

Abstract
Motivation
Global analysis of translation regulation has recently been enabled by the development of Ribosome Profiling, or Ribo-seq, technology. This approach provides maps of ribosome activity for each expressed gene in a given biological sample. Measurements of translation efficiency are generated when Ribo-seq data is analyzed in combination with matched RNA-seq gene expression profiles. Existing computational methods for identifying genes with differential translation across samples are based on sound principles, but require users to choose between accuracy and speed.
Results
We present Riborex, a computational tool for mapping genome-wide differences in translation efficiency. Riborex shares a similar mathematical structure with existing methods, but has a simplified implementation. Riborex directly leverages established RNA-seq analysis frameworks for all parameter estimation, providing users with a choice among robust engines for these computations. The result is a method that is dramatically faster than available methods without sacrificing accuracy.
Availability and Implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Regulation of translation has been shown to play an essential role in many biological processes, and misregulation of translation is associated with disease states (Bazzini et al., 2012; Lu et al., 2007). The recent development of Ribosome Profiling technology, also called Ribo-seq, now enables accurate global analysis of translation (Ingolia et al., 2009). Ribo-seq involves deep sequencing of ‘ribosome-protected fragments’ (RPFs) which are presumed to exist in proportion to the fraction of total translation that is associated with a given gene. RPFs can also provide information on the modes by which cells regulate translation, and to date Ribo-seq has been applied in a broad variety of biological contexts (Ingolia, 2016).
Often the goal of a Ribo-seq experiment is to generate genome-wide maps of translation efficiency. The number of RPFs associated with a given gene or transcript is determined both by gene-specific translation activity and mRNA abundance. Without accounting for background mRNA levels, one cannot distinguish differences associated with translation from those arising via regulation of other process (e.g. transcription). Ribo-seq data is therefore usually accompanied by RNA-seq conducted in a matched biological sample. The key analysis problem then becomes identifying genes whose difference in RPF abundance cannot be explained by differences in background mRNA abundance. Several methods were developed for this purpose, including anota (Larsson et al., 2011), Babel (Olshen et al., 2013), RiboDiff (Zhong et al., 2016) and Xtail (Xiao et al., 2016). These methods differ in details of parameter estimation and implementation, but are all founded on a similar model for the differential translation problem.
We developed Riborex as a simple method to identify genes exhibiting differential translation from Ribo-seq data. The name Riborex is derived from the objective of analyzing Ribo-seq ratios with expression. Similar to previous approaches, ours involves modeling a natural dependence of translation on mRNA levels as a generalized linear model (GLM). Unlike previous methods, we directly leverage existing model-based frameworks for gene expression analysis, using them to simultaneously estimate all parameters of our model. As a consequence, Riborex is significantly faster than all existing approaches, supports general experimental designs, and employs robust and mature software implementations for the underlying statistical calculations.
2 Approach
Established frameworks for RNA-seq data analysis, e.g. edgeR (Robinson et al., 2010) and DESeq2 (Love et al., 2014), model the read count ygi from gene g in sample i as following a negative binomial distribution. Expected counts satisfy
where Ni is the total counted reads, xj is a covariate vector associated with the treatment condition j, and βg is the corresponding vector of gene-specific coefficients. The expected proportion λgj of reads from gene g may then be regarded as the expression level of gene g in condition j. The negative binomial distribution includes a dispersion parameter for each gene. The need to consider dispersion parameter is clear, but how best to estimate dispersion remains an active area of research. Our strategy for detecting differentially translated genes models read counts rgi from Ribo-seq in a similar way:
where Ri is the total counted reads for sample i. The parameter αg may be thought of as representing how the translation of g differs from its background expression level. One interpretation is that any regulation specifically at the level of translation will be captured by the coefficients vector αg. Identifying differential translation between conditions then amounts to testing contrasts concerning α. We combine RNA-seq and Ribo-seq read counts by constructing a specific design matrix (details in supp. info.). Then all coefficients α and β can be fit simultaneously in existing RNA-seq data analysis frameworks (i.e. the ‘engine’ used by Riborex). The dispersion for each gene is estimated using an approach specific to the engine used.
3 Results
We evaluated Riborex in comparison with existing methods using a simulation approach based on modifying real data to introduce differences and examining accuracy in identifying those differences. We compared Riborex with a representative set of existing methods: Xtail (Xiao et al., 2016), RiboDiff (Zhong et al., 2016) and Babel (Olshen et al., 2013), acknowledging that more comprehensive comparisons already exist in the literature (Xiao et al., 2016). Briefly, from a published dataset with 5 replicates (Schafer et al., 2015), we randomly selected 4 and divided those into two groups of two replicates. We randomly sampled 1% of ∼15k expressed genes and assigned them a fold change of 4 in both RNA-seq and Ribo-seq data; these, along with unchanged genes, are considered true negatives. As positive control, we randomly sampled 1% of genes and assigned a different fold change to RNA-seq and Ribo-seq to introduce 4-fold change in translation efficiency (details in supp. info.). Riborex, Xtail, RiboDiff and Babel were applied to this semi-simulated data. We set a FDR cutoff at 0.05 and calculated the F scores for different methods. We repeated the simulations with larger control sets (10% of genes) and also on a second dataset (Diaz-Muñoz et al., 2015). The results of 100 such simulations are summarized in Figure 1. The methods give extremely similar performance, with Riborex (using all three engines) and Xtail having a slight advantage over RiboDiff and Babel. Repeating the simulation with relatively small differences (2×) in translation efficiency yielded decreased accuracy for most methods (details in supp. info.).
Fig. 1.
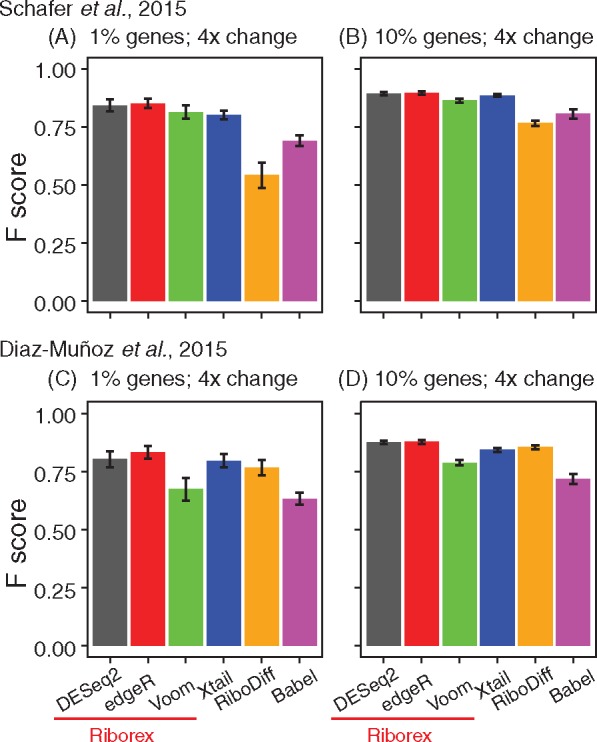
Accuracy identifying differentially translated genes in simulated data (see supp. info.) comparing Riborex (using DESeq2, edgeR and Voom (Law et al., 2014)), Xtail, RiboDiff and Babel. Values are averages of 100 simulations and error bars indicate standard deviation of F scores. (A) and (B) Results based on rat heart data (Schafer et al., 2015) with 1% and 10% implanted true differentially translated genes, respectively, and simulated 4-fold change in translation efficiency. (C) and (D) Results from simulations based on mouse HuR data (Diaz-Muñoz et al., 2015)
We measured running time of each method based on 4 datasets differing in numbers of genes (mouse vs. human) and numbers of replicates (data in supp. info.) (Bennett et al., 2016; Hsieh et al., 2012; Schafer et al., 2015; Zur et al., 2016). Riborex finishes in seconds; all other methods take substantially longer, with Xtail requiring > 4 hours (Table 1).
Table 1.
Running time of Riborex (using DESeq2, edgeR and Voom as engine), Xtail, RiboDiff and Babel on four published Ribo-seq datasets
| Riborex engine | ||||||
|---|---|---|---|---|---|---|
| Dataset | DESeq2 | edgeR | Voom | Xtail | RiboDiff | Babel |
| MSI2 | 39 s | 12 s | 7 s | 4.34 h | 29.60 min | 23.53 min |
| Hela S3 | 49 s | 16 s | 7 s | 5.31 h | 32.55 min | 25.47 min |
| mTOR | 23 s | 8 s | 4 s | 4.16 h | 34.07 min | 15.27 min |
| Liver | 56 s | 19 s | 8 s | 5.69 h | 26.22 min | 54.30 min |
In summary, Riborex is implemented as an open source R package, and has accuracy on par with the most accurate existing methods, but is significantly faster.
Funding
This work has been supported by NIH grant R01 HG006015.
Conflict of Interest: none declared.
Supplementary Material
References
- Bazzini A.A. et al. (2012) Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science, 336, 233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett C.G. et al. (2016) Genome-wide analysis of Musashi-2 targets reveals novel functions in governing epithelial cell migration. Nucleic Acids Res., 1, gkw207.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Muñoz M.D. et al. (2015) The RNA-binding protein HuR is essential for the B cell antibody response. Nat. Immunol., 16, 415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh A.C. et al. (2012) The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature, 485, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T. (2016) Ribosome footprint profiling of translation throughout the genome. Cell, 165, 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T. et al. (2009) Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science, 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson O. et al. (2011) anota: analysis of differential translation in genome-wide studies. Bioinformatics, 27, 1440–1441. [DOI] [PubMed] [Google Scholar]
- Law C.W. et al. (2014) Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol., 15, R29.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I. et al. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P. et al. (2007) Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol., 25, 117–124. [DOI] [PubMed] [Google Scholar]
- Olshen A.B. et al. (2013) Assessing gene-level translational control from ribosome profiling. Bioinformatics, 29, 2995–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D. et al. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer S. et al. (2015) Translational regulation shapes the molecular landscape of complex disease phenotypes. Nat. Commun., 6, 7200.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z. et al. (2016) Genome-wide assessment of differential translations with ribosome profiling data. Nat. Commun., 7, 11194.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y. et al. (2016) RiboDiff: detecting changes of translation efficiency from ribosome footprints. bioRxiv, 1, 017111.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zur H. et al. (2016) Complementary post transcriptional regulatory information is detected by punch-p and ribosome profiling. Sci. Rep., 6, 21635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.