

Abstract
Motivation: Not only sequence data continue to outpace annotation information, but also the problem is further exacerbated when organisms are underrepresented in the annotation databases. This is the case with non-human-pathogenic viruses which occur frequently in metagenomic projects. Thus, there is a need for tools capable of detecting and classifying viral sequences.
Results: We describe VIRALpro a new effective tool for identifying capsid and tail protein sequences, which are the cornerstones toward viral sequence annotation and viral genome classification.
Availability and implementation: The data, software and corresponding web server are available from http://scratch.proteomics.ics.uci.edu as part of the SCRATCH suite.
Contact: clovis.galiez@inria.fr or pfbaldi@uci.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
As of April 2015, 77% of the protein sequences referenced in UniProtKB/TrEMBL (Magrane and Consortium, 2011) are predicted protein sequences. For the non-predicted sequences, only 2% have been confirmed through biological evidence while the remaining 21% is inferred by homology and this imbalance will continue to worsen in the foreseeable future. The situation is even worse for specific classes of important organisms, in particular viruses the majority of which lack proper annotation. For instance, it is typical to encounter only 10% of annotated sequences in marine viral samples (Holmfeldt et al., 2013; Hurwitz and Sullivan, 2013; Suttle, 2007), although these viruses play key roles in the ecosystem. It is for instance estimated that marine viruses kill up to 20% of the living biomass in the oceans daily, and this organism turnover may play a major role in the global carbon cycle (Lehahn et al., 2014). In short, new computational tools are needed to help identify and annotate unknown viral sequences.
One criterion for virus classification is the genomic organization and sequence of the genes coding for structural proteins—i.e. the proteins that compose the virion, in particular capsid and tail proteins. Capsid proteins in particular are present in all viral genomes and, as suggested in Seguritan et al. (2012), the capsid genes may be used as an equivalent of 16S rRNA for prokaryote identification in genomes and metagenomes.
Previously, a tool called iVireons (Seguritan et al., 2012) has been introduced to detect structural proteins in phages—i.e. viruses that infect bacteria. The tool uses a single input—the average amino acid composition of the query sequence—which is fed into three classification neural networks: one for all structural proteins, one for tail proteins and one for capsid proteins. As stated by the authors, the tool performs well at detecting phage capsids, but its performance degrades when used to detect capsids in other viruses. The structural protein predictor has a reasonable sensitivity to all capsids, but its specificity is too low. To address these problems we develop VIRALpro using Support Vector Machines (SVM) and extended set of features to identify capsid (CAPSIDpro) and tail (TAILpro) sequences.
2 Methods
Data
We built a dataset of, respectively, 2648 and 483 non-redundant capsid and nucleocapsid sequences. The non-capsid sequences were randomly chosen from the phage non-structural sequences (Seguritan et al., 2012) and from the NCBI protein database by querying for non-phage, non-structural, proteins (see Supplementary materials). When merging this set of sequences with the training set of iVireons MCP1:1 denoted by iVcapsid, we obtained 3888 positive and 4071 negative sequences. We refer to the resulting dataset as Cprotrain. Using the same process, we built a dataset of 1719 positives tail sequences and when merged with the training set of iVireons tail 1:1 ANN, we obtained 2574 positive and 4095 negative tail sequences. We refer to the resulting dataset as Tprotrain. We denote by iVtest the test set of phage protein sequences described in Seguritan et al. (2012). We used 10-fold cross validation on the training sets to assess performance. For each training set, one of the folds is used as the validation set to produce the plots in Figure 1.
Fig. 1.
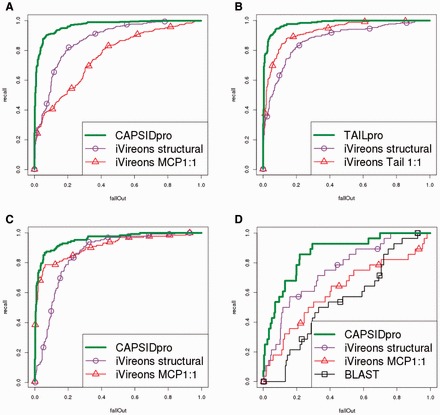
ROC curves on the validation sets of (A) CAPSIDpro; (B) TAILpro; (C) CAPSIDpro when removing all non-phage sequences from the positive set; and (D) CAPSIDpro when keeping only sequences that have no homology to the training set
Features
To identify capsid or tail protein sequences, we use the average amino acid composition (20 features) as well the average secondary structure composition (three features), as predicted by SSpro (Magnan and Baldi, 2014). In addition, we built 3380 Profile Hidden Markov Models (HMMs) to locally probe the sequences. These HMMs are built using HMMER (Eddy, 2009) from multiple sequence alignment of contact fragments of capsid proteins—which are essentially pairs of fragments that are close in the tertiary structure (see Supplementary materials) and whose structure has been shown to be well conserved even for distantly related homologs (Galiez and Coste, 2015). The e-values of the different HMMs are linearly combined using coefficients that are obtained using the RankBoost algorithm (Freund et al., 2003). Finally, the HMMs yield three features: the boosted linear combination of HMM e-values, the e-value of the best HMM hit and the number of HMM hits (e-value of 10 or better). Finally, we train an SVM (Chang and Lin, 2011) with these 26 features.
3 Results
Figure 1. shows the receiver operating characteristic (ROC) curves of VIRALpro and iVireons on various validation sets. VIRALpro provides a significant detection improvement in all cases, even when restricted to phage proteins only. The improvement is particularly pronounced in the case of difficult sequences with no homology to the training sets (e-value ≤0.001) where the performance of BLAST or iVireons degrades to levels close to random—the area under the curve (AUC) of the ROC curve is 54.9 and 61.5%, respectively. These results are further confirmed using various performance metrics given in Table 1 for CAPSIDpro. Similar results are obtained for TAILpro (see Supplementary materials).
Table 1.
Performance metrics for iVireons and CAPSIDpro
| Test | Measure | iVireons MCP 1:1 | iVireons Structural | CAPSIDpro |
|---|---|---|---|---|
| iVtest | Accur. | 90% | 80% | 97.3%* |
| 10-fold iVcapsid | Accur. | 91.3% | – | 96.8* ± 2.5% |
| 10-fold Cprotrain | AUC | 73.9 ± 2.4% | 85.1 ± 2.1% | 95.9 ± 0.8% |
| F-Meas. | 51.1 ± 2.6% | 77.9 ± 2.5% | 89.5 ± 1.5% | |
| Validation Set | AUC | 76% | 87.1% | 96.8% † |
| F-Meas. | 51.7% | 79.5% | 91.3% † |
Since the fold distribution for iVireons is not publicly available, a * indicates that CAPSIDpro was trained with iVcapsid to be as close as possible to the training of iVireons MCP1:1. Tests on the validation set, where CAPSIDpro has been trained only on the remaining 90% of the training data, are indicated by a †. Best results are shown in bold.
Finally, we tested VIRALpro on the unannotated portion of three metagenomic sequence datasets: (1) RNA viruses from coastal seawater in British Columbia (Culley et al., 2006; RnaCoastal); (2) marine phages from Baltic seawater (Oresund); and (3) marine phages sequenced by the Broad Institute under the Gordon and Betty Moore Foundation’s ‘Marine Phage, Virus and Virome Sequencing’ project (Moore) (see Supplementary materials). RnaCoastal and Oresund-Struct have 0% of their sequences that share any homology with our positive training set. Oresund-Hypo, Oresund-NonStruct and Moore have, respectively, 0.2, 1.3 and 1.8% of their sequences that share some homology (e-value ≤0.001) with our positive training set. Table 2 provides the recall obtained using VIRALpro. Interestingly, TAILpro detects some sequences in RnaCoastal, suggesting that these may be fiber proteins that may confirm the presence of rotaviruses in the sample, as suggested in Culley et al. (2006). In short, VIRALpro clearly outperforms existing tools for predicting capsid and tail proteins and can be used as a screening tool in metagenomic and other projects.
Table 2.
Recall on the metagenomic sequences annotated as unknown
| Dataset | Recall CAPSIDpro (%) | Recall TAILpro (%) |
|---|---|---|
| RnaCoastal (86) | 40.7 | 22.1 |
| Oresund-Struct (85) | 36.5 | 47.1 |
| Oresund-Hypo (524) | 22.0 | 10.1 |
| Oresund-NonStruct (156) | 12.8 | 8.3 |
| Moore (1172) | 33.8 | 27.4 |
The total number of sequences is given in parentheses
Funding
The work of C.G. was completed at UCI and has been supported by travel grants from Rennes Metropole and Inria Explorer. The work of C.M. and P.B. has been supported by grants NIH LM010235 and NSF-IIS-1321053 to PB. We also thank Yuzo Kanomata for helping set up the VIRALpro server.
Conflict of interest: none declared.
Supplementary Material
References
- Chang C.C., Lin C.J. (2011) Libsvm: a library for support vector machines . ACM Trans. Intell. Syst. Technol., 2, 27:1–27:27. [Google Scholar]
- Culley A.I. et al. (2006) Metagenomic analysis of coastal RNA virus communities. Science, 312, 1795–1798. [DOI] [PubMed] [Google Scholar]
- Eddy S.R. (2009) A new generation of homology search tools based on probabilistic inference In: Proceedings of the 20th International Conference on Genome Informatics 2009, Imperial College Press, pp. 205–211. [PubMed] [Google Scholar]
- Freund Y. et al. (2003) An efficient boosting algorithm for combining preferences. J. Mach. Learn. Res., 4, 933–969. [Google Scholar]
- Galiez C., Coste F. (2015) Structural conservation of remote homologues: better and further in contact fragments In: ISMB/ECCB 2015 Satellite Meeting—3DSIG: Structural Bioinformatics and Computational Biophysics, Dublin, Ireland. [Google Scholar]
- Holmfeldt K. et al. (2013) Twelve previously unknown phage genera are ubiquitous in global oceans. Proc. Natl Acad. Sci., 110, 12798–12803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz B.L., Sullivan M.B. (2013) The pacific ocean virome (pov): a marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLoS One, 8, e57355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehahn Y. et al. (2014) Decoupling physical from biological processes to assess the impact of viruses on a mesoscale algal bloom. Curr. Biol., 24, 2041–2046. [DOI] [PubMed] [Google Scholar]
- Magnan C.N., Baldi P. (2014) SSpro/ACCpro 5: almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics, 30, 2592–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane M., Consortium U. (2011) Uniprot knowledgebase: a hub of integrated protein data. Database, 2011, bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seguritan V. et al. (2012) Artificial neural networks trained to detect viral and phage structural proteins. PLoS Comput. Biol., 8, e1002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttle C.A. (2007) Marine viruses major players in the global ecosystem. Nat. Rev. Microbiol., 5, 801–812. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.