

Abstract
Motivation
The accurate interpretation of genetic variants is critical for characterizing genotype–phenotype associations. Because the effects of genetic variants can depend strongly on their local genomic context, accurate genome annotations are essential. Furthermore, as some variants have the potential to disrupt or alter gene structure, variant interpretation efforts stand to gain from the use of individualized annotations that account for differences in gene structure between individuals or strains.
Results
We describe a suite of software tools for identifying possible functional changes in gene structure that may result from sequence variants. ACE (‘Assessing Changes to Exons’) converts phased genotype calls to a collection of explicit haplotype sequences, maps transcript annotations onto them, detects gene-structure changes and their possible repercussions, and identifies several classes of possible loss of function. Novel transcripts predicted by ACE are commonly supported by spliced RNA-seq reads, and can be used to improve read alignment and transcript quantification when an individual-specific genome sequence is available. Using publicly available RNA-seq data, we show that ACE predictions confirm earlier results regarding the quantitative effects of nonsense-mediated decay, and we show that predicted loss-of-function events are highly concordant with patterns of intolerance to mutations across the human population. ACE can be readily applied to diverse species including animals and plants, making it a broadly useful tool for use in eukaryotic population-based resequencing projects, particularly for assessing the joint impact of all variants at a locus.
Availability and Implementation
ACE is written in open-source C ++ and Perl and is available from geneprediction.org/ACE
Supplementary information
Supplementary information is available at Bioinformatics online.
1 Introduction
The accurate interpretation of genetic variants and their impact on gene function is central to modern genetics, with implications for both disease studies and elucidation of basic biology. However, the complexities of eukaryotic gene structure and function challenge our ability to predict the effects of genetic variants on the products of expressed genes. The context of a variant—whether in an exon, intron or intergenic region—directly impacts the interpretation of likely variant effects. A number of bioinformatic tools are available for interpretation of individual variants, including ANNOVAR (Wang et al., 2010), SnpEff (Cingolani et al., 2012), VEP (McLaren et al., 2016), PolyPhen (Adzhubei et al., 2010), and SIFT (Kumar et al., 2009). These tools typically assume that gene structures are fixed and that multiple variants do not act in combination. A recent analysis of exome sequencing data of more than 60 000 individuals highlighted the importance of interpreting variants in the context of the entire haplotype, particularly in the case of variants that alter the annotated reading frame (Lek et al., 2016). In addition, while a number of high-quality gene annotation sets are available for humans and other species, including GENCODE (Harrow et al., 2012), RefSeq (Pruitt et al., 2014), and Ensembl (Yates et al., 2016), it has been demonstrated that variant interpretation results can be sensitive to the gene structures used in the analysis (McCarthy et al., 2014; Frankish et al., 2015).
A productive step toward improving our understanding of how genetic variants can impact gene function in an individual is to characterize the potential changes to gene structure that may be induced by sequence variants. Methods for computational modeling and prediction of eukaryotic gene structures have been well-disseminated (Allen and Salzberg, 2005; Burge and Karlin, 1997; Guigo et al., 1992; Korf et al., 2001; Lukashin and Borodovsky, 1998; reviewed in Majoros, 2007; Stanke et al., 2006) and applied to the problem of annotating reference genomes, both human and non-human (Adams et al., 2000; Haas et al., 2008; Holt and Yandell, 2011; Lander et al., 2001; Parra et al., 2007; Venter et al., 2001; reviewed in Yandell and Ence, 2012). However, traditional gene-finding approaches make several assumptions that limit their application to predicting deleterious effects on gene structure in individuals. Specifically, they assume that genes are well formed, have typical codon usage statistics, and ultimately produce functional proteins. Many approaches also take into account evolutionary conservation between species. Those assumptions enable gene-finding models to achieve high levels of accuracy in elucidating the structures of protein-coding genes in reference genomes. However, such assumptions also limit the ability of gene-finders to identify functional changes to gene structure between individuals of a species.
As an example, traditional de novo gene finders struggle to correctly model the ABO gene that determines human blood group. The allele that gives rise to the O blood group contains an early frameshift inducing a premature stop codon believed to result in either mRNA degradation or translation to a different protein lacking enzymatic activity (Yamamoto et al., 1990). Probabilistic gene finders predict an incorrect gene structure for the O allele that modifies the reading frame in order to avoid the in-frame stop codon (Supplementary Fig. S1), as doing so allows a downstream exon to be annotated as coding, resulting in a higher probability according to the gene-finder’s objective function. In this way, traditional gene finders conflate multiple molecular and evolutionary processes in order to integrate diverse signals and maximize predictive accuracy in identifying functional genes in reference genomes, and in doing so are hampered in their ability to identify changes to gene structure that result in loss of function in an individual.
Here we describe a novel approach (ACE—Assessing Changes to Exons) that aids the elucidation of differences in gene structure between individuals of a species. In contrast to traditional gene-finding models, ACE does not assume that genes are fully functional in every individual. In particular, by considering within-species changes to gene structure without regard to possible downstream effects, ACE is able to identify changes to gene structure that may alter the function of the resulting protein, even if that protein is highly conserved between species. ACE can therefore predict individualized gene isoforms having altered—and possibly deleterious—protein function relative to the reference.
We demonstrate the use of ACE by generating personalized human transcriptome references for >2000 people sequenced as part of the Phase 3 1000 Genomes Project (The 1000 Genomes Project Consortium, 2015). We then quantify transcript expression using RNA-seq data from matched individuals for a subset of the 1000 Genomes Project sample. That analysis reveals that predicted cases of complete or partial loss of function in protein-coding genes via nonsense-mediated decay (NMD) are detectable as a reduction in transcript levels, albeit with much variation in the degree of reduction. That analysis also validates the use of ACE for identifying novel splice forms that may result when annotated splice sites are disrupted via sequence variants. In addition, we show that transcripts predicted to suffer loss of function in healthy adults are significantly depleted in genes found to be intolerant to mutation across the human population.
We designed ACE to be broadly applicable across eukaryotes. For that reason, we minimized the burden of extensive retraining for use on nonhuman species. We demonstrate that feature by confirming known phenotype-causing differences in gene structures between plant varieties.
2 Methods
2.1 Reconstructing haplotype sequences from a VCF file
ACE begins by reconstructing explicit haplotype sequences based on variants given in a phased VCF file (Fig. 1A), including all single-nucleotide variants, insertions, deletions, and short copy-number variants. VCF files may contain one or more individuals; ACE processes each sample independently. ACE left-normalizes all variants (Tan et al., 2015) and disambiguates overlapping variants by computing the transitive closure of the overlap relation and applying the longest variant, provided all other overlapping variants are properly nested and call for consistent substitutions. ACE provides two warning levels corresponding to overlapping variants that are compatible versus those that are incompatible. Those warnings are provided in an easily parsed format to allow filtering of sequences by confidence level prior to downstream analyses. ACE uses tabix (Li, 2011) for efficient extraction of variants in pre-specified intervals (Supplementary Methods), thus reducing the memory requirements for genome sequencing studies across large populations and facilitating parallelization on cluster compute environments. Detailed tracking of insertions and deletions allows ACE to efficiently compute a coordinate transformation to map reference annotations to haplotype sequences without the need to perform explicit sequence alignment (Supplementary Methods).
Fig. 1.

(A) ACE reconstructs explicit haplotype sequences from a phased VCF file, projects reference annotations onto them, detects possible gene structure changes, and interprets changes in terms of possible loss of function. (B) When a disrupted splice site is encountered, ACE enumerates possible alternate splice forms resulting from cryptic splicing, exon skipping, intron retention or any combination resulting from multiple variants
2.2 Identifying changes to splice patterns and reading frames
ACE requires that all reference gene models contain valid splice site consensus sequences as defined in a user-supplied configuration file. Similarly, ACE requires that reference protein-coding gene models contain valid start and stop codons in a consistent reading frame. Reference genes that violate those constraints are reported as possible mis-annotations and removed from further consideration. For all noncoding and coding genes, ACE identifies splice sites in the reference that change in the individualized genome. Such changes may either be absolute, by disrupting a valid consensus splice site, or may weaken the splice site at flanking nucleotides. ACE evaluates the latter possibility by aligning to a probabilistic weight matrix, or PWM. Models of human splice sites are provided (Supplementary Methods), and scripts to re-train for other organisms are also provided.
For each isoform of a gene in which a splice site is disrupted, ACE enumerates possible alternate splicing patterns for the isoform, including those in which an exon is skipped, an intron is retained, or a cryptic splice site is activated. By default, ACE identifies cryptic sites within 70 nucleotides (nt) of a disrupted site via a PWM thresholded to admit ∼98% of known human splice sites. The default distance was selected after observing that ∼75% of cryptic sites in DBASS, the Database of Aberrant Splice Sites (Buratti et al., 2007), are within that distance (Supplementary Fig. S2). For isoforms with multiple disrupted splice sites, ACE enumerates all combinations, corresponding to the set of paths through a splice graph for the gene (Fig. 1B). The splice graph is constrained to include only annotated splice sites and putative cryptic sites proximal to a disrupted annotated site.
ACE also identifies possible changes to reading frames. In cases in which the original start codon of a protein-coding gene is absent in the alternate sequence, ACE searches for the first downstream start codon of sufficient strength via a PWM. Changes to 5′ untranslated regions trigger a scan for upstream start codons that may be created as a result. For transcripts annotated as noncoding, ACE searches for reading frames longer than a configurable minimum length (default: 150 nt), and reports whether the reading frame exists in both the reference and alternate sequence (suggesting possible mis-annotation of the gene as noncoding) or only the alternate sequence (suggesting possible gain of function in the alternate sequence, or loss of function in the reference individual).
2.3 Identifying loss of function
For protein-coding genes, ACE identifies instances of protein truncation or nonsense-mediated decay (NMD), either in the mapped transcript or in alternate transcripts proposed when a splice site is disrupted. NMD is predicted based on the linear nucleotide distance between an in-frame stop codon and the most 3’ exon junction in the spliced mRNA. Distances greater than 50 nt have been shown to trigger NMD (Nagy and Maquat, 1998), and this phenomenon appears to be conserved between vertebrates and plants (Nyiko et al., 2013). ACE also reports loss of function (LOF) due to lack of either a valid in-frame stop codon or lack of a start codon scoring above the PWM threshold. Scans for start/stop codons are performed on spliced transcripts, so that start/stop codons straddling an intron are not overlooked. To enable filtering at arbitrary similarity thresholds, protein alignment scores (Supplementary Methods), defined as the percent sequence match between the reference and alternate proteins, are reported. Protein sequences are also emitted to allow detailed downstream analysis of amino acid changes by programs such as PolyPhen (Adzhubei et al., 2010), SIFT (Kumar et al., 2009), or VAAST (Hu et al., 2013).
2.4 Configuration and structured output
ACE is fully configurable in all of the parameters described above, via a simple configuration file (Supplementary Methods).
ACE produces a highly structured output file (Supplementary Fig. S3) describing gene structures in the reference and alternate sequences and results of their detailed comparison. The variants incorporated into the haplotype sequences are listed and classified as to their context within gene elements. Classification of variants is performed separately for both mapped isoforms and putative novel splice forms to highlight changes to a variant’s context between isoforms. We provide scripts for querying and filtering outputs and for converting to XML or GFF for use with other software.
2.5 Computational validation
To demonstrate the utility of ACE for large-scale genome sequencing projects, we used ACE to fully annotate the genomes of 2504 human samples sequenced by the Thousand Genomes Project. The analysis was parallelized across 500 compute nodes, and required two weeks to complete. GENCODE version 19 (Harrow et al., 2012) annotations were used as reference annotations for that analysis. To validate predicted novel isoforms, we aligned RNA-seq data from lymphoblastoma cell lines from 445 of the same individuals to the individualized genomes generated by ACE, using TopHat 2 (Kim et al., 2013). RNA data was obtained from the Geuvadis project (Montgomery et al., 2011). We used StringTie (Pertea et al., 2015) to quantify transcript abundance. Recent benchmarks have shown StringTie’s accuracy to be competitive with other state-of-the-art methods, though it is also clear that transcript abundance estimation is still an inaccurate process (Hayer et al., 2015). Thus, for validation of putative novel splice forms we rely primarily on finding spliced reads that map precisely to the putative splice junctions. We provided TopHat 2 and StringTie with both reference annotations mapped to the individualized genomes, as well as novel transcripts predicted by ACE (Supplementary Methods). For the analyses of human genes, we disabled intron retention as it has been found to be present in the Geuvadis data at lower levels than cryptic splicing and exon skipping (Lappalainen et al., 2013; Monlong et al., 2014), and has been shown to be overwhelmingly likely to lead to loss of function in human coding genes (Braunschweig et al., 2014; Jung et al., 2015).
To quantify the effect of predicted NMD events, we analyzed the relationship between transcript abundance and the number of NMD alleles in an individual, under the hypothesis that each additional NMD allele in an individual would result in a proportionate decrease in transcript abundance for a given gene isoform. We fit a linear mixed-effects model, log2(FPKM) ∼ Xβ + Zu, to the transcript abundance estimates provided by StringTie, where FPKM (fragments per kilobase of transcript per million reads mapped) measures transcript abundance, X is the number of functional (non-NMD) alleles, and Z is an indicator variable encoding the transcript identifier. The random-intercept term Zu incorporates a different intercept for each transcript, accounting for differences in expression between different transcripts and genes. Values of BETA were estimated after removing transcripts with a mean FPKM across all samples for each transcript below a chosen threshold. That threshold was then varied to assess stability of BETA estimates to threshold choice. Estimates of β were transformed (Supplementary Methods) into relative abundance ratios r0/2 = FPKM0/FPKM2, where FPKMk denotes mean FPKM among individuals predicted to have k functional alleles of a transcript. Thus, 1-r0/2 is the proportionate reduction in NMD homozygotes relative to individuals with two functional alleles.
As the 1000 Genomes Project individuals were reportedly healthy adults, we expected isoforms with LOF in at least one individual to be enriched for genes tolerant of functional mutations. We expected this effect to be stronger for the genes that are found as a homozygous LOF because they will exhibit both recessive and dominant effects. To test this, we analyzed the distributions of RVIS (Residual Variant Intolerance Score—Petrovski et al., 2013) and ncRVIS (nocoding RVIS—Petrovski et al., 2015) percentiles for genes in which ACE predicts LOF for at least one annotated isoform of the gene in 1000 Genomes Project samples. RVIS reflects the intolerance of genes to functional mutations affecting amino acids in protein-coding genes, while ncRVIS reflects intolerance to mutations in noncoding portions of genes.
To demonstrate the applicability of ACE to nonhuman species, we also analyzed 30 rice samples with fully sequenced genomes (The 3000 Rice Genomes Project, 2014).
3 Results
3.1 ACE predicts changes to gene structure
In the 1000 Genomes Project samples, ACE predicted a modest number of alternative splice forms for each disrupted splice site: 80% of cases involve at most three alternate patterns per disrupted site (median = 2, mode = 1) (Fig. 2A). When the alternate structures predicted in the Geuvadis samples are provided as annotations (in addition to mapped reference annotations), TopHat 2 is able to assign spliced reads to significantly more of the putative novel junctions than if TopHat 2 is provided only mapped reference annotations (cryptic-site isoforms: Fig. 2B, Wilcoxon W = 513660, P < 2.2 × 10−16; exon-skipping isoforms: Supplementary Fig. S4A, W = 537900, P < 2.2 × 10−16). Similarly, StringTie assigns nonzero FPKM values to significantly more of these putative novel splice patterns when they are provided as annotations than when they are not provided (cryptic sites: Fig. 2C, W = 198020, P < 2.2 × 10−16; exon-skipping: Supplementary Fig. S4B; W = 198020, P < 2.2 × 10−16). As such, ACE improves the sensitivity of both spliced read mapping and transcript quantification for putative novel isoforms when an annotated splice site is disrupted, and it is able to do so while predicting conservative numbers of such alternate splice patterns per disrupted site.
Fig. 2.
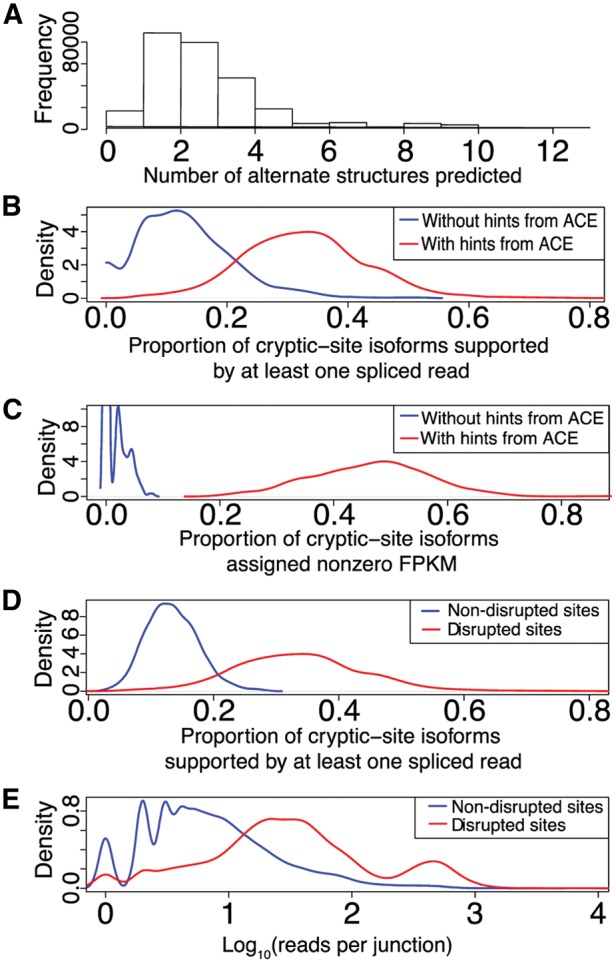
(A) Distribution of number of alternate structures predicted per disrupted splice site. (B) Distribution of proportions of predicted cryptic-site isoforms supported by at least one spliced read, when predicted isoforms are not provided to TopHat 2 (blue) and when they are provided (red). (C) Distribution of proportions of predicted cryptic-site isoforms assigned nonzero FPKM by StringTie when predicted isoforms are not provided to StringTie (blue) and when they are provided (red). (D) Distribution of proportions of predicted cryptic-site isoforms supported by at least one spliced read for splice sites simulated to be disrupted (blue) and for those that are disrupted (red). (E) Distribution of spliced reads per junction, on log10 scale, supporting sites simulated to be disrupted (blue) versus those that are disrupted
We also applied transcript quantitation methods Salmon (Patro et al., 2016) and Kallisto (Bray et al., 2016) to the Geuvadis data and quantified the number of ACE-predicted novel transcripts that were assigned expression values above a range of thresholds (Supplementary Fig. S5). Due to the substantial differences between expression estimates by the three approaches, we instead used raw counts of spliced reads aligning exactly to predicted novel splice junctions to investigate the specificity of ACE’s predictions. As a negative control, we randomly sampled 3.25 × 106 non-disrupted, annotated splice sites from the Geuvadis samples, and used ACE to generate putative novel splice patterns that could result if the splice site had been disrupted. We then quantified support for these negative control splicing events via the number of spliced reads assigned by TopHat 2 to the junctions.
Due to the stochastic nature of eukaryotic splicing, some splicing at non-annotated sites is expected (Pickrell et al., 2010; Stepankiw et al., 2015). The proportion of ACE predictions of cryptic splicing or of exon skipping at disrupted splice sites that are supported by at least one spliced read is significantly greater than that of randomly selected non-disrupted sites (Fig. 2D and Supplementary Fig. S4C, respectively; P < 2.2 × 10−16 in both cases). Similar results for all of the above comparisons were obtained when applying higher read-count or FPKM thresholds (Supplementary Figs S6 and S7). Furthermore, the numbers of spliced reads supporting predicted novel splice junctions are significantly greater in the case of disrupted splice sites than for non-disrupted sites (raw read counts: Fig. 2E, W = 785190, P < 2.2 × 10−16; normalized read counts: Supplementary Fig. S8, W = 791430, P < 2.2 × 10−16). Among those transcripts with disrupted splice sites for which ACE predicted at least one alternate splice form, in 55.5% of cases at least one ACE prediction was supported by at least three spliced reads mapped to the novel splice junction. Possible outcomes that may comprise the remaining cases but that we did not investigate include intron retention, use of cryptic sites further than the 70bp limit, failure to sequence spliced products due to low intrinsic expression levels, accelerated degradation of aberrant transcripts by RNA surveillance pathway, and sampling error. When multiple cryptic sites were available and at least one site was supported by at least three spliced reads, support for more than one site was found in only 13.4% of cases, suggesting possible discrimination among available cryptic sites by the splicing machinery.
As an additional negative control, we quantified mean cryptic splicing activity in the vicinity of all annotated splice sites that were disrupted in some individuals but not in others. We found that cryptic splicing levels were higher in individuals with disruption of the annotated splice site (Supplementary Fig. S9). That result illustrates that stochastic splicing does result in occasional use of cryptic sites, but that cryptic splicing is enriched near functional sites that have been disrupted.
3.2 ACE identifies thousands of annotated human splice sites as being potentially robust to disruption
In order to further explore the utility of ACE in identifying alternate splice forms that may arise when an annotated splice site is disrupted, we simulated disruption to every annotated splice site in every protein-coding gene in the human reference and classified each site as to whether there existed an alternate splice pattern found by ACE that could produce a highly similar protein product. Only alternate splice forms that did not result in a prediction of NMD, did not lack a start or stop codon, and encoded a protein differing by no more than ten amino acids (aa) from the reference protein were accepted as potentially retaining function.
Nearly 80000 human splice sites (78226/377278 = 20.7%) in 15134 genes were deemed by ACE to be potentially robust to disruption. A more conservative PWM threshold that would reject ∼20% of annotated human splice sites still results in over 30000 (32465/377278 = 8.6%) splice sites being identified as potentially robust to disruption. These results indicate that there may be ample opportunities to reduce false positives in disease studies in which splicing defects are suspected by applying ACE for interpretation of these altered gene structures. When tissue samples are available, putative splice forms proposed by ACE can be validated against RNA-seq data by providing them as annotations to a transcript quantification pipeline as described in the previous section, or by validating protein presence via western blot.
Among 1000 Genomes Project samples, the mean proportion of transcripts with disrupted splicing for which ACE was able to identify at least one alternate structure with no predicted LOF according to the above criteria was 0.46 (SD = 0.08; Supplementary Fig. S10A). This represents a substantial enrichment compared to the 0.21 estimated for the genome-wide scan, possibly reflecting the effects of natural selection on this control population.
3.3 ACE confirms previous estimates of the effect of nonsense-mediated decay on transcript levels
Nonsense-mediated decay accounted for over two-thirds (69%) of the loss-of-function predictions in the 2504 1000 Genomes Project samples. To better understand the impact of NMD on expression of target genes, we used the Geuvadis RNA-seq data and the transcript quantification pipeline described above to quantify the effect of NMD in terms of the average reduction in transcript levels per NMD allele, relative to individuals with two functional alleles. We first restricted our analysis to heterozygous individuals.
Based on the results of earlier in vitro experiments showing that NMD achieves a halving of transcript levels in episomal mini-gene constructs (Rosenberg et al., 2015), we hypothesized that each additional NMD allele at a diploid locus would reduce total transcript levels by 25% and that the homozygous NMD state would therefore result in a halving of mean FPKM. In Figure 3A, we show, on a log2 scale, the distribution of effect sizes E = FPKM1/FPKM2 for autosomal transcripts expressed in LCLs, where FPKM1 is the mean FPKM pooled among heterozygous individuals (having one NMD allele and one functional allele), and FPKM2 is the mean FPKM pooled among individuals having two functional alleles. The observed distribution matches our expectation of a 25% reduction (denoted by the dashed line) among heterozygotes, albeit with much variability, as also noted previously based on a subset of this data from 119 individuals (MacArthur et al., 2012). Applying higher FPKM thresholds produced similar results (Supplementary Fig. S11).
Fig. 3.

(A) Distribution of log2 effect sizes of N = 578 heterozygous NMD events as measured via RNA-seq transcript quantification. Dashed line at -0.42 denotes a 25% reduction in total transcript quantity. Data were filtered to improve power (sample size ≥ 30, mean FPKM ≥ 1). (B) Percentiles of Residual Variant Intolerance Scores (RVIS) for N = 633 genes in which at least one individual was predicted to be homozygous for gene loss of function
To extend the analysis to include homozygotes, we fit a linear mixed-effects model to the Geuvadis data. Utilizing a linear mixed model with random intercepts allows us to account for differences in expression levels between genes and isoforms because each isoform can have a different (random) intercept. After filtering to only include transcripts expressed in at least 30 individuals and that have both NMD and non-NMD predictions, we were left with 578 heterozygous and 38 homozygous observations. All estimates of coefficient β were significantly different from zero (all P < 2 × 10−27), and estimates were robust to filtering of the data at different minimum FPKM thresholds (Supplementary Fig. S12). The largest estimated β = 0.37 (SE = 0.01) a halving of transcript levels (r0/2 = 0.60) for homozygotes. The low sample size for homozygous alleles after filtering, variability in the efficiency of NMD for different transcripts, and general noise in RNA-seq quantification are likely contributors to the divergence of estimated r0/2 from an exact halving.
NMD events resulting from the creation of new upstream start codons were omitted from the above analysis, as many of these likely constitute so-called uORFs (upstream open reading frames), which can affect gene expression in myriad ways that are not fully understood (Barbosa et al., 2013). Indeed, fitting the above model to these uORF NMD predictions at various FPKM thresholds consistently results in an estimated β ≤ 0, indicating that NMD in uORFs is not predictable using the established methods for downstream reading frames, possibly due to their position near the 5’ cap site on the circularized RNA (Peixeiro et al., 2012; Silva et al., 2008) or to the potential for reinitiation of translation downstream (Neu-Yilik et al., 2011). As such, ACE marks all NMD predictions in uORFs as hypothetical and provides position and length information for the uORF, enabling users to interpret them on a case-by-case basis.
3.4 ACE’s loss-of-function predictions in healthy individuals are highly enriched for genes tolerant to mutation
All 2504 individuals in the 1000 Genomes Project sample harbored alleles predicted to suffer loss of function (LOF). Using ACE we estimated a median of 148 LOF genes per individual (range: 115-192), which is higher than the estimate of 97 based on experimental validation and stringent filtering of variants in a single European individual (MacArthur et al., 2012), but similar to the estimate of 149–182 truncation events found by the more recent Phase 3 1000 Genomes Project study (The Thousand Genomes Consortium, 2015).
For healthy adults we expect LOF predictions to be enriched for genes not critical to survival, and thus to have elevated tolerance to functional mutation. We assessed tolerance to mutation by computing RVIS and ncRVIS percentiles for all autosomal protein-coding genes predicted to suffer LOF in at least one individual. LOF was presumed if a transcript that was well-formed in the reference was predicted in the individual’s genome to suffer NMD (69% of all predicted LOF cases), to lack a start or stop codon (8% of cases), to have a disrupted splice site in a terminal exon with no viable alternative splice forms (2% of cases), or to encode a protein differing by at least 50% of its amino acids from the reference protein (21% of cases).
Loss-of-function predictions were enriched for genes tolerant to mutation according to both RVIS and ncRVIS scores. The distribution of RVIS percentiles for homozygous LOF genes was highly biased toward genes tolerant to mutation (higher RVIS scores), as expected (Fig. 3B). The observed distribution differs significantly from the distribution of all RVIS scores (Supplementary Fig. S13A) (median = 80th percentile, versus 50th percentile for all genes; Wilcoxon rank-sum test: W = 7378700, P < 2.2 × 10−16). Random sets of genes having similar lengths, numbers of exons, or G + C nucleotide composition resulted in distributions that could not be distinguished from uniform (Wilcoxon rank-sum, all P > 0.6; Supplementary Fig. S13B–E). The bias toward tolerance was significantly higher for homozygous LOF genes than for heterozygous LOF (W = 2214700, P < 2.2 × 10−16; Supplementary Fig. S14A–B), though heterozygous LOF genes were also significantly enriched for tolerance (median = 62nd percentile; W = 53451000, P < 2.2 × 10−16). Percentiles for ncRVIS were also significantly biased toward tolerance to mutation in these genes (homozygous: median = 59th percentile, W = 6032200, P < 5.5 × 10−11; heterozygous: median = 56th percentile, W = 48724000, P < 2.2 × 10−16), and that bias was again higher for homozygotes than heterozygotes (W = 1812500, P = 0.005; Supplementary Fig. S14C and D).
Because RVIS and ncRVIS scores are assigned to genes rather than to individual isoforms, they may not indicate intolerance levels for every isoform equally. Indeed, genes with a predicted homozygous LOF in at least one individual for at least one isoform that are classified as intolerant to variation (RVIS percentile < 0.20) were found to have significantly elevated numbers of isoforms compared to all of GENCODE (Wilcoxon rank-sum, W = 1932000, P < 2.2 × 10−16) (Supplementary Fig. S15). This observation is consistent with the possibility that the gene-level intolerance detected by RVIS might not indicate intolerance for the particular isoforms found to suffer LOF in these samples. Indeed, among the LOF predictions in 1000 Genomes Project samples, a majority of the genes were predicted to suffer LOF in some, but not all, of their isoforms (mean proportion among individuals was 0.59, SD = 0.03; Supplementary Fig. S10B), indicating that many LOF variants do not affect all isoforms equally.
3.5 ACE aids interpretation of insertion and deletion variants within genes
Insertions and deletions of short sequences can substantially alter gene structures, through their effect on translation reading frames, splice sites, or start or stop codons. Proper interpretation of such variants requires analysis of the resulting sequence within the context of the correct gene structure.
For example, in the CTU2 gene (Ensembl gene ENSG00000174177), which is involved in post-transcriptional modification of transfer RNAs, variant rs11278302 deletes an entire donor splice site (Fig. 4A), suggesting a possible effect on splicing. Indeed, the Ensembl variant effect predictor, VEP (McLaren et al., 2016) classifies this common variant (minor allele frequency in 1000 Genomes Project samples = 0.22) as having ‘high impact’ (Supplementary Fig. S16A). However, ACE discovers that the resulting sequence after deletion contains a valid donor consensus at the same location relative to the preceding exon, that the new splice site scores more highly under the donor-site PWM than the original donor site (−18.82 versus −19.77), and that the coding sequence remains unchanged, producing an identical protein. Furthermore, while sample HG00096 is homozygous for the alternate allele, TopHat 2 assigns 33 and 35 spliced reads respectively to the new splice junctions in the two haplotypes, consistent with ACE’s predictions.
Fig. 4.

(a) Deletion of an entire splice site (top: hg19 reference sequence; bottom: haplotypes 1 and 2 of 1000 Genomes Project sample HG00096). The resulting allele appears to retain a functional splice site despite the deletion, as concluded by ACE and supported by spliced RNA-seq reads. (B) Compensatory frameshift variants: the second variant corrects the change to the reading frame introduced by the first variant (top: hg19 reference sequence, bottom: haplotype 2 of 1000 Genomes Project sample HG00096)
An important class of insertion/deletion variants are frameshift mutations, which are insertions or deletions of a length not divisible by three in a coding sequence. These have the potential to radically alter encoded proteins by shifting the reading frame. Frameshifts typically induce premature in-frame stop codons resulting in truncated proteins and, often, a reduction in transcript levels via NMD. In the 1000 Genomes Project population, frameshifts were the largest contributor to predictions of NMD, accounting for 60% of predicted cases. Frameshifts were also the largest contributor to LOF predictions stemming from large protein changes, accounting for 71% of cases. When multiple frameshifts are present in a coding segment, however, their combined effect may be less severe than the predicted effect of any one frameshift if a downstream variant restores the original reading frame. Because ACE analyzes sequences after simultaneously applying all variants present, combinations of frameshifts that mutually cancel each other by restoring the original reading frame can be detected.
One example of compensatory frameshifts detected by ACE occurs in the ZFPM1 gene (ENSG00000179588), which plays a key role in erythroid differentiation. Within the coding segment of this gene are three common deletion variants, all within 10 nt of each other (Fig. 4B). The first two deletions (rs67712719, rs67322929) induce frameshifts, while the third (rs67873604) maintains the reading frame. Either rs67712719 or rs67322929 in isolation would result in premature termination and a large change to the amino acid sequence (Supplementary Fig. S17). Consequently, VEP classifies both rs67712719 and rs67322929 as having ‘high impact’ (Supplementary Fig. S16B). However, rs67712719 and rs67322929 commonly occur together in the 2504 1000 Genomes Project samples (4869/5008 = 97% of haplotypes), and the combination results in only two amino acid changes, as rs67322929 corrects the reading frame change introduced by rs67712719; the three variants together modify only four amino acids due to their proximity.
Every individual in the 1000 Genomes Project sample harbored one or more (median = 7 per individual) compensatory frameshifts affecting ≤30 amino acids. In this sample, the observed lengths of affected intervals (in amino acids) are very short on average (Supplementary Fig. S18), with a median length of only 1 aa, as compared to a null expectation of 260 aa for uniformly random, non-compensated frameshifts (Supplementary Fig. S19). This bias toward short affected lengths may reflect selection against large functional changes in proteins.
3.6 ACE accurately reconstructs human blood-group alleles at the ABO locus
The human ABO gene (ENSG00000175164) is responsible for human blood types. It encodes a glycosyltransferase that modifies carbohydrate content of red blood cell antigens, with the A allele producing the A antigen, the B allele the B antigen, and the O allele being non-functional (Yamamoto et al., 1990). In the non-functional O allele, a deletion of a single guanine in exon 6 creates a frameshift resulting in an in-frame stop codon in the same exon, so that only alleles A and B have a seventh coding exon.
The ABO locus is highly diverse in human populations and has assembly issues in both the GRCh37 and GRCh38 human reference genomes. The annotated allele in GRCh37 was the result of improper assembly of two different O alleles, while GRCh38 combined A and O alleles, producing a sequence identical to the known O1.01 allele (Yamamoto et al., 1990; Yip, 2002). Both assemblies now contain a patch as an alternate contig that represents an A allele. GENCODE version 19, the reference annotation for all of our analyses, annotates this gene as a processed transcript, and identifies no reading frame.
In 1000 Genomes Project sample HG00096, ACE identifies a start codon and open reading frame in both haplotypes (Fig. 5A), and proposes that the gene might be mis-annotated as noncoding. In haplotype 1 ACE identifies a coding gene structure that precisely matches the known O allele. In haplotype 2 ACE identifies a structure matching both the A and B alleles; translation of this structure reveals that the amino acid sequence is identical to the known B allele (Yamamoto et al., 2014). Thus, ACE has identified this individual as being heterozygous for the O and B alleles, and thus likely has a B blood type.
Fig. 5.

(A) Blood-group alleles of the ABO gene (ENSG00000175164). Black: coding segment; gray: untranslated region (UTR). Reference genome hg19 has the O allele; GENCODE version 19 annotates this gene as a processed transcript with no reading frame. ACE identifies the coding segment for the O and B alleles in heterozygous individual HG00096. (Coordinates have been transformed and mapped to the forward strand). (B) Complex differences in gene structure between alleles of the waxy gene in rice, due to a single G-to-T variant in a donor splice site. ACE detects a 1 nt shift in the donor splice site in the Wxb allele, resulting in a new start codon straddling the first intron. The new start codon alters the reading frame, leading to a premature stop codon and NMD
As noted in the Introduction, applying a state-of-the-art gene finder to this locus results in very different results. This stark difference highlights the importance of ACE’s method of modeling splicing decisions as independent of downstream translation effects when analyzing gene structures in re-sequencing data.
3.7 ACE identifies complex gene-structure changes in a plant gene influencing flavor and nutritional content
The waxy gene in domestic rice provides a test case for ACE’s ability to discover complex alterations to gene structure involving simultaneous changes to both splicing patterns and translation reading frames. Different alleles of waxy produce different ratios of amylose to amylopectin, leading to very different tastes and textures. Moreover, as these polysaccharide starches result in substantially different glycemic indices, their relative expression in different rice varieties has nutritional relevance.
We provided ACE with the annotated Wxa allele as reference annotation and projected this to the Wxb allele (Fig. 5B) using variants provided by the 3000 Rice Genomes Project. ACE recognizes that the G to T substitution caused by variant id12648080 causes a disruption to the donor splice consensus at the end of the first exon in the 5′ untranslated region. It then scans for and detects a new splice site scoring above PWM threshold in the vicinity of the annotated site. The new site is 1 nt upstream of the annotated site. This 1 nt shift in the donor site results in a new splice junction in which an A at the end of the first exon joins with a TG at the beginning of the second exon. ACE recognizes the spliced ATG as a valid start codon consensus. Together with its flanking bases this putative start codon scores above PWM threshold. ACE then proposes that the Wxb allele preferentially begins translation at this upstream start codon, and traces the resulting open reading frame, finding that it ends in a premature stop codon resulting in a prediction of NMD.
These conclusions match current understanding of how the Wxb allele functions (Cai et al., 1998; Isshiki et al., 1998; Tian et al., 2009). The differences between Wxa and Wxb would be particularly challenging for a traditional gene finder to identify, as gene finders based on generalized hidden Markov models (GHMMs) utilize discrete states to represent multi-nucleotide features such as start codons. GHMM-based gene finders are therefore unable to predict a start codon straddling an intron using standard decoding algorithms (e.g., Majoros et al., 2005). The approach taken by ACE to separate modeling of transcription from translation simplifies the task because splicing decisions are made first. Only after introns are removed does ACE apply the ribosome scanning model to search for a start codon. In this way, ACE more closely models the way splicing and translation are believed to occur in the cell.
4 Discussion
The accurate detection and interpretation of gene structure differences in the genomes of individuals or strains is an important and unsolved problem, with clear relevance to genetic studies of disease and other phenotypes. As we have shown, individual variants disrupting splice sites or reading frames do not necessarily result in LOF. Correct disambiguation of the effects of such variants, particularly within the context of individual genomes harboring combinations of variants that may interact, has the potential to substantially reduce false positives in burden testing. We have also demonstrated that traditional gene-finding models are not suited for such applications without modification, as such models make assumptions incongruous to the task of detecting possibly deleterious changes that violate conservation patterns in genes.
Here we have proposed an alternate framework for identifying and interpreting gene structure changes, in which the potentially deleterious downstream effects of changes to gene structure are not considered when proposing such changes. By withholding information regarding possible downstream effects when considering changes to gene structures, we enable ACE to identify changes that may result in a loss or change of function, and to do so in a minimally biased manner. Because ACE has very few parameters, it is more readily applicable to other species than traditional gene finding models that utilize tens of thousands of parameters and need to be retrained for each new species (Korf, 2004). Moreover, when the intended application is to provide plausible novel gene structures to an RNA-seq pipeline, the use of a minimally biased approach favoring sensitivity over specificity may be desirable. However, as noted previously, interpretation of transcript abundance estimates for these putative isoforms should be undertaken with caution (Hayer et al., 2015).
The example of the ABO gene is particularly instructive, as it demonstrates a case of different gene structures in different individuals with different and medically important phenotypes (blood type). As we have shown, state-of-the-art de novo gene finders have difficulty correctly identifying the gene structures of individual alleles of this gene. In the case of the O allele, which has fewer coding exons than the A and B alleles, there is a potential for misinterpretation of variants occurring in the gene. As the final coding exon of the A and B alleles is not present in the O allele, correct interpretation of variants in that exon depends on knowing which allele is present in an individual. Furthermore, as the O allele is likely nonfunctional, accumulation of variants in that allele is likely underway (Yamamoto et al., 1990) and may lead to false positives in identification of deleterious variants when incorrect annotations are used. The waxy gene in rice provides another example of allelic differences in gene structure precipitated by a simple sequence variant. We speculate that there may be numerous other genes in which the correct interpretation of variants differs between alleles in a way that depends on knowing the correct gene structure for each allele.
ACE’s predictions of loss of function in the 1000 Genomes Project samples are highly enriched for genes tolerant of functional mutation, indicating a low false positive rate for identification of loss-of-function alleles. Furthermore, our analyses of the Geuvadis data have confirmed that the nonsense-mediated decay pathway in humans typically does not result in complete loss of transcripts, but rather achieves a quantitative reduction on the order of a halving, albeit with much variation, often leaving many copies of NMD target isoforms undegraded. Such transcripts escaping degradation will encode truncated proteins that, if they escape further checkpoints during folding, can in some cases result in a deleterious gain-of-function and poison products (Balasubramani et al., 2015). Because ACE reports truncation products for all putative NMD targets, downstream analyses may be thereby enabled to infer deleterious effects directly or via association with phenotypes.
There is much room for enhancement of our method, for example through detailed modeling of the splicing regulatory landscape and its influence on splice site selection (e.g., Rosenberg et al., 2015). It is also important to note that the accuracy of ACE’s predictions depends on the accuracy of genotype phasing. In the case of the 1000 Genomes Project data used here, much effort has gone into ensuring that data are accurately phased (Delaneau et al., 2013). As sequencing costs decrease and read lengths increase, we expect phasing accuracy to continue to improve in newer resequencing studies, which will further increase ACE’s accuracy.
The use of phased haplotypes is important for joint interpretation of variants that may interact in cis, as highlighted recently by Lek et al. (2016) using exome sequencing data from ∼60 000 individuals. Those authors reported an average of 23 multinucleotide polymorphisms (multiple variants that affect the same codon) per individual, and lament the lack of tools that can interpret variants in the context of a haplotype. The true mean number of compensatory variants will likely be higher than 23 when other compensatory mechanisms are considered, including frame-restoring indels and generation and/or use of alternate splice sites. These scenarios support a shift away from variant-centric analysis pipelines to tools such as ACE that generate haplotype-aware gene annotations as a way of understanding genetic variation in populations.
In summary, ACE is an initial step towards modeling gene structure differences among individuals of a single species. Importantly, ACE makes fewer assumptions than traditional gene-finding techniques to improve prediction of deleterious differences in gene structure. The abundance of human splice sites with possible robustness in the form of alternate splicing solutions that result in minimal changes to the encoded protein suggests that ACE may have ample opportunities to reduce false positives in disease studies in which splicing defects are identified but have unknown significance. ACE is equally applicable to identifying differences between lines of economically important animal or crop species, and it may have utility for RNA-seq analyses and for detecting possible gain-of-function variants in cancer genomes. The design of ACE’s computational model makes it directly applicable to nonhuman species with minimal re-training, enabling studies of other model and non-model animal and plant species.
Funding
W.H.M. and T.E.R. were supported by grants from the National Institutes of Health [R01 DK099820 and R01 DK097534]. M.S.C. was supported by a Postdoctoral Research Fellowship in Biology from the National Science Foundation [PRFB-1523793]. W.H.M., C.H., and M.Y were supported by grants from the National Science Foundation [EAGER IOS-1561337 and IOS-1126998]. E.D. was supported by a grant from the National Science Foundation [REU-1559816].
Conflict of Interest: none declared.
Supplementary Material
References
- Adams M.D. et al. (2000) The genome sequence of Drosophila melanogaster. Science, 287, 2185–2195. [DOI] [PubMed] [Google Scholar]
- Adzhubei I.A. et al. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen J.E., Salzberg S.L. (2005) JIGSAW: integration of multiple sources of evidence for gene prediction. Bioinformatics, 21, 3596–3603. [DOI] [PubMed] [Google Scholar]
- Balasubramani A. et al. (2015) Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1–BAP1 complex. Nat. Commun., 6, 7307.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa C. et al. (2013) Gene expression regulation by upstream open reading frames and human disease. PLoS Genet., 9, e1003529.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig U. et al. (2014) Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res, 10, 1101/gr.177790.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray N.L. et al. (2016) Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol., 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Buratti E. et al. (2007) Aberrant 5' splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res., 35, 4250–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge C., Karlin S. (1997) Prediction of complete gene structures in human genomic DNA. J. Mol. Biol., 268, 78–94. [DOI] [PubMed] [Google Scholar]
- Cai X. et al. (1998) Aberrant splicing of intron 1 leads to the heterogeneous 5’ UTR and decreased expression of waxy gene in rice cultivars of intermediate amylose content. Plant J, 14, 459–465. [DOI] [PubMed] [Google Scholar]
- Cingolani P. et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin), 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O. et al. (2013) Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat. Commun., 5, 3934.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish A. et al. (2015) Comparison of GENCODE and RefSeq gene annotation and the impact of reference geneset on variant effect prediction. BMC Genomics, 16(Suppl 8), S2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigo R. et al. (1992) Prediction of gene structure. J. Mol. Biol., 226, 141–157. [DOI] [PubMed] [Google Scholar]
- Haas B.J. et al. (2008) Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol., 9, R7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J. et al. (2012) GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res., 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayer K.E. et al. (2015) Benchmark analysis of algorithms for determining and quantifying full-length mRNA splice forms from RNA-seq data. Bioinformatics, 31, 3938–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt C., Yandell M. (2011) MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics, 12, 491.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H. et al. (2013) VAAST 2.0: improved variant classification and disease-gene identification using a conservation-controlled amino acid substitution matrix. Genet. Epidemiol., 37, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isshiki M. et al. (1998) A naturally occurring functional allele of the rice waxy locus has a GT to TT mutation at the 5’ splice site of the first intron. Plant J., 15, 133–138. [DOI] [PubMed] [Google Scholar]
- Jung H. et al. (2015) Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat. Genet., 47, 1242–1248. [DOI] [PubMed] [Google Scholar]
- Kim D. et al. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol., 14, R36.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. et al. (2001) Integrating genomic homology into gene structure prediction. Bioinformatics, 17, S140–S148. [DOI] [PubMed] [Google Scholar]
- Korf I. (2004) Gene finding in novel genomes. BMC Bioinformatics, 5, 59.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P. et al. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc., 4, 1073–1081. [DOI] [PubMed] [Google Scholar]
- Lander E.S. et al. (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- Lappalainen T. et al. (2013) Transcriptome and genome sequencing uncovers functional variation in humans. Nature, 501, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M. et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2011) Tabix: fast retrieval of features from generic TAB-delimited files. Bioinformatics, 27, 718–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashin A.V., Borodovsky M. (1998) GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res., 26, 1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur D.G. et al. (2012) A systematic survey of loss-of-function variants in human protein-coding genes. Science, 335, 823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros W.H. et al. (2005) Efficient decoding algorithms for generalized hidden Markov model gene finders. BMC Bioinformatics, 6, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros W.H. (2007) Methods for Computational Gene Prediction. Cambridge University Press, Cambridge, UK. [Google Scholar]
- McCarthy D.J. et al. (2014) Choice of transcripts and software has a large effect on variant annotation. Genome Med., 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W. et al. (2016) The Ensembl Variant Effect Predictor. Genome Biol., 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monlong J. et al. (2014) Identification of genetic variants associated with alternative splicing using sQTLseekeR. Nat. Commun., 5, 4698.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery S.B. et al. (2011) Rare and common regulatory variation in population-scale sequenced human genomes. PLoS Genet., 7, e1002144.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy E., Maquat L.E. (1998) A rule for termination-codon position within intron-containing genes: when nonsense affects mRNA abundance. Trends Biochem. Sci., 23, 198–199. [DOI] [PubMed] [Google Scholar]
- Neu-Yilik G. et al. (2011) Mechanism of escape from nonsense-mediated mRNA decay of human b-globin transcripts with nonsense mutations in the first exon. RNA, 17, 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyiko T. et al. (2013) Plant nonsense-mediated mRNA decay is controlled by different autoregulatory circuits and can be induced by an EJC-like complex. Nucleic Acids Res., 41, 6715–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G. et al. (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Patro R. et al. (2016) Salmon provides accurate, fast, and bias-aware transcript expression estimates using dual-phase inference. bioRxiv.
- Peixeiro I. et al. (2012) Interaction of PABPC1 with the translation initiation complex is critical to the NMD resistance of AUG-proximal nonsense mutations. Nucleic Acids Res., 40, 1160–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M. et al. (2015) StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotech. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski S. et al. (2013) Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet., 9, e1003709.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski S. et al. (2015) The intolerance of regulatory sequence to genetic variation predicts gene dosage sensitivity. PLoS Genet., 11, e1005492.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell J.K. et al. (2010) Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet., 6, e1001236.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt K.D. et al. (2014) RefSeq: an update on mammalian referenced sequences. Nucleic Acids Res., 42(Database), D756–D763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg A.B. et al. (2015) Learning the sequence determinants of alternative splicing from millions of random sequences. Cell, 163, 698–711. [DOI] [PubMed] [Google Scholar]
- Silva A.L. et al. (2008) Proximity of the poly(A)-binding protein to a premature termination codon inhibits mammalian nonsense-mediated mRNA decay. RNA, 14, 563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M. et al. (2006) Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics, 7, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepankiw N. et al. (2015) Widespread alternative and aberrant splicing revealed by lariat sequencing. Nucleic Acids Res., 43, 8488–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan A. et al. (2015) Unified Representation of Genetic Variants. Bioinformatics, 31, 2202–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Z. et al. (2009) Allelic diversities in rice starch biosynthesis lead to a diverse array of rice eating and cooking qualities. pnas, 106, 21760–21765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 3000 Rice Genomes Project (2014) The 3000 rice genomes project. GigaScience, 3, 7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter J.C. et al. (2001) The sequence of the human genome. Science, 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- Wang K. et al. (2010) ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data Nucleic Acids Research, 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto F. et al. (1990) Molecular genetic basis of the histo-blood group ABO system. Nature, 345, 229–233. [DOI] [PubMed] [Google Scholar]
- Yamamoto F. et al. (2014) An integrative evolution theory of histo-blood group ABO and related genes. Sci. Rep., 4, 6601.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandell M., Ence D. (2012) A beginner's guide to eukaryotic genome annotation. Nat. Rev. Genet., 13, 329–342. [DOI] [PubMed] [Google Scholar]
- Yates A. et al. (2016) Ensembl 2016. Nucleic Acids Res., 44(Database), D710–D716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip S.P. (2002) Sequence variation at the human ABO locus. Ann. Hum. Genet., 66(Pt 1), 1–27. 2002, [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.