

Abstract
Motivation: We introduce PRINCESS, a privacy-preserving international collaboration framework for analyzing rare disease genetic data that are distributed across different continents. PRINCESS leverages Software Guard Extensions (SGX) and hardware for trustworthy computation. Unlike a traditional international collaboration model, where individual-level patient DNA are physically centralized at a single site, PRINCESS performs a secure and distributed computation over encrypted data, fulfilling institutional policies and regulations for protected health information.
Results: To demonstrate PRINCESS’ performance and feasibility, we conducted a family-based allelic association study for Kawasaki Disease, with data hosted in three different continents. The experimental results show that PRINCESS provides secure and accurate analyses much faster than alternative solutions, such as homomorphic encryption and garbled circuits (over 40 000× faster).
Availability and Implementation: https://github.com/achenfengb/PRINCESS_opensource
Contact: shw070@ucsd.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
The past decade has witnessed rapid advances of human genomic sequencing technologies and their wide applications to healthcare and biomedicine (Sudmant et al., 2015). The effective and efficient utilization of human genomic data in biomedical research, in particular for devising novel diagnostic, therapeutic, and prognostic tools tailored to individual patient was laid out in the Precision Medicine Initiative (Collins and Varmus, 2015). An important consideration in using these data is the privacy concern of data donors, because genomic data carry sensitive information that may reveal identity (Gymrek et al., 2013), predisposition to diseases (McGuire et al., 2008) and even facial features (Claes et al., 2014) of data donors. Such disclosure of human genomic data may have negative impact on people beyond the individual from whom the data were collected, and may propagate the privacy risks to blood relatives (Bloss, 2013). Individuals have marked differences in the way they want their data utilized for research (Kim et al., 2015, 2016). More importantly, data are irrevocable once they are disseminated, and new privacy threats may emerge over time with new discoveries of human genetics and the advance of attack methods (Shringarpure and Bustamante, 2015). Due to these concerns (Homer et al., 2008), many aggregate results have been removed from the public domain hosted by NIH (2007).
Security researchers have made significant progress on cryptographic techniques to support secure genomic data analyses (Ayday et al., 2014; He et al., 2014; Kamm et al., 2013; McLaren et al., 2016; Shimizu et al., 2016b; Xie et al., 2014; Zhao et al., 2015) as well as on approaches that compute with horizontally (Wang et al., 2013; Wu et al., 2012) and vertically partitioned data (Li et al., 2016). For example, homomorphic encryption (HME) (Halevi and Shoup, 2014) allows data owners to securely outsource genomic data analysis. However, HME-based techniques are demanding in both computation and storage. It is also challenging for HME to handle complicated genomic analysis tasks, as it only supports a limited set of arithmetic operations (i.e. addition and multiplication). Several HME-based approaches have been developed using HElib (Halevi and Shoup, 2014) for association tests (Lauter et al., 2014) and exact logistic regression model learning (Wang et al., 2016), but these methods are only applicable to small datasets. Another popular approach, secure multiparty computation (SMC), was proposed more than three decades ago (Yao, 1982), primarily for secure collaboration. Despite its great promise in various applications, very few garbled circuit-based methods have been implemented in practice (e.g. garbled circuit-based FlexSC (Wang et al., 2015)) and even fewer have been applied to support privacy-preserving genomic data analysis (Constable et al., 2015; Zhang et al., 2015). Despite the lower computational complexity, garbled circuit-based solutions usually require sophisticated circuit design and optimization for each specific task, which limits flexibility. Finally, secret sharing-based secure genome-wide association methods (Kamm et al., 2013) show better performance than HME and garbled circuit-based methods. However, they still impose significant computation overhead when compared to the same analyses over plaintext.
Preserving privacy in genomic data analysis while enabling scientific discovery via cross-institutional collaborations remains a big challenge. Collaborations among countries are further complicated by governmental policies that may prohibit sharing of individual-level genomic data. A key challenge is how to perform the necessary analyses on large genomic data and satisfying security and policy constraints. More importantly, recent efforts in secure genomic data analysis have only been tested on simulated data and environments (Chen et al., 2016; Lauter et al., 2014; Wang et al., 2016; Zhang et al., 2015), but not in the real-world settings. In this study, we introduce the PRINCESS framework for Privacy-protecting Rare disease International Network Collaboration via Encryption through Software guard extensionS. We have evaluated PRINCESS in a real-life study of family-based transmission tests used to understand the genetic basis of Kawasaki Disease (KD) (Khor et al., 2011). KD is a self-limited acute vasculitis that is the most common cause of acquired pediatric heart disease. Up to 25% of untreated children will develop coronary artery aneurysms. While the etiology is unknown, it is possible that an environmental factor initiates an immunologic reaction in genetically susceptible hosts. Although epidemiologic studies have shown that the relative risk of KD is two times higher in African American (AA) children than the risk in European descent individuals (Abuhammour et al., 2005), no KD genetic study to date has included AA children. Unlike KD children of Asian or European descent, limited sample size (especially from trios) has been a major bottleneck for research on the increased susceptibility of AA children to KD. In collaboration with the International Kawasaki Disease Genetic Consortium (IKDGC), we utilized cohorts associated with AA children from the U.S., the UK and Singapore to conduct jointly analysis using the TDT (transmission disequilibrium test, see Section 2.3 for details).
Unlike existing solutions, which rely on heavyweight cryptographic techniques, our PRINCESS framework takes advantage of Software Guard Extensions (SGX) (Anati et al., 2013), a suite of hardware and software architectures (publicly released in December 2015) that provide isolation of sensitive data analysis within a protected enclave. Solutions based on SGX are not expected to introduce significant computational overhead or big restrictions on data analysis operations that are common to software-based techniques such as the garbled circuit-based FlexSC framework (Wang et al., 2015) and homomorphic encryption based HElib framework (Halevi and Shoup, 2014), and thus are expected to make secure large-scale, inter-continental, genetic analysis feasible in practice. Our proposed framework has three key contributions (Fig. 1). First, within our framework for secure collaboration for genomic data analysis, we implemented a secure TDT module for studying KD and demonstrated that it can be highly efficient. Second, by jointly utilizing the SGX suite lightweight cryptographic primitives and data compression techniques, our framework achieves efficient secure computation and communication of sensitive genomic data. For example, PRINCESS was more than 40 000 times faster than HElib (Halevi and Shoup, 2014) and FlexSC (Wang et al., 2015) based methods we implemented for protecting data privacy, and provided accurate outcomes (see Fig 4(b) and Section 3.3 for details). Third, we designed and executed real life experiments involving family trio genetic data from three international institutions (UC San Diego, Imperial College London and Genome Institute of Singapore) to demonstrate how well PRINCESS enables a secure international collaboration to conduct TDT analysis of KD without compromising individual participants’ privacy. The individuals whose data were used provided broad consent for use of these data.
Fig. 1.
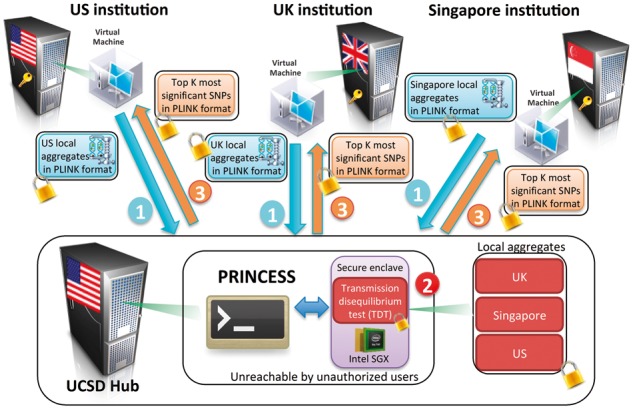
Overview of the PRINCESS framework that supports secure communication of several sites to an untrusted server to conduct secure genomic data analysis through SGX. (1) The inputs from each participating site in the PRINCESS framework are local aggregates in the PLINK format (Purcell et al., 2007), compressed and encrypted before transfer to the untrusted server. (2) All local aggregates are securely processed within an enclave to compute global TDT statistics on the untrusted server. (3) Only the top most significant SNPs (encrypted) will be returned to each site in the PLINK format and only authorized clients with the secret keys can decrypt the results (Color version of this figure is available at Bioinformatics online.)
Fig. 4.

Experimental performance of PRINCESS. (a) The performance with or without data compression using a synthetic dataset with 5 million SNPs in a three-site setting. Based on an average of 10 trials, we measured the average client execution time (including encryption and compression, if applicable); enclave execution time at the server (including decryption and decompression when applicable, TDT statistics computation, and sorting/encryption for the top most significant SNPs); average data transmission time among three clients; total data volume received by the server. (b) The total time spent (red line plot) and data transferred (bar plots) by each method (PRINCESS, FlexSC and HElib) to compute securely with different input sizes based on a two-site setting. As HME does not support division and sorting operations, our HElib based implementation only refers to the time cost for addition and multiplication operations in TDT (Halevi and Shoup, 2014). As FlexSC supports division, we included division cost, but without sorting operations. In addition, we compared the total amount of transferred data (green bar plot in plaintext and blue bar plot in PRINCESS) and the computation time (red diamond-dash line plot in plaintext and solid-dot line in PRINCESS), with the differences are also shown on top of the bars. (c) The percentage of time costs, including average time costs of encryption/compression (in green) and data transmission (in cyan) among all clients and server in 3, 6, 9 and 12-sites setups. The encryption and compression time as well as data transmission time at the clients are illustrated in boxplots (Color version of this figure is available at Bioinformatics online.)
2 Methods
In this section, we introduce the proposed SGX based framework, data source and statistical model.
2.1 Overview
Fig. 2 illustrates the system architecture of the proposed PRINCESS framework for secure collaboration using the SGX model (See Supplementary Note 1 for an overview). Our architecture supports secure transmission and analyses of sensitive genomic data, and joint analyses without compromising either control over personally identifiable data (privacy) or disclosure of intermediary results (confidentiality), whether deliberate or accidental. The PRINCESS system is designed to be scalable and easy to extend with support of plug-in modules for new features/new tasks. These modules include analysis algorithms, data management tools, compression methods, etc. In PRINCESS, we provide base classes for these modules and define common application programming interfaces (APIs). Thus, when new modules are implemented based on APIs, they can be easily integrated into the system. In PRINCESS, the data management module on the client side recognizes the standard PLINK format in the input file (See Supplementary Note 2 for details of data format) and can parse input data into memory so that the compression module or encryption module can process parsed data accordingly. The compression module provides an interface to client sites (data owners) to transfer compressed data using range coding (Martin, 1979) to the service provider, which has a static decompression library running inside the enclave (since the range-coding algorithm is lightweight, it is feasible to implement it inside the enclave). Compression performance is good (See Fig. 4(a) and Section 3.3 for details). When the enclave at the server receives the encrypted data from all client sites, it can decrypt the data inside the enclave (this step is blind even to the platform at the service provider). Then, the TDT is performed securely according to data owners’ instructions.
Fig. 2.
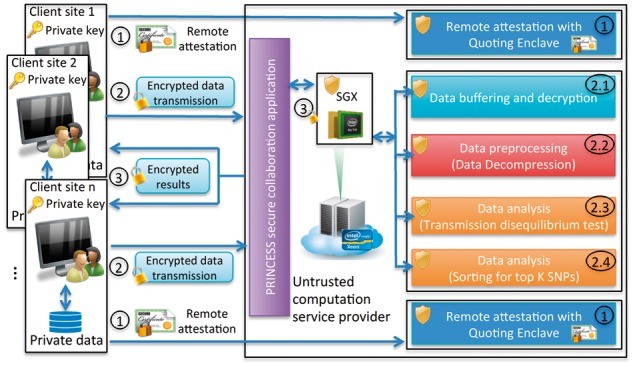
PRINCESS system architecture for secure collaboration. The steps are described in the Section 2.2 (Color version of this figure is available at Bioinformatics online.)
2.2 Security framework
(1) The remote attestation protocol allows multiple data owners and an enclave to verify each other’s authenticity and integrity. After an enclave is built, each data owner can obtain a unique signature of the enclave (a 256-bit hash), in which any falsification (e.g. the enclave has been modified unwillingly before the execution) on the enclave will result in an invalid signature. An enclave can also challenge the data owners’ identity through the Elliptic Curve Digital Signature Algorithm (ECDSA) (Locke and Gallagher, 2009), which can prevent the server from receiving fake or malicious data from unauthorized clients. In addition, PRINCESS allows Secure Sockets Layer (SSL) protection to enable an encrypted link between server and clients.
(2) After attestation, data owners send encrypted data obtained by the Advanced Encryption Standard (AES) in Galois Counter Mode (GCM) (Dworkin, 2007) to the PRINCESS data processing enclave hosted by an untrusted computation service provider. The encryption key of AES-GCM is derived from the key obtained by the Elliptic Curve Diffie–Hellman (ECDH) protocol (Barker et al., 2007) between the enclave and each data owner. To maximize security protection, we incorporated a time varying initialization vector; this ensures that even the same plaintext inputs in different encryption phases cannot generate the same ciphertext outputs, to avoid replay attacks (Syverson, 1994). One of the big challenges in developing the analysis module for the TDT algorithm is memory; the SGX enclave has a memory restriction so a limited amount of data can be processed inside the enclave. Unfortunately, the size of genomic data can be much larger, especially when a large number of sites are involved in a study. As a result, the required memory to process multi-million SNPs from multiple sites can easily exceed this memory restriction. To address this limitation, we performed batch evaluation and kept a global queue for the top SNPs to reduce unnecessary memory footprint in the enclave. Large genomic data also pose challenges to secure data transmission over the network. Data compression algorithms can be applied to reduce the data size and speed up the transmission process. An overall description of step (2) where data compression and batch evaluation schemes are applied within the PRINCESS framework is shown in Fig. 3. Data from each client site are first segmented into small chunks. The range coding-based compression algorithm is then used to reduce the data size of each small chunk. Following this, the compressed data are encrypted and securely transmitted to the data enclave hosted by an untrusted computation service provider through multiple threads and secure communication channels. When the encrypted data are received, the data processing enclave decrypts each segment and decompresses the data to recover the original data segment (e.g. 8000 SNPs). This is followed by a batch evaluation over each segment while keeping an updated global queue for the top SNPs in the enclave. All the operations within the enclave are secured by SGX. The proposed secure TDT algorithm in the PRINCESS framework running inside the enclave is completely self-contained and does not leave the enclave for system services, ensuring good secure computing performance.
Fig. 3.
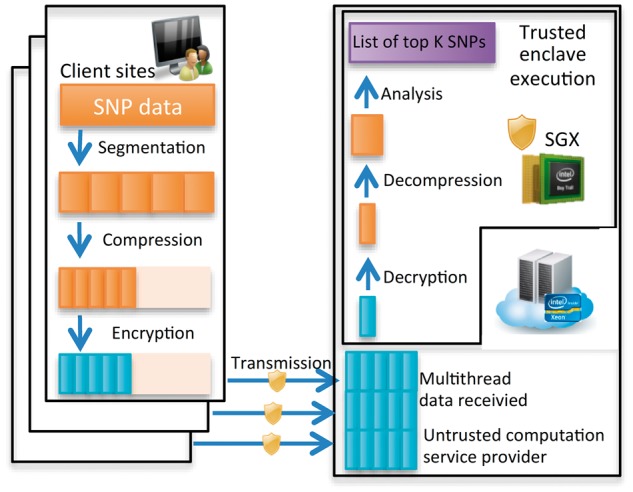
Streaming compression algorithms to improve secure communication and improve data analysis efficiency (Color version of this figure is available at Bioinformatics online.)
(3) Finally, the results are returned to the data owners in a secure manner, through an AES-GCM encrypted format at the 128-bit security level, ensuring their confidentiality and integrity.
2.3 Experimental setup
The following experimental environment was used to generate our results.
The PRINCESS server was hosted on a machine with 6th Generation Core i7-6820HQ CPU at 3.60 GHz with SGX support and 48 GB Memory. We used SGX Windows software development kit (SDK) v1.1. All the clients were connected to the PRINCESS server hosted at UCSD through a 1 GB Ethernet connection.
For simulated data analysis in PRINCESS, we launched up to 12 instances on Amazon Web Services (AWS) of type ‘t2.micro’ with 3.3 GHz CPU and 1 GB memory and single core virtual CPUs running a 64-bit window 2012 server to act as clients. We generated synthetic datasets based on the distribution of real KD data. We enabled Secure Sockets Layer (SSL) transmission, setting the data segmentation size to 8000 and reporting the top K = 100 most significant SNPs in these experiments.
For real KD data analysis in PRINCESS, three institutions, the University of California San Diego (UCSD), the Imperial College in London (ICL) and the Genome Institute of Singapore (GIS) participated. The top SNPs were returned. For client sites, we used Xeon E5-1680 v2 CPUs at 3.00 GHz with 16 GB memory at UCSD, i7-3770 CPU at 3.90 GHz with 16 GB memory at ICL, and i7-6700HQ at 3.50 GHz with 16 GB memory at GIS.
Our HElib-based HME implementation was based on the following parameter settings: p = 401,r = 2,d = 1,c = 2,L = 38,s =0,m = 38677 to allow a potential large circuit depth for further computation over HME encrypted data, which resulted in 1172 slots for parallel computation in SIMD mode. The computation time includes key generation time, encryption/decryption times, ciphertext operation time and compression/decompression times. As HME does not support division and sorting operations, the computation time only measured the addition and multiplication operations in TDT. The performance was evaluated on a Ubuntu 14.04 machine with Xeon E5-2687W CPU at 3.10 GHz with 96 GB memory.
For FlexSC-based implementation, communication and computation costs were averaged over two parties. There was no data compression for FlexSC. The performance was evaluated on a MAC OS 10.11 machine with I7-4870HQ CPU at 2.5 GHz with 16 GB Memory. In addition, there were also no sorting of top SNPs in the FlexSC implementation.
All results for PRINCESS using simulated and real data were based on the average of 10 and 3 trials, respectively. All results for HME and FlexSC implementations were based on the average of 5 trials, as both methods were very time consuming.
All above implemented algorithms are availabe at https://github.com/achenfengb/PRINCESS_opensource.
2.4 Statistical models
A genome wide study of KD, using TDT, was used to illustrate the practical use of our proposed framework. TDT is a family-based test for disease traits that uses the genotype information from both parents and a child. TDT measures the transmitted and untransmitted allele counts from heterozygous parents (denoted by and , respectively) to their affected child. The TDT statistic can be expressed as , which will approximately follow a chi-square distribution with one degree of freedom (Spielman et al., 1993). The advantage of the TDT, when compared to a case/control study design, is to avoid errors due to the population stratification. This is important to our study, which has a large degree of genetic admixture (see Supplementary Table 1). The TDT data used in this study were derived from complete parent-child trios.
Suppose that sites across the world plan to jointly compute global TDT statistics over their own confidential SNP data. Let us denote by , and the transmitted and untransmitted allele counts from a site for a SNP with rs number . Then, the global TDT statistic can be evaluated as follows:
| (1) |
Because of the horizontal data splitting, alleles 1 and 2 may differ among local sites in distributed TDT. For this reason, we also collected allele frequencies to determine globally the minor allele frequency (MAF) for data integration in a secure manner using SGX. After securely calculating all SNPs, a list of encrypted top (most significant) SNPs is returned to each site. The P-value is calculated using a χ2 distribution with one degree of freedom. The odds ratio is obtained by dividing B by C.
3 Results
3.1 Subjects and samples
Seventy two Kawasaki disease (KD) children and their biological parents were recruited from Rady Children’s Hospital San Diego (RCHSD) (), Emory University () in Atlanta, and Imperial College in London (); here, indicates the number of families. The ancestry analysis of 216 individuals from 72 trios can be found in Supplementary Note 3. Subjects had previously provided consent for reuse of data. Kawasaki disease was diagnosed according to the criteria of the American Heart Association (AHA), as previously described (Khor et al., 2011).
3.2 Genotyping
For the KD families, the blood or mouthwash/Oragene samples from each subject were used to generate the genomic data with Illumina Human OmniExpress 24 BeadChips following the manufacturer’s instructions. Genotype data from RCHSD were hosted at University of California, San Diego (UCSD) and the UK data were hosted at the Imperial College London (ICL). Genotype data from Emory University were hosted at the Genome Institute of Singapore (GIS).
3.3 Experimental results
The experiments were designed in four phases (a–d) (Supplementary Note 4), where we set in experiments (a–c) using simulated data and in experiment (d) using real data. We first compared the performance of PRINCESS by securely computing on 5 million SNPs from three geographically distributed locations with or without compression (Fig. 4(a)). With compression, PRINCESS reduced the data volume to 54.6% of the original size and saved 45.2% of the transmission time in communications (see Supplementary Table 2). Data compression at the client and decompression at the server introduced a small computational overhead. As a result, the overall average execution time was reduced from 126.1 s (without compression) to 74.8 s (with compression). Fig. 4(b) shows a comparison of PRINCESS, HElib (Halevi and Shoup, 2014) and FlexSC (Wang et al., 2015) implementations done by our team and running in the hardware, network and software environment described in Section 2.2, using synthetic data with sizes varying from to in a two-site setting. The proposed PRINCESS framework was >40 000 times faster than our HElib and FlexSC implementations (see Supplementary Table 3). In addition, the cost in terms of data transmission and execution time increased linearly with the size of input, taking less than 3.6 s to securely compute 5 million SNPs from two sites. The plaintext protocol with data compression is also illustrated in Fig. 4(b), where the PRINCESS framework shows a small overhead on data transmission (0.02%) and in computation (17.92%). To demonstrate scalability, we evaluated PRINCESS with communication and computation costs (Fig. 4(c)) using 1 million SNPs and varying number of participating sites (i.e. 3, 6, 9 and 12 sites across different countries as illustrated in Supplementary Fig. 2). The data transmission between clients and the server was the most time consuming part (i.e. 62–77%) in all settings, when compared to the execution time at the clients or at the server. Table 1 shows the top 10 SNPs identified by PRINCESS.
Table 1.
The top 10 SNPs identified by TDT analysis of genetic variants and KD susceptibility in the International Kawasaki Disease Genetic Consortium (IKDGC) cohorts for a total of 72 trios among UC San Diego (UCSD), the Imperial College in London (ICL) and the Genome Institute of Singapore (GIS). Lists of top 252 SNPs can be found in Supplementary tables 6.1 to 6.3
| GENE | CHR | SNP ID | BP | Allele 1/2 | T | U | OR | CHISQ | -value |
|---|---|---|---|---|---|---|---|---|---|
| 75kb 5' of PLEKHA5 | 12 | rs7976757 | 19207948 | A/G | 9 | 44 | 0.20 | 23.11 | 1.53E-06 |
| 144kb 5' of RP11-347L18.1 | 6 | rs10455943 | 164914089 | G/A | 7 | 38 | 0.18 | 21.36 | 3.82E-06 |
| CNTNAP5 | 2 | rs1504016 | 125368982 | G/A | 10 | 42 | 0.24 | 19.69 | 9.10E-06 |
| 131kb 5' of GNPDA2 | 4 | rs10517120 | 44859761 | A/G | 15 | 51 | 0.29 | 19.64 | 9.37E-06 |
| FMN1 | 15 | rs12592701 | 33307752 | G/A | 36 | 8 | 4.50 | 17.82 | 2.43E-05 |
| FBN1 | 15 | rs6493328 | 48819502 | G/A | 34 | 7 | 4.86 | 17.78 | 2.48E-05 |
| ZNF280D | 15 | rs7168178 | 57012816 | A/G | 45 | 13 | 3.46 | 17.66 | 2.65E-05 |
| LAMC3 | 9 | rs869457 | 133924451 | A/G | 39 | 10 | 3.90 | 17.16 | 3.43E-05 |
| FBN1 | 15 | rs683282 | 48847733 | A/G | 35 | 8 | 4.38 | 16.95 | 3.83E-05 |
| FBN1 | 15 | rs668842 | 48891965 | G/A | 35 | 8 | 4.38 | 16.95 | 3.83E-05 |
CHR: Chromosome; BP: base position; T: Transmitted, U: Untransmitted; OR: Odds Ratio; CHISQ: χ2statistics.
4 Discussion
4.1 KD associated genetic variations
PRINCESS applies lightweight cryptographic technologies to tackle secure genomic data computation, providing computation efficiency for real-world, secure, international collaboration. Using 72 trios and 695 784 SNPs across three continents, PRINCESS identified the top 10 SNPs from 5 genes (CMNTMAP5, FMN1, FBN1, ZNF280D, LAMC3), which are displayed in Table 1. Lists of top 252 SNPs can be found in Supplementary Tables 6.1 to 6.3. The discussion of KD associated genetic variations can be found in (Shimizu et al., 2016a).
4.2 Efficiency of the proposed PRINCESS framework
The proposed PRINCESS framework uses the SGX computing architecture. This hardware architecture provides isolation of the sensitive data analysis within a protected enclave. Our results indicate that this architecture did not introduce significant computational overhead and restrictions on data analysis in this study. We performed efficiency analysis with respect to varying segment lengths during batch evaluation in the enclave. In Table 2, we show the steps and execution time of the TDT analysis algorithm with and without the enclave. In both experiments, we conducted the computation of TDT statistics over 1 million SNPs from 3 local sites to identify the top 100 SNPs. We can see that the proposed framework imposed a small computation overhead (i.e. additional 0.0258 s on average) in our study.
Table 2.
Tasks and run time of the TDT algorithm with and without enclave where we used 1 million SNPs from 3 local sites and identified the top 100 SNPs
| Steps | Enclave | Non-enclave |
|---|---|---|
| AES-based decryption | ✓ | ✗ |
| Batch execution (segment length of 8000) | ✓ | ✗ |
| TDT computation | ✓ | ✓ |
| Computing the top SNPs | ✓ | ✓ |
| Average run time over 10 trials (s) | 0.1303 | 0.1045 |
In addition, we set up 3 local sites, each with a dataset of 5 million SNPs, to study the efficiency in terms of memory footprint and run time using different segment lengths (Table 3). When we executed batch analyses inside the enclave, buffers containing encrypted and decrypted segment data were allocated in the enclave heap. Different segment lengths resulted in different peak memory usage. With the compression flag turned on, extra buffers containing compressed data were allocated. During the decompression of each segment data in the enclave, we built a lookup table of size 500 KB, which is reflected by an increased heap space usage when the compression flag is turned on. The lookup table was designed to speed up the decompression process. Supplementary Figure 3 compared the performances between an ‘on-the-fly’- based implementation and a lookup table-based implementation using different number of sites (varying from one to four sites) and different number of SNPs (ranging from 1 to 5 million). Without using the lookup table, the program needs to perform repeated computation on the fly, which led to 41.8% computational overhead on average over different number of sites (see Supplementary Figure 3). The lookup table-based implementation shows the computational advantage over the on-the-fly implementation.
Table 3.
Memory footprint and run time using different segment length over 10 trials, where we used a 3-site setup with 5 million SNPs
| Enclave Computation | Segment length (SNPs) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 100 | 1000 | 5000 | 8000 | 10 000 | 100 000 | 1 million | |||
| Compression | On | Memory (KB) | 554 | 586 | 714 | 814 | 870 | 3822 | 28 442 |
| Run time (s) | 19.435 | 6.145 | 4.980 | 4.801 | 4.764 | 4.768 | 4.746 | ||
| Off | Memory (KB) | 46 | 58 | 154 | 222 | 270 | 2378 | 23 474 | |
| Run time (s) | 0.936 | 0.637 | 0.608 | 0.603 | 0.650 | 0.720 | 0.722 | ||
We have enabled lookup table for the setup with compression on.
4.3 Comparison with existing genomic data protection methods
The existing genome privacy-preserving technologies can be categorized into protection of (1) genomic data dissemination and (2) genomic data analyses. Differential privacy (Dwork, 2006) (DP) is a popular perturbation technology with provable privacy guarantees and has been widely used in protection genomic data dissemination. It generally requires adding noise to the data in order to satisfy the DP requirement, which will reduce data utility. For the genomic data analyses, security researchers have made significant progress in cryptographic techniques such as HME and SMC to facilitate secure outsourcing and secure collaboration in genomic research. HME methods allow data owners to directly analyze encrypted data using public cloud computing resources. However, HME-based techniques (Halevi and Shoup, 2014) are heavy-weighted in both computation and storage. In addition, it is challenging to handle complicated genomic analysis tasks, as HME only supports limited arithmetic operations (i.e. addition and multiplication). SMC-based methods such as garbled circuits (Chen et al., 2014; Wang et al., 2015) and secret sharing (Chen et al., 2015; Kamm et al., 2013) can securely perform collaborative computation among multiple parties, as long as the underlying algorithms can be expressed by logical operations. However, the high computational and design complexity of garbled circuits and secret sharing is still a bottleneck for the corresponding applications to handle large-scale data and complicated algorithms (see Table 4 for the side-by-side comparison among HME, SMC and SGX frameworks). In addition, Canim et al. also studied hardware-based protection on biomedical data analysis (Canim et al., 2012), where genomic data were securely joined and queried. This framework demonstrated that oblivious algorithms could be developed for secure genomic data analysis on secure hardware without requiring generic oblivious RAM solutions. However, it required additional cryptographic coprocessors with relative low computational power and memory. In contrast, the proposed PRINCESS framework can be directly utilized on SGX-enabled CPUs without requiring additional coprocessors.
Table 4.
Comparison of various secure approaches
| Methods | Hardware Requirement | Time | Types of Operations | Risk of failure |
|---|---|---|---|---|
| HME* | Any CPU | Slow | Addition and multiplication | Malicious (Active) attacker (Zhang et al., 2011) |
| SMC** | Any CPU | Slow | Boolean operations (High complexity) | Malicious (Active) attacker (Yao, 1982) |
| SGX | SGX-enabled CPU | Fast | Almost any operation | Controlled side channel attack (Xu et al., 2015) |
*HME: Homomorphic Encryption; **SMC: Secure Multi-party Computation.
In the SGX framework, the operating system (OS) or other hypervisors cannot detect the precise memory access patterns within one page (i.e. 4KB). However, the OS may be able to detect the page access pattern by observing page-fault through a controlled-channel attack (CCA) (Xu et al., 2015). To achieve efficient oblivious memory access, we have forced the data used for updating the global queue of top SNPs to reside within one page (4 KB memory). This was achieved by blocking larger input data, such that both the global queue and segment of data could be restricted within one page. However, within a 4KB page, we can only handle the sorting of 1024 SNPs at most using a 4-byte single precision floating format.
4.4 Limitations
PRINCESS has some limitations. For example, the current framework does not support secure storage outsourcing, which means all clients need to be synchronized during the collaborative analysis. This restriction might be mitigated by adopting a data sealing process in SGX to enable asynchronized secure collaboration. In the current experiment, we only developed the secure TDT enclave module. Extending this to additional genomic data analysis methods will broaden the impact and adoption by the biomedical research community. In the current version of PRINCESS, we used the same compression scheme for different fields of the PLINK inputs. The compression performance could be further improved by adopting different compression schemes based on the characteristics of the different fields in PLINK inputs. The adoption of compression scheme significantly reduced the amount of data to be transferred, and also reduced transmission time and overall processing time. The secure decompression module in the enclave also increased the enclave memory footprint as well as the server side execution time. . To protect against the controlled side channel attack, we needed to implement all the sorting operations within a 4 KB page, which restricted the maximum number of top K results to 1024. SGX has limitations on the total amount of secure memory, which may prevent its application in large-scale datasets. To mitigate this bottleneck, we can use data paging or sealing technologies to securely store additional data through encryption in the untrusted memory or disk space, respectively. However, the data paging or sealing process will impose additional computational overhead, which might slow down certain data intensive applications. We plan to investigate efficient solutions for such applications in future work. Finally, SGX is a proprietary solution that may or may not be affordable to users. The costs and benefits of running the same analyses in other hardware while maintaining the same level of protections will also be investigated in future work.
5 Conclusion
In this paper, we presented the PRINCESS framework, which ensures a high security level for privacy-preserving international collaboration on rare disease analysis. In PRINCESS, all genomic data are encrypted with AES-GCM. We utilized a time-varying initialization vector to enhance the protection of encrypted data, where the attacker cannot gain more information based on multiple encrypted messages than a single encrypted message by using the same encryption key. In addition, our framework ensures that the same message under different encryption instances will yield completely different ciphertexts. As both the client and the server have a synchronized time-varying initialization vectors, the proposed framework can detect replay attacks (Syverson, 1994). Furthermore, encrypting data using AES-GCM supports authenticity and integrity checks. When compared with other state-of-the-art trustworthy computation schemes (i.e. homomorphic encryption and garbled circuits), PRINCESS took advantage of a software-and-hardware based hybrid solution to achieve more than 40 000 times performance gain in our specific example.
Supplementary Material
Acknowledgements
We would like to thank Mona Vij, Simon Jahnson, Vinay Phegade, Anand Rajan from the Intel® SGX team for reviewing the technical parts of this manuscript and for helpful discussions and suggestions, and Dr. Michael Levin from Imperial College London for helpful suggestions.
Funding
This work has been supported by NIH through grants R00HG008175, NLM R01HG007078, R21LM012060 and U54HL108460.
Conflict of Interest: F.C., S.W., X.J., S.D. and Y.L received an outstanding achievement award from Intel for secure genomic data analysis using SGX for the proposed research.
References
- Abuhammour W.M. et al. (2005) Kawasaki disease hospitalizations in a predominantly African-American population. Clin. Pediatr. (Phila), 44, 721–725. [DOI] [PubMed] [Google Scholar]
- Anati I. et al. (2013) Innovative technology for CPU based attestation and sealing. In: Proceedings of the 2nd International Workshop on Hardware and Architectural Support for Security and Privacy., p. 10.
- Ayday E. et al. (2014) Privacy-preserving processing of raw genomic data. Data Priv. Manag. Auton. Spontaneous Secur., 8247, 133–147. [Google Scholar]
- Barker E. et al. (2007) NIST special publication 800-56A: Recommendation for pair-wise key establishment schemes using discrete logarithm cryptography (revised). In: Comput. Secur. Natl. Inst. Stand. Technol. (NIST), Publ. by NIST.
- Bloss C.S. (2013) Does family always matter? Public genomes and their effect on relatives. Genome Med., 5, 107.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canim M. et al. (2012) Secure management of biomedical data with cryptographic hardware. IEEE Trans. Inf. Technol. Biomed., 16, 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F. et al. (2014) PRECISE: PRivacy-prEserving Cloud-assisted quality Improvement Service in hEalthcare. IEEE Int. Conf. Syst. Biol. [Proceedings]. IEEE Int. Conf. Syst. Biol, 2014, 176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F. et al. (2015) Cloud-assisted distributed private data sharing. In: Proceedings of the 6th ACM Conference on Bioinformatics, Computational Biology and Health Informatics - BCB ’15 ACM Press, New York, New York, USA, pp. 202–211.
- Chen F. et al. (2016) PREMIX: PRivacy-preserving EstiMation of Individual admiXture. In: American Medical Informatics Association Annual Symposium. [PMC free article] [PubMed]
- Claes P. et al. (2014) Modeling 3D facial shape from DNA. PLoS Genet., 10, e1004224.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins F.S., Varmus H. (2015) A new initiative on precision medicine. N. Engl. J. Med., 372, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constable S. et al. (2015) Privacy-preserving GWAS analysis on federated genomic datasets. BMC Med Inf. Decis Mak., 15, S2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwork C. (2006) Differential privacy. Int. Colloq. Autom. Lang. Program, 4052, 1–12. [Google Scholar]
- Dworkin,M.J. (2007) NIST Special Publication 800-38D: Recommendation for Block Cipher Modes of Operation: Galois/Counter Mode (GCM) and GMAC. Comput. Secur. Natl. Inst. Stand. Technol. (NIST), Publ. by NIST.
- Gymrek M. et al. (2013) Identifying personal genomes by surname inference. Science (80), 339, 321–324. [DOI] [PubMed] [Google Scholar]
- Halevi S., Shoup V. (2014) Algorithms in HElib. Adv. Cryptology–CRYPTO, 2014, 554–571. [Google Scholar]
- He D. et al. (2014) Identifying genetic relatives without compromising privacy. Genome Res., 24, 664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homer N. et al. (2008) Resolving individuals contributing trace amounts of DNA to highly complex mixtures using high-density SNP genotyping microarrays. PLoS Genet., 4, e1000167.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamm L. et al. (2013) A new way to protect privacy in large-scale genome-wide association studies. Bioinformatics, 29, 886–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor C.C. et al. (2011) Genome-wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat. Genet., 43, 1241–1246. [DOI] [PubMed] [Google Scholar]
- Kim K.K. et al. (2015) Comparison of consumers’ views on electronic data sharing for healthcare and research. J. Am. Med. Inform. Assoc., 22, 821–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. et al. (2016) iCONCUR: informed consent for clinical data and bio-sample use for research. J. Am. Med. Inf. Assoc, doi: 10.1093/jamia/ocw115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauter K. et al. (2014) Private computation on encrypted genomic data. In: 14th Privacy Enhancing Technologies Symposium, Workshop on Genome Privacy. Amsterdam, The Netherlands.
- Li Y. et al. (2016) VERTIcal Grid lOgistic regression (VERTIGO). J Am Med Inf. Assoc., 23, 570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke G., Gallagher P. (2009) FIPS PUB 186-3: Digital Signature Standard (DSS). Comput. Secur. Natl. Inst. Stand. Technol. (NIST), Publ. by NIST. [Google Scholar]
- Martin G.N. (1979) Range encoding: an algorithm for removing redundancy from a digitalized image. In: Proceedings, of Video and Data Compression Conference.
- McGuire A.L. et al. (2008) Confidentiality, privacy, and security of genetic and genomic test information in electronic health records: points to consider. Genet. Med., 10, 495–499. [DOI] [PubMed] [Google Scholar]
- McLaren P.J. et al. (2016) Privacy-preserving genomic testing in the clinic: a model using HIV treatment. Genet. Med., 18, 814–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIH. (2007) Policy for Sharing of Data Obtained in NIH Supported or Conducted Genome-Wide Association Studies (GWAS).
- Purcell S. et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet., 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu C. et al. (2016a) Genetic variation in the SLC8A1 calcium signaling pathway is associated with susceptibility to Kawasaki disease and coronary artery abnormalities. Circ. Cardiovasc. Genet., (in press). [DOI] [PubMed] [Google Scholar]
- Shimizu K. et al. (2016b) Efficient privacy-preserving string search and an application in genomics. Bioinformatics, 32, 1652–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shringarpure S.S., Bustamante C.D. (2015) Privacy leaks from genomic data-sharing beacons. Am. J. Hum. Genet., 97, 631–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman R.S. et al. (1993) Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am. J. Hum. Genet., 52, 506.. [PMC free article] [PubMed] [Google Scholar]
- Sudmant P.H. et al. (2015) An integrated map of structural variation in 2,504 human genomes. Nature, 526, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syverson P. (1994) A taxonomy of replay attacks [cryptographic protocols]. In: Computer Security Foundations Workshop VII, 1994. CSFW 7. Proceedings., pp. 187–191.
- Wang S. et al. (2013) EXpectation Propagation LOgistic REgRession (EXPLORER): distributed privacy-preserving online model learning. J. Biomed. Inform., 46, 1–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. et al. (2015) Circuit ORAM: On Tightness of the Goldreich-Ostrovsky Lower Bound. In: Proceedings of the 22nd ACM SIGSAC Conference on Computer and Communications Security.
- Wang S. et al. (2016) HEALER: homomorphic computation of ExAct Logistic rEgRession for secure rare disease variants analysis in GWAS. Bioinformatics, 32, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. et al. (2012) Grid Binary LOgistic REgression (GLORE): building shared models without sharing data. J. Am. Med. Inform. Assoc., 2012, 758–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W. et al. (2014) SecureMA: protecting participant privacy in genetic association meta-analysis. Bioinformatics, 30, 3334–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. et al. (2015) Controlled-channel attacks: Deterministic side channels for untrusted operating systems. In: 2015 IEEE Symposium on Security and Privacy (SP), pp. 640–656.
- Yao A.C. (1982) Protocols for secure computations. In: 23rd Annual Symposium on Foundations of Computer Science (sfcs 1982). IEEE, pp. 160–164.
- Zhang Z. et al. (2011) Reaction attack on outsourced computing with fully homomorphic encryption schemes. In: International Conference on Information Security and Cryptology., pp. 419–436.
- Zhang Y. et al. (2015) Secure distributed genome analysis for GWAS and sequence comparison computation. BMC Med Inf. Decis Mak, 15, S4. [DOI] [PMC free article] [PubMed]
- Zhao Y. et al. (2015) Choosing blindly but wisely: differentially private solicitation of DNA datasets for disease marker discovery. J. Am. Med. Inform. Assoc., 22, 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.