – can one size fit all? In: Proceedings of the 2008 IEEE, CIBEC'08. Cairo, Egypt.
- DePristo M.A. et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet., 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endel F. (2015) Data Wrangling: Making data useful again. 8th Vienna Int. Conf. Math. Modell., 48, 111–112. [Google Scholar]
- Feldman S.I. (1979) Make—a program for maintaining computer program. Software, 9, 255–265. [Google Scholar]
- Friedman-Hill E. (2003) Jess in Action. Manning Publications, Greenwich, CT. [Google Scholar]
- Gatza M.L. et al. (2010) A pathway-based classification of human breast cancer. Proc. Natl. Acad. Sci. U. S. A., 107, 6994–6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil Y. et al. (2011) A semantic framework for automatic generation of computational workflows using distributed data and component catalogs. J. Exp. Theor. Artif. Intell., 23, 389–467. [Google Scholar]
- Gil Y. et al. (2013) Using Semantic Workflows to Disseminate Best Practices and Accelerate Discoveries in Multi-Omic Data Analysis. In: Conference of the Association for the Advancement of Artificial Intelligence. Bellevue, WA.
- Goecks J. et al. (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol., 11, R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub T.R. et al. (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science, 286, 531–537. [DOI] [PubMed] [Google Scholar]
- Goodstadt L. (2010) Ruffus: a lightweight Python library for computational pipelines. Bioinformatics, 26, 2778–2779. [DOI] [PubMed] [Google Scholar]
- Hoon S. et al. (2003) Biopipe: a flexible framework for protocol-based bioinformatics analysis. Genome Res., 13, 1904–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis J.P. et al. (2009) Repeatability of published microarray gene expression analyses. Nat. Genet., 41, 149–155. [DOI] [PubMed] [Google Scholar]
- Koster J., Rahmann S. (2012) Snakemake–a scalable bioinformatics workflow engine. Bioinformatics, 28, 2520–2522. [DOI] [PubMed] [Google Scholar]
- Kroigard A.B. et al. (2016) Evaluation of nine somatic variant callers for detection of somatic mutations in exome and targeted deep sequencing data. PLoS One, 11, e0151664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek J.T. et al. (2010) Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet., 11, 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh Y.H. et al. (2006) The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet., 38, 431–440. [DOI] [PubMed] [Google Scholar]
- Lohr S. (2014) For Big-Data Scientists, ‘Janitor Work’ Is Key Hurdle to Insights. The New York Times (August 17).
- McKenna A. et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble W.S. (2006) What is a support vector machine? Nat. Biotechnol., 24, 1565–1567. [DOI] [PubMed] [Google Scholar]
- Oinn T. et al. (2004) Taverna: a tool for the composition and enactment of bioinformatics workflows. Bioinformatics, 20, 3045–3054. [DOI] [PubMed] [Google Scholar]
- Reich M. et al. (2006) GenePattern 2.0. Nat. Genet., 38, 500–501. [DOI] [PubMed] [Google Scholar]
- Russell S., Norvig P. (2009) Artificial Intelligence: A Modern Approach. Prentice Hall, Upper Saddle River, NJ.
- Van Driessche N. et al. (2005) Epistasis analysis with global transcriptional phenotypes. Nat. Genet., 37, 471–477. [DOI] [PubMed] [Google Scholar]
- Wang Q. et al. (2013) Detecting somatic point mutations in cancer genome sequencing data: a comparison of mutation callers. Genome Med., 5, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]