

Abstract
Motivation
Despite significant efforts in expert curation, clinical relevance about most of the 154 million dbSNP reference variants (RS) remains unknown. However, a wealth of knowledge about the variant biological function/disease impact is buried in unstructured literature data. Previous studies have attempted to harvest and unlock such information with text-mining techniques but are of limited use because their mutation extraction results are not standardized or integrated with curated data.
Results
We propose an automatic method to extract and normalize variant mentions to unique identifiers (dbSNP RSIDs). Our method, in benchmarking results, demonstrates a high F-measure of ∼90% and compared favorably to the state of the art. Next, we applied our approach to the entire PubMed and validated the results by verifying that each extracted variant-gene pair matched the dbSNP annotation based on mapped genomic position, and by analyzing variants curated in ClinVar. We then determined which text-mined variants and genes constituted novel discoveries. Our analysis reveals 41 889 RS numbers (associated with 9151 genes) not found in ClinVar. Moreover, we obtained a rich set worth further review: 12 462 rare variants (MAF ≤ 0.01) in 3849 genes which are presumed to be deleterious and not frequently found in the general population. To our knowledge, this is the first large-scale study to analyze and integrate text-mined variant data with curated knowledge in existing databases. Our results suggest that databases can be significantly enriched by text mining and that the combined information can greatly assist human efforts in evaluating/prioritizing variants in genomic research.
Availability and implementation
The tmVar 2.0 source code and corpus are freely available at https://www.ncbi.nlm.nih.gov/research/bionlp/Tools/tmvar/
Contact
zhiyong.lu@nih.gov
1 Introduction
Understanding the associations of genomic variants with diseases or conditions and assessing their clinical significance is a key step towards precision medicine. For example, G6PD (glucose-6-phosphate dehydrogenase) deficiency is a human genetic disease which may cause red blood cells to break down in response to certain medications, infections, or other stressors. It is caused by one of several single nucleotide polymorphisms (SNPs) [e.g. G202A (rs1050828), A376G (rs1050829), T968C (rs76723693) and C563T (rs5030868) (Zamorano-Jiménez et al., 2015)].
The U.S. National Center for Biotechnology Information (NCBI) provides web access to biomedical and genomic information. Several of its resources archive rich information related to genomic sequence variants. For example, the Database for Short Genetic Variations (dbSNP) (Sherry et al., 2001), despite its name, archives a broad collection of common and rare simple genetic variations including SNPs, short deletion/insertion, short tandem repeats and multi-nucleotide polymorphisms. Each SNP record in dbSNP is assigned a stable and unique variant accession identifier, known as RS number (RSID), which is linked to aggregated information such as the associated gene, functional consequences and allele frequency. Using an RSID for referring to a genomic variation provides several advantages: (i) The record is updated regularly by dbSNP with precise genomic locations and aggregated information from multiple submitters; (ii) it is accepted as an unambiguous variant identifier in publication; and (iii) it is convenient for data sharing and for integration with other data sources.
ClinVar (Landrum et al., 2016) is another NCBI database which collects and reports relationships between human variation and phenotype for a subset of dbSNP variations that may be medically relevant (ClinVar also has a subset of variants dbVar for large structural variations). The ClinVar database provides information about the variant, characterization of the phenotype and supporting evidence for clinical significance (pathogenic, benign, etc.). Variants with clinical significance in dbSNP are linked to ClinVar information.
In addition to the NCBI databases, several other variation-disease/phenotype/trait resources are publicly available. The Online Mendelian Inheritance in Man (OMIM) (Amberger et al., 2015) database is continuously updated with catalog of human genes and genetic traits. The NHGRI-EBI GWAS Catalog is a collection of a genome-wide set of genetic variants in different individuals associated with a trait (Coordinators, 2016). For genomic variant information in cancer, COSMIC contains expert-curated data of somatic mutations (Forbes et al., 2017) while CIViC is an expert-crowdsourced database focusing on clinical implications (Griffith et al., 2017). DisGeNET (Piñero et al., 2017) is a recent platform integrating information on gene-variation-disease associations from several public data sources and the literature.
Despite these efforts in manual curation and information cumulated over past decades, the clinical relevance of most of the 154 million dbSNP reference variants (RS) remains unknown. On the other hand, a wealth of human knowledge about the variant biological function and disease impact is buried in unstructured literature data (Jensen et al., 2006). Integrating literature data is challenging because: (i) the increasing volume of publication is outpacing manual literature curation (Rodriguez-Esteban, 2015); and (ii) despite the HGVS standards for variant nomenclature and the use of dbSNP RSID, a large number of genetic variants are mentioned with different names in the literature, making it difficult to assemble all known information (e.g. associated diseases) for a given variant.
In the past, a few attempts have been made to harvest such information using text-mining techniques, but their results are of limited use because the automatically extracted variant mentions are not standardized or normalized to standard database identifiers such as RSIDs (Bonis et al., 2006; Burger et al., 2014; Caporaso et al., 2007; Doughty et al., 2011; Hofmann-Apitius et al., 2008; Klinger et al., 2007; Krallinger et al., 2009; Laurila et al., 2010; Naderi and Witte, 2012; Ravikumar et al., 2015; Thomas et al., 2016; Wei et al., 2013a). For instance, automatic mutation detection tools such as MutationFinder (Caporaso et al., 2007) and EMU (Doughty et al., 2011) return their extraction results in the wNm format (e.g. A42G), which is difficult to integrate with existing genomic information in databases. Previously, we developed tmVar (Wei et al., 2013a), a high-performance software tool (see Lee et al., 2016; Yepes and Verspoor, 2014a,b for external evaluations comparing tmVar with other tools) for identifying mutations asserted in various formats in the PubMed article and re-writing them in HGVS formats (e.g. p.Pro12Ala). However, HGVS names can still be ambiguous: one can often be linked to multiple RSIDs (e.g. rs767209585 and rs773973301 are both associated with p.Pro12Ala). Indeed, on average, one protein mutation in HGVS name maps to more than ten RSIDs.
Hence, in this work we first extended tmVar to automatically normalize the variant mentions and map them to standard dbSNP RS numbers (see Fig. 1 for such an example). Our proposed approach uses multiple resources to resolve the ambiguity in search of the correct identifier. When evaluated against a human gold standard, our improved tmVar method (tmVar 2.0) shows high performance (nearly 90% in F-measures) and compares favorably to SETH (Thomas et al., 2016), the only other automated tool that also returns normalized mutation ids. Next, we applied our automatic approach over the entire PubMed (downloaded on 2017/2/24) and validated our results with dbSNP and ClinVar for data consistency and knowledge discovery. Our analysis includes: (i) comparing the text-mined PMID-RSID pairs with annotated dbSNP data, (ii) analyzing variants curated in ClinVar and (iii) discovering novel connections between variants, gene and diseases. Our investigation revealed 161 178 missing RSID-PMID links in dbSNP and 41 889 RSIDs not found in ClinVar. Moreover, our results also include over 120 000 rare variants (MAF ≤ 0.01) in nearly 4000 genes across the genome which are presumed to be deleterious and are not frequently found in the general population.
Fig. 1.

An example of detecting variation mentions, rewriting in HGVS forms, and normalizing to a unique RS number
To our best knowledge, we are the first to analyze and integrate literature-mined variant information with existing data in dbSNP and ClinVar. Our PubMed-scale analysis shows that automatically computed information combined with existing database annotations can significantly aid human efforts to prioritize and curate variants for interpretation in personal genomes and precision medicine.
2 Materials and methods
2.1 Detecting mentions of genomic variants
As described in Figure 2, tmVar applies a machine learning approach to tag mutation mentions in free text. Given a text input, it detects terms that represent variants of multiple types (SNV, insertion, deletion, etc.) and sequence context (genomic, transcript and protein) and returns its results in HGVS form. For instance, when applied to the example sentence in Figure 2, tmVar would tag E429A as a mutation in PMID: 22023786 and return p.Glu429Ala in its output. The tmVar model was trained on a set of PubMed abstracts and was reported to achieve over 90% accuracy in F-measure in benchmarking results (we refer readers to Wei et al., 2013a for more details).
Fig. 2.

The overall workflow of our mutation normalization process
2.2 Normalizing genomic variants
In this work, we developed a new module for mapping each previously detected mutation mention to a corresponding RS number as shown in Figure 1. Before we performed normalization, we first built a comprehensive lexicon containing all possible mappings between variant mentions and RSIDs, harvested from three difference sources: dbSNP, ClinVar and PubMed. Specifically, we first downloaded the dbSNP data [ftp://ftp.ncbi.nlm.nih.gov/snp/organisms/human_9606/database/organism_data/SNP_HGVS.bcp.gz (downloaded on 8/16/2016)] where each RSID maps to a unique gene and a list of variations of different sequence types [e.g. genomic (g.), transcript CDS (c.) and protein sequences (p.)]. Second, mappings from ClinVar [ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/tab_delimited/hgvs4variation.txt.gz (downloaded on 8/16/2016)] were downloaded. Finally, we collected mappings from the entire PubMed by extracting adjacent pairs of mutations and RSIDs in the text [e.g. Pro12Ala (rs1801282) in PMID: 27581934]. In total, we collected over 93 million mappings which correspond to 38 657 human genes with on average 2431 RSIDs per gene.
Next, for each extracted mention, we use two main strategies to find its corresponding RSID (see Fig. 2):
(1) Pattern Matching: We developed a set of patterns (e.g. ‘[Gene/Protein] ([DNAMutation] with [RSID])’) to detect a pair of mutation and RSID co-occurring in the same sentence. When matched, the RSID would be directly assigned to the corresponding variation. For example, the sentence ‘The less common Thr164Ile polymorphism (rs1800888, Ile allele frequency 1.5%)’ in PMID: 16935688 matches one of our patterns. Thus, Thr164Ile is normalized to rs1800888 without further processing.
(2) For the remaining mentions, we generated a list of candidate RSIDs by searching our lexicon. Since one variation is commonly mapped to multiple RSIDs, a disambiguation step is required to select the correct RSID from the list of candidates. For disambiguation, we used global information in the entire article and/or variant-associated gene information. Specifically, if any of the candidate RSIDs appeared in the article, we would return that RS number for the target mutation and consider the normalization process finished. Otherwise, we searched for gene(s) in the abstract that could be associated with the candidate RSIDs. For this, we first applied our state-of-the-art gene tagger, GNormPlus (Wei et al., 2015) to locate all genes mentioned in the text. Then for each gene, we obtained its list of associated RSIDs from dbSNP [ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b147_GRCh38p2/database/organism_data/b147_SNPContigLocusId_107.bcp.gz (downloaded on 8/16/2016)]. Next, we evaluated each gene in order of their proximity to the target mutation mention by comparing the two lists of RSIDs (see Fig. 3). Any RSID found in both lists would then be returned as the answer for the target mention.
Fig. 3.

An example of the dictionary-lookup step
2.3 Integrating dbSNP and ClinVar information and analysis
Matched RS and gene were loaded into MSSQL 2014 database server for comparison with dbSNP human build 149 data and ClinVar release (March 2, 2017) based on GRCh38.p7 assembly and human RefSeq annotation 108. SQL queries were used for subsequent comparisons and analysis.
The RS-gene ID pairs found by the tmVar tool were compared to existing RS-gene ID pairs in dbSNP and/or ClinVar and then annotated with existing data. The RSIDs were analyzed across several metrics including protein functional consequences and clinical significance. The frequency data used to assess population frequency comes from the 1000 Genomes Phase 3, the Exome Aggregation Consortium (ver 0.3), the NHLBI GO Exome Sequencing Project and gnomAD (Lek et al., 2016). The RS found by the tmVar tool which exist in dbSNP, but not in ClinVar, are denoted as ‘novel RS’ in this work.
3 Results
3.1 Results of automatic mutation normalization by tmVar 2.0
Following the lead of (Thomas et al., 2016), we evaluated our normalization method on two external corpora: OSIRIS (Furlong et al., 2008) and Thomas (Thomas et al., 2011). The first corpus (Furlong et al., 2008) consists primarily of protein mutations—among its 258 mentions, only 32 are DNA variations—while the (Thomas et al., 2011) corpus is more balanced including 283 protein variations and 244 DNA variations. In addition, we manually annotated a new corpus of 158 PubMed abstracts. We collected these articles from the original tmVar corpus (Wei et al., 2013a) and manually added associated RSIDs for each mutation mention when applicable. The resulting annotations contain 435 protein and 190 DNA variations.
The results of our method are shown in Table 1 and compared with those of SETH (Thomas et al., 2016), the only other method, to our best knowledge, that produces normalized mutation IDs. As can be seen, our method achieved consistently higher accuracy than SETH on these datasets: 10.2, 10.3 and 24.1% relative improvement in F-scores respectively. This further suggests (also confirmed by our manual examination of the results) that the normalization performance of our method on DNA variation is in particular more accurate than that of the previous method.
Table 1.
The performance comparison with SETH on the revised tmVar corpus, OSIRIS (Furlong et al., 2008) corpus and Thomas et al. (2011) corpora
| Corpus | method | TP | FP | FN | Precision | Recall | F-score |
|---|---|---|---|---|---|---|---|
| tmVar2 | tmVar | 565 | 16 | 60 | 97.25% | 90.40% | 93.70% |
| SETH | 466 | 5 | 159 | 98.94% | 74.56% | 85.04% | |
| OSIRIS | tmVar | 208 | 6 | 50 | 97.20% | 80.62% | 88.14% |
| SETH | 179 | 11 | 79 | 94.21% | 69.38% | 79.91% | |
| Thomas | tmVar | 465 | 52 | 62 | 89.94% | 88.24% | 89.08% |
| SETH | 303 | 14 | 224 | 95.58% | 57.50% | 71.80% |
Note: The highest performance value for each corpus is shown in bold.
3.2 PubMed-scale analysis: enriching literature links for variants in dbSNP and ClinVar
Performing normalization allows us to compare text-mined PMID-RSID linkages with existing annotated linkages in dbSNP. After applying our method to the entire PubMed, we obtained a total of 230 147 unique PMID-RSID pairs (62 452 unique RSIDs and 122 903 PMIDs, respectively). On average, each article is associated with 1.88 RS numbers, though this distribution is highly skewed and some PMIDs are associated with over 25 RSIDs; see Figure 4. Conversely, each SNP is associated with an average of 3.65 PubMed articles. This distribution is also highly skewed, with some RSIDs, like rs77375493, appear in over 1000 articles; see Figure 4.
Fig. 4.
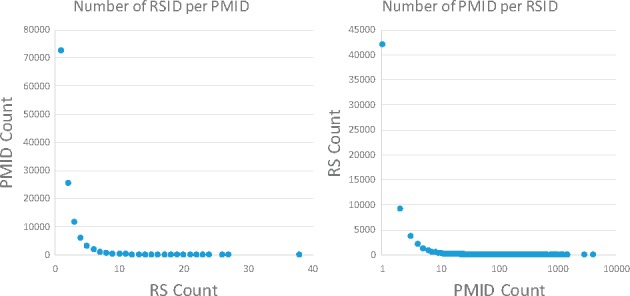
The distribution of PMID and RSID links
We compared these PMID-RSID pairs with the 310 323 unique pairs in dbSNP that were collected through automatic extraction of explicitly mentioned RS numbers in full text using regular expressions. The dbSNP annotated PMID-RS pairs are updated regularly and made available publicly as part of dbSNP build release (Sherry et al., 2001). As shown in Figure 5, the overlap is only approximately 30% (68 969 pairs) of total text-mined pairs by tmVar. To understand the discrepancies, we randomly reviewed 100 pairs that were not found by our automatic method. We observed that these are exclusively present in the full text (as opposed to abstracts), with many found in the tables or supplementary files.
Fig. 5.

The overlap between tmVar, dbSNP and ClinVar results. The numbers refer to the pairs of PMID-RSID
We also compared tmVar results with the 151 368 unique PMID-RSID pairs in ClinVar where PMIDs were provided along with the variants submitted to ClinVar by the submitters. As shown in Figure 5, we find even fewer overlapping pairs. In addition to the same abstract versus full text issue (as in dbSNP), this is also because submitted PMIDs may not actually describe the variant in text but rather the guidelines used to interpret the clinical assertion (e.g. PMID: 11280952, RSID: 38493).
Our automatic method produced a total of 161 178 novel PMID-RSID pairs that are not in dbSNP (Fig. 5), representing a potential 52.2% increase over existing literature links for dbSNP. For these pairs, they do not have their RSIDs explicitly mentioned in the PubMed abstract. Instead, the authors describe the mutations in various forms, making the normalization process necessary for their recognition (e.g. 74 Asp replaced by His; Pro-40-Leu; -1067G>A). tmVar added 161 178 new PMID-RSID links to dbSNP that include 33 661 unique RSIDs not previously linked to any PMID and 93 200 unique PMIDs not linked to an RS in dbSNP.
3.2.1 Variant analysis using dbSNP and ClinVar annotations
To access the biological and clinical impacts of the text-mined RS found by tmVar (tmVar RS), we compared them with existing annotations in dbSNP and ClinVar. The tmVar RS results (62 452 RS numbers in 9782 genes) were categorized using dbSNP and ClinVar annotations along multiple facets, including functional consequences (synonymous, non-synonymous, etc.) based on RefSeq mRNA annotations, minor allele frequency (MAF), and clinical significance in order to prioritize their biological significance and assess their clinical impact.
Functional consequence. Variations that alter the protein coding—including missense, stop-gain, stop-loss and frameshifts—are more likely to have deleterious effects (see supporting evidence with known variants in ClinVar; Fig. 6a). These coding variations (summarized by the non-redundant sets of RS from CDS-missense, CDS-synon, CDS-stop gain, CDS-indel and CDS-stop loss in Table 2) account for 50% (31 868/62 452) of the total tmVar RS identified. The next largest categories are the intronic variants which may affect mRNA binding motifs, expression regulatory regions and splicing.
Fig. 6.

The total counts of pathogenic and benign of ClinVar in different (a) functional consequence and (b) population frequency (MAF)
Table 2.
Summary of the intersection of text-mined RSIDs by tmVar with existing dbSNP and ClinVar annotations
| RS attributes | tmVar RS |
||
|---|---|---|---|
| Total RS | Novel RS | ||
| Variant | Number of RSIDs (in Gene) | 62 452 (9782) | 41 889 (9151) |
| Functional Class | CDS-missense | 27 865 | 12 854 |
| CDS-synon | 4608 | 2985 | |
| CDS-stop gain | 2810 | 694 | |
| CDS-stop loss | 46 | 13 | |
| CDS-frameshift | 1024 | 275 | |
| CDS-indel | 123 | 41 | |
| Intron | 24 021 | 21 399 | |
| nearGene-5 | 4981 | 4424 | |
| nearGene-3 | 1128 | 870 | |
| UTR-5 | 3495 | 2160 | |
| UTR-3 | 3274 | 2390 | |
| splice-5 | 296 | 103 | |
| splice-3 | 207 | 71 | |
| ncRNA | 9236 | 4631 | |
| ClinVar Clinical Significance | All clinical RS: | 20 563 | N/A in ClinVar |
| Pathogenic | 11 555 | ||
| Untested | 1901 | ||
| Benign | 1782 | ||
| Uncertain significance | 1764 | ||
| Other | 1646 | ||
| Likely Benign | 1484 | ||
| Likely pathogenic | 1132 | ||
| Drug-response | 170 | ||
| MAF | Common in any population (≥0.01) | 31 699 | 27 772 |
| Rare in any population (<0.01) | 21 719 | 12 520 | |
| Not reported in any population | 10 333 | 1765 | |
Note: Column ‘Total RS’ lists all text-mined variations in dbSNP & ClinVar and ‘Novel RS’ shows those in dbSNP only (i.e. not found in ClinVar). The total counts of tmVar RS in Row 1 are further broken down in Rows 2–4 by various attributes: (1) functional consequences (Functional class); (2) ClinVar clinical significance (Benign, Pathogenic, etc.) when known; and (3) Minor allele frequency (MAF).
Clinical significance. Of 62 452 tmVar RS, a total of 20 563 are known in ClinVar. These are spread across all possible clinical significance categories, as shown in Table 2. 19 709 RS have a single clinical significance assertion (Benign, Pathogenic, etc.) and 854 RS have multiple clinical significance assertions; each assertion is added to the total for each class. tmVar RS found in ClinVar with most severe clinical significance, pathogenic (11 555) and likely pathogenic (1132), accounted for 12 538 unique RS and 62% of the total (Table 2). This demonstrates that tmVar can find variants with clinical impact from publications to use for validating or enriching ClinVar annotations (see also Variants and Disease Connection, below).
Allele frequency. We also used population allele frequency to prioritize variants with impact under the general assumption that deleterious mutations are selected against during human evolution and are rare in the global population (see supporting evidence with known variants in ClinVar in Fig. 6b). To identify common and rare variants in the tmVar results we used the minor allele frequency (MAF) data from 1000 Genomes, GO-ESP, ExAC and gnomAD (Lek et al., 2016). RS with MAF equal or greater than 0.01 (MAF ≥ 0.01) is common if reported in any of the three populations and RS with MAF less than 0.01 (MAF < 0.01) is rare. Although many of the RS identified are common variants (total = 31 699), a significant number are rare (total = 21 719). In addition, 10 333 variants have no frequency (non-MAF) data reported in any of the three large populations. These may be rare or private mutations belonging to individuals or patient cohorts that are not represented in any of these major populations of presumed normal individuals. In support of this hypothesis, out of the 10 333 RS without MAF more than half (7847) are annotated by dbSNP as missense mutation and 6492 are classified as pathogenic in ClinVar with 5007 having both missense and pathogenic annotations (see Table 2). This result suggests, while frequency data is not available, the non-MAF variants are deleterious and perhaps extremely rare and not detected in the general normal populations.
To demonstrate the enrichment of RS attributes in tmVar results (over dbSNP), we plotted the relative ratio of percentages of tmVar versus dbSNP results having the above attributes (e.g. CDS-missense, Pathogenic, or Rare MAF). Figure 7 shows an increase enrichment of CDS encoding RS and those in defined features (splice sites and ncRNA) and less enrichment for other functional classes (intron, nearGene, etc.). Likewise, tmVar RS is more likely than dbSNP RS to be pathogenic and having a rare allele or unknown MAF.
Fig. 7.

Enrichment of RS attributes in tmVar versus dbSNP RS set. The percentages, % tmVar or % dbSNP, of total RS having the above attributes (i.e. CDS-missense, Pathogenic, or Rare MAF) were first calculated for each tmVar and dbSNP RS dataset separately. The ratio of enrichment for each attribute was then computed by dividing the tmVar percentage over the corresponding dbSNP percentage and plotted along the Y-axis
3.2.2 Analysis of variants in ACMG genes
To analyze variants that may be associated with known diseases and investigate their deleterious effects, we examined a subset of ClinVar variants in the 56 American College of Medical Genetics and Genomics (ACMG) genes known to be associated with lethal or severe genetic disorders and cancer. The ACMG has published recommendations for reporting incidental findings for variations in these genes with medically actionable information. In total 3727 RS are found in 57 of 58 ACMG genes with 694 RS novel to ClinVar for 55 genes. We then analyzed the functional class and allele frequency for this subset of variants associated with the ACMG genes and found most of them, similar to the overall results in Table 2, to be missense and rare as well.
Figure 8 shows the total number of known variants present in ClinVar (orange) for each of the 58 ACMG genes and the total number of (from 1 to 51) novel variants (blue) for each gene (except for two genes MYL3 and TMEM43). The TP53 gene is frequently observed to have alterations with diverse types and positions that predisposes to a wide spectrum of early onset cancers [17]. In accordance, our result shows TP53 has the highest number of novel variants (51). Nonetheless, the function and clinical impact of the novel variants are not known without further analysis and may be deleterious, neutral, or of little clinical importance. Assuming variants with the most biological impact would alter protein coding (missense, stop-gain, etc.) and also be rare, we prioritized the novel variants with functional class missense and stop-gain and with the lowest MAF for further analysis to identify novel connections not in ClinVar, between the variation, gene and disease with the publication.
Fig. 8.

The total counts of tmVar RS that already existed in ClinVar (Yellow) and additional novel variants (Blue) for each ACMG Gene. All genes, except two MYL3 and TMEM43, shown contain from 1 to 51 novel variants (Color version of this figure is available at Bioinformatics online.)
3.2.3 Variants and disease connection
We manually examined a subset of 10 ultra-rare variants novel to ClinVar, including those in ACMG genes, with the functional class missense, frameshift or nonsense variations. By virtue of being rare variations that altered the protein sequence, they are top candidates for having the strongest biological effects and strongest connection to diseases. Eight variants with allele frequency show they are extremely rare occurring only 1:10 000 and 1:100 000 individuals (Table 3). Our manual analysis showed all 10 variants to be described in the literature as associated with diseases or cancer, as shown in Table 3. These associations include: (i) a stop-gain mutation (rs764082747) for the gene HAX1 involved in severe congenital neutropenia (SCN), (ii) an insertion mutation (rs772720024, c.1155dupC) for the ATP6V1B1 gene that is believed to cause distal renal tubular acidosis (DRTA) and hearing loss syndrome in patients from Turkey and (iii) a stop-gain mutation in the ACMG-recommended tumor suppressor gene STK11, which causes Peutz-Jeghers Syndrome (PJS), resulting in a high risk for many different cancers, including breast, colon, pancreatic, stomach, small bowel, cervical and endometrial. These findings demonstrate that our approach can identify high impact variants from publications and that our results can be combined with dbSNP and ClinVar data to prioritize the variants by functional consequence, allele frequency, gene annotation and clinical significance for further manual or automated analysis and interpretation of effects on biological functions and diseases to enrich ClinVar database.
Table 3.
Manually reviewed tmVar RS not in ClinVar and associated diseases
| RSID | Functional consequence | MAF | PMID | Gene | ACMG gene | Mutation | Associate disease |
|---|---|---|---|---|---|---|---|
| 794728162 | Missense | N/A | 22262941 | FBN1 | Yes | NP_000129.3: p.Gly214Se | Marfan Syndrome |
| NM_000138.4: c.640G>A | |||||||
| 141414377 | Missense | 7.70E-05 | 12974739 | MYH7 | Yes | NP_000248.2: p.Arg807His | Cardiomyophthy |
| NM_000257.3: c2420G>A | Hypertrophic | ||||||
| 369067856 | Missense | 7.70E-05 | 19762784 | PCSK9 | Yes | NP_777596.2: p.Arg104Cys | Diabetes Mellitus |
| NM_174936.3: c.310C>T | Hypobetalipoproteinemias | ||||||
| 749864465 | Missense | 9.00E-06 | 19406494 | SCN5A | Yes | NP_000326.2: p.Arg808Cys | Brugada Syndrome |
| NM_198056.2: c.2422C>T | |||||||
| 121913317 | stop_gained | N/A | 27081308 | STK11 | Yes | NP_000446.1: p.Glu199Lys | Cancer Neoplasms |
| splice | NM_000455.4: c.595G>A | ||||||
| 748789700 | frameshift | 2.47E-05 | 16189622 | AGL | No | NM_000028.2: c.750_753delAGAC | Glycogen storage disease 111a |
| 781969081 | frameshift | 8.25E-06 | 25285676 | ATP6V1B1 | No | NP_001683.2: p.Ile386Hisfs | Distal renal tubular acidosis |
| 23923981 | |||||||
| 780955130 | splice-3 | 8.70E-06 | 24003324 | ATP7B | No | NM_000053.3: c.3061-1G>A | Wilson Disease |
| 23159873 | |||||||
| 764082747 | Frameshift | 1.37E-04 | 19499579 | HAX1 | No | XM_005244828.1: c.91delG | Severe congenital neutropenia |
| stop_grained | 19036070 | XP_005244885.1: p.Glu31Lysfs | |||||
| 772720024 | frameshift | 8.24E-06 | 27806791 | NPHP1 | No | NM_000272.3: c.1298delA | Nephronophthisis |
Note: The 10 variants were selected as top candidates from a list prioritized by functional consequences and either having the lowest MAF or no MAF (not observed in any populations). Shown are the dbSNP RS numbers, the functional consequences, MAF, the PMID containing descriptions of the variant and disease and confirmed by manual review, the effected gene, mutation description and the disease cited in the PMID.
4 Discussion
4.1 tmVar 2.0 error analysis
To understand the causes of errors made by tmVar 2.0, we manually reviewed the errors from the evaluation datasets and classified them into three broad categories. As shown in Table 4, one category of errors is due to the newly developed normalization procedure. In pattern matching, tmVar errors can either be due to imprecise patterns or the extracted RSID from the text was merged to a different RSID in the gold standard (in the Thomas corpus). For instance, rs17015215 in PMID: 17318851 was annotated as rs2235371 in the gold standard. There are also errors made by the dictionary-lookup step. For example, tmVar failed to map ‘(ACG –> ACA) in codon 594’ to its correct RS number. The second group of errors is due to imperfect NER performance, which includes both imperfect boundary detection and completely missing mutations (e.g. ‘-33 + 16 TG’ was missed by tmVar because of the lack of ‘>’ symbol between T and G). Together with in Table 1, these results also suggest inherent differences between these datasets, depended on how the articles were selected and annotated in each set.
Table 4.
Mutation normalization error types in Thomas et al. (2011) and OSIRIS (Furlong et al., 2008) Corpus
| Thomas |
OSIRIS |
tmVar2 |
||||
|---|---|---|---|---|---|---|
| FP | FN | FP | FN | FP | FN | |
| Normalization | ||||||
| –Pattern matching | 46 | 50 | 0 | 2 | 0 | 0 |
| –Dictionary lookup | 4 | 1 | 0 | 6 | 10 | 24 |
| Recognition | ||||||
| –Boundary issue | 1 | 10 | 6 | 30 | 6 | 36 |
| –Incomplete variation mentions | 0 | 0 | 0 | 12 | 0 | 0 |
| Others | 1 | 1 | 0 | 0 | 0 | 0 |
| Total | 52 | 62 | 6 | 50 | 16 | 60 |
Note: False positive (FP) and negative (FN) errors are shown respectively.
4.2 Further applications of this research
We believe our analysis demonstrates our text mining results to be useful for enriching literature links in existing databases and assisting the curation and prioritization of clinically important variants. We can provide literature links to 16 820 SNPs (e.g. rs587783048 (c.2398delC) in PMID: 24366306) that are otherwise not possible to find in any existing databases (i.e. ClinVar, dbSNP, or PubMed). For 18 838 SNPs, we are able to increase the number of their associated studies in PubMed. By combining other variant information such as functional consequences and MAF, we show that the list of computed variants can be further prioritized for assessing their biological and clinical impacts. As previously reported, automated text-mining systems are playing increasingly important roles in the manual curation process, especially during the triage process (Wei et al., 2012).
In addition, our results could be used by other computational methods in bioinformatics research such as connecting genotypes with phenotypes and/or modeling gene-disease-variant relations (Piñero et al., 2017; Singhal et al., 2016). We are therefore making our PubMed-scale text-mined data publicly available to the entire scientific community through multiple channels. Upon publication, the complete data can be downloaded from our ftp set (ftp://ftp.ncbi.nlm.nih.gov/pub/lu/PubTator/). Additionally, like our other work, we plan to incorporate our results in PubTator (Wei et al., 2013b), an NCBI web server for visualizing and retrieving pre-computed biomedical entities and relations in the entire literature, where data of individual articles can be retrieved through web APIs (Wei et al., 2016). Finally, tmVar is already open source; its new normalization module and revised corpus with normalized RS annotations will be made publicly available upon publication. We are also integrating these computed data into dbSNP to enrich variant annotation and to improve variant reporting and searching.
4.3 Limitations of the approach and future work
In this work, we focused on text mining and analysis of genomic variation information with the abstract data in PubMed. As shown in Figure 5, a number of existing RSID-PMID links in dbSNP appear only in the body of full text. As found by the previous research (Kafkas et al., 2015; Yepes and Verspoor, 2014a,b), both full text and supplementary materials are rich for reporting genomic variation information. All existing mutation extraction tools, including tmVar, were first developed based on abstract data in PubMed. A recent study (Lee et al., 2016) demonstrates that processing full text can be more challenging because ‘compared with abstracts, there are more texts in Alphabet-Number-Alphabet form (in full text) that are not variants.’ Instead, they may represent figure numbers, chemical formulae and other unrelated data. Furthermore, in our preliminary analysis we observed that a significant proportion of errors arise from processing table contents when text data are not extracted properly (e.g. ill-formed or truncated). Fortunately, along with their investigation the authors of [12] produced a dataset consisting of 108 full-length papers with 403 annotated variants and their relations with genes, diseases, drugs and cell lines. In the future, we plan to use this data to re-train tmVar, given it is based on machine learning, to improve its performance on processing full texts.
Other limitations of this work include that extraction and analysis are currently limited to small, simple genetic variants with precise locations. We are not looking at complex or large structural variations such as deletions and insertions. Neither did we investigate any functional implications of variant-associated genes in this work. Also, our MAF analysis was based on a global population data so the ‘common’ and ‘rare’ variant classifications cannot be generalized to ethnic subpopulations such as Caucasians, African, Asian and Hispanics where the variant alleles may be common in one subpopulation and rare in another. Finally, due to limited resources we only manually reviewed ten variants with regard to their potential connection to diseases from the variant list. To scale up such an investigation in the future, one could apply an automatic tool for disease detection and compare its results with curated disease data in ClinVar.
5 Conclusion
In this work, we demonstrate that genomic variation information in the literature is a valuable source for direct use in expert-curated knowledge bases and data-driven research in precision medicine, despite the unstructured format. We show that a large fraction of mutations are described in free style (by the authors) and thus mutation normalization is critical for integrating literature data. The improved tmVar algorithm demonstrated high performance in benchmarking experiments, and its utility is validated through comparing its results with curated data in dbSNP and ClinVar. Finally, we demonstrate that the automatically extracted information, when combined with structured database content in a complementary manner, provides fundamental insights into the inter-relationships of gene, variants and diseases.
Acknowledgements
The authors would like to thank Dr Ayush Singhal for discussing the normalization step and thank Dr Robert Leaman for proofreading our manuscript.
Funding
This work was supported by the National Institutes of Health intramural research program, National Library of Medicine.
Conflict of Interest: none declared.
References
- Amberger J.S. et al. (2015) OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders, Nuclear. Acids Res., 43, D789–D798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonis J. et al. (2006) OSIRIS: a tool for retrieving literature about sequence variants. Bioinformatics, 22, 2567–2569. [DOI] [PubMed] [Google Scholar]
- Burger J.D. et al. (2014) Hybrid curation of gene-mutation relations combining automated extraction and crowdsourcing. Database J. Biol. Datab. Cur., 2014, bau094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J.G. et al. (2007) MutationFinder: a high-performance system for extracting point mutation mentions from text. Bioinformatics, 23, 1862–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coordinators N.R. (2016) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res., 44, D7–D19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doughty E. et al. (2011) Toward an automatic method for extracting cancer-and other disease-related point mutations from the biomedical literature. Bioinformatics, 27, 408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes S.A. et al. (2017) COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res., 45, D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong L.I. et al. (2008) OSIRISv1.2: a named entity recognition system for sequence variants of genes in biomedical literature. BMC Bioinformatics, 9, 84.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith M. et al. (2017) CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet., 49, 170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann-Apitius M. et al. (2008) Knowledge environments representing molecular entities for the virtual physiological human. Philos. Trans. R. Soc. Lond. A Math. Phys. Eng. Sci., 366, 3091–3110. [DOI] [PubMed] [Google Scholar]
- Jensen L.J. et al. (2006) Literature mining for the biologist: from information retrieval to biological discovery. Nat. Rev. Genet., 7, 119–129. [DOI] [PubMed] [Google Scholar]
- Kafkas Ş. et al. (2015) Database citation in supplementary data linked to Europe PubMed Central full text biomedical articles. J. Biomed. Seman., 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger R. et al. (2007) Identifying gene-specific variations in biomedical text. J. Bioinf. Comput. Biol., 5, 1277–1296. [DOI] [PubMed] [Google Scholar]
- Krallinger M. et al. (2009) Extraction of human kinase mutations from literature, databases and genotyping studies. BMC Bioinformatics, 10, S1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum M.J. et al. (2016) ClinVar: public archive of interpretations of clinically relevant variants. Nuclear Acids Res., 44, D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurila J.B. et al. (2010) Algorithms and semantic infrastructure for mutation impact extraction and grounding. BMC Genomics, 11, S24.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. et al. (2016) BRONCO: Biomedical entity Relation ONcology COrpus for extracting gene-variant-disease-drug relations. Database J. Biol. Datab. Cur., 2016, baw043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M. et al. (2016) Analysis of protein-coding genetic variation in 60, 706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naderi N., Witte R. (2012) Automated extraction and semantic analysis of mutation impacts from the biomedical literature. BMC Genomics, 13, S10.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñero J. et al. (2017) DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nuclear Acids Res., 45, D833–D839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar K.E. et al. (2015) Text mining facilitates database curation - extraction of mutation-disease associations from Bio-medical literature. BMC Bioinformatics, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Esteban R. (2015) Biocuration with insufficient resources and fixed timelines. Database J. Biol. Datab. Cur., 2015, bav116.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry S.T. et al. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res., 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal A. et al. (2016) Text mining genotype-phenotype relationships from biomedical literature for database curation and precision medicine. PLoS Computat. Biol., 12, e1005017.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P. et al. (2016) SETH detects and normalizes genetic variants in text. Bioinformatics, 32, 2883–2885. [DOI] [PubMed] [Google Scholar]
- Thomas P.E. et al. (2011) Challenges in the association of human single nucleotide polymorphism mentions with unique database identifiers. BMC Bioinformatics, 12, S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C.-H. et al. (2012) Accelerating literature curation with text-mining tools: a case study of using PubTator to curate genes in PubMed abstracts. Database J. Biol. Datab. Cur., 2012, bas041.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C.-H. et al. (2013a) tmVar: A text mining approach for extracting sequence variants in biomedical literature. Bioinformatics, 29, 1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C.-H. et al. (2013b) PubTator: a Web-based text mining tool for assisting Biocuration. Nucleic Acids Res., 41, W518–W522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C.-H. et al. (2015) GNormPlus: an integrative approach for tagging genes, gene families, and protein domains. BioMed Res. Int., 2015, 918710.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C.-H. et al. (2016) Beyond accuracy: creating interoperable and scalable text-mining web services. Bioinformatics, 32, 1907–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepes A.J., Verspoor K. (2014a) Literature mining of genetic variants for curation: quantifying the importance of supplementary material. Database J. Biol. Datab. Cur., 2014, bau003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepes A.J., Verspoor K. (2014b) Mutation extraction tools can be combined for robust recognition of genetic variants in the literature. F1000Research, 3, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamorano-Jiménez C.A. et al. (2015) Molecular identification of glucose-6-phosphate dehydrogenase (G6PD) detected in neonatal screening. Gaceta Medica De Mexico, 151. [PubMed] [Google Scholar]