

Abstract
Summary
Integrative omics is a central component of most systems biology studies. Computational methods are required for extracting meaningful relationships across different omics layers. Various tools have been developed to facilitate integration of paired heterogenous omics data; however most existing tools allow integration of only two omics datasets. Furthermore, existing data integration tools do not incorporate additional steps of identifying sub-networks or communities of highly connected entities and evaluating the topology of the integrative network under different conditions. Here we present xMWAS, a software for data integration, network visualization, clustering, and differential network analysis of data from biochemical and phenotypic assays, and two or more omics platforms.
Availability and implementation
https://kuppal.shinyapps.io/xmwas (Online) and https://github.com/kuppal2/xMWAS/ (R)
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Technological advances have led to a major paradigm shift where multi-assay molecular profiling of biological samples is increasingly being used to understand molecular mechanisms for diseases and host responses to environmental exposures (Hawkins, 2010). Most cellular processes in a biological system are dependent on complex molecular interactions (Barabasi, 2011). Integrative omics allows researchers to address such complexity and answer challenging biological questions, such as function of genetic variants and unknown metabolites, mechanisms of gene regulation, signaling and metabolic pathway responses to infection and toxicity (Chandler, 2016; Hawkins, 2010; Uppal, 2016).
Numerous data-driven/unsupervised and knowledge-based tools allow integration of data from different omics technologies and other molecular assays (Meng, 2016; Wanichthanarak, 2015). Most existing data integration tools allow integration of only two datasets and do not allow identification of community structure and evaluation of network changes between different conditions (Supplementary Table S1). Community detection reveals topological modules comprised of functionally related biomolecules (Barabasi, 2011; Yang, 2016). Differential network analysis allows characterization of nodes that undergo changes in topological characteristics between different conditions, e.g. healthy versus disease (Lichtblau, 2016).
To advance these capabilities, we present, xMWAS, which provides an automated workflow for integrative analysis, differential network analysis and community detection to improve our understanding of complex molecular interactions and disease mechanisms.
2 Implementation
xMWAS utilizes existing algorithms and provides an automated framework for integrative and differential network analysis of up to four datasets from unpaired (two or more classes) or paired (repeated measures with one or two factors) study designs (Supplementary Fig. S1A). Pairwise data integration is performed using Partial Least Squares (PLS), sparse PLS and multilevel sparse PLS methods (Le Cao, 2009; Liquet, 2012; Gonzalez, 2012). The igraph package in R is used to generate a multi-data integrative network (Csardi 2006). Community detection is performed using the multilevel community detection method (Blondel, 2008). Eigenvector centrality measure (ECM) and betweenness centrality measure are used to evaluate and compare the importance of nodes between different conditions (Lichtblau, 2016; Odibat and Reddy, 2012). Supplementary Section S1 provides details for different stages of xMWAS. xMWAS is available as an R package and a web-based application. A tutorial is provided in Supplementary Section S2 that includes information related to installation and usage.
3 Example and conclusion
We tested xMWAS using cytokine, transcriptome and metabolome datasets from a recently published case-control study to examine H1N1 influenza virus infection-altered metabolic response in mouse lung (Chandler, 2016). For comparisons, we used data from only control samples (Fig. 1A) and only H1N1 influenza samples (Fig. 1B). More information related to Methods is provided in Supplementary Section S3.
Fig. 1.
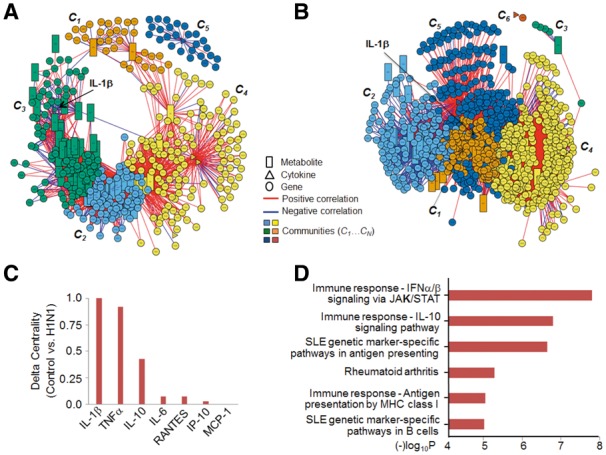
Integrative network analysis of cytokine, metabolome and transcriptome datasets from a study of H1N1 virus infection of mice. (A). Using only control samples. Five communities (C1…C5; represented by different colors) were detected using the multilevel community detection algorithm (Supplementary Table S2). (B) Using only H1N1 infected samples. Six communities (C1…C6) were detected (Supplementary Table S3). (C) Delta centrality based on the ECM, |ECMcontrol – ECMH1N1|, for the cytokines in the integrative networks in (A) and (B). (D) Pathway analysis of genes with delta centrality (|ECMcontrol – ECMH1N1|) > 0.1 using MetaCore (https://portal.genego.com/); only top 5 pathways are shown). Note: (rectangle: metabolite, circle: gene, triangle: cytokine)
The results show that xMWAS allows identification and visualization of associations between genes, cytokines, and metabolites (Fig. 1A and B). In the control group, 5 cytokines, 348 genes, and 71 metabolites were selected in the sPLS analysis that was grouped into five communities in the three-way integrative network (Fig. 1A and Supplementary Table S2). The five cytokines were assigned to communities C3 (IL-1beta), C4, (IL-6, RANTES, IP-10) and C5 (MCP-1) and had significant associations (P < 0.05) with genes related to immune response, cell signaling, bacterial infections, cell death and survival, serotonin-melatonin biosynthesis and amino acids metabolism (Supplementary Table S2). Cytokines are known to be involved in the recruitment of the inflammatory cells and influence the adaptive immune response during influenza (Chandler, 2016). In the H1N1 infected group, 4 cytokines, 806 genes, and 55 metabolites were selected in the sPLS analysis that was grouped into six communities in the three-way integrative network (Fig. 1B and Supplementary Table S3). The cytokines were assigned to communities C5 (IL-1beta, IL-10, and TNF-alpha) and C6 (MCP-1) and had significant associations (P < 0.05) with genes related to immune response, antiviral actions, and cell damage (Supplementary Table S3).
The analysis also allows identification of nodes that undergo network changes, which is determined based on the eigenvector centrality (importance) measure (Supplementary Table S4). IL-1beta, TNF-alpha and IL-10 had the largest change in eigenvector centrality between control and H1N1 groups (Fig. 1C, Supplementary Table S4). Overall, the centrality of the cytokines was much higher in the H1N1 network, which is expected based on the current knowledge about their role in H1N1 infection (Chandler, 2016). Pathway analysis of genes with |ECMcontrol – ECMH1N1|> 0.1 showed enrichment of pathways related to immune response, autoimmune disease, and other inflammatory disease (Fig. 1D). Metabolites associated with cytokines (P < 0.05) in H1N1 sub-group were evaluated using Mummichog (Li, 2013), which showed enrichment of pathways related to leukotrienes, steroids, and lipids (Supplementary Table S5). These pathways have previously been associated with influenza infection (Chandler, 2016; Le Bel, 2014).
In summary, xMWAS provides a platform-independent framework for integrative network analysis, identification of communities of functionally related biomolecules, and differential network analysis. The results show that xMWAS can improve our understanding of disease pathophysiology and complex molecular interactions.
Supplementary Material
Acknowledgement
We acknowledge members of the Clinical Biomarkers Laboratory, Emory University for testing and suggesting improvements to the software.
Funding
This work was supported by National Institutes of Health (USA) ES025632, ES023485, ES019776, HL095479, EY022618 and the US National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under contract no. [HHSN272201200031C].
Conflict of Interest: none declared.
References
- Barabasi A.L. et al. (2011) Network medicine: a network-based approach to human disease. Nat. Rev. Genet., 12, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel V.D. et al. (2008) Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp., 2008, P10008. [Google Scholar]
- Chandler J.D. et al. (2016) Metabolic pathways of lung inflammation revealed by high-resolution metabolomics (HRM) of H1N1 influenza virus infection in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol., 311, R906–R916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csardi G. (2006) The igraph software package for complex network research. InterJ., Complex Syst., 1695, 1695. [Google Scholar]
- Gonzalez I. et al. (2012) Visualising associations between paired ‘omics’ data sets. BioData Min, 5, 19.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins R.D. et al. (2010) Next-generation genomics: an integrative approach. Nat. Rev. Genet., 11, 476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Cao K.A. et al. (2009) integrOmics: an R package to unravel relationships between two omics datasets. Bioinformatics, 25, 2855–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bel M. et al. (2014) Leukotriene B4, an endogenous stimulator of the innate immune response against pathogens. J. Innate Immun., 6, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. et al. (2013) Predicting network activity from high throughput metabolomics. PLoS Comput. Biol., 9, e1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtblau Y. et al. (2016) Comparative assessment of differential network analysis methods. Brief Bioinform., 2016, 837–850. [DOI] [PubMed] [Google Scholar]
- Liquet B. et al. (2012) A novel approach for biomarker selection and the integration of repeated measures experiments from two assays. BMC Bioinformatics, 13, 325.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng C. et al. (2016) Dimension reduction techniques for the integrative analysis of multi-omics data. Brief Bioinform., 17, 628–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odibat O., Reddy C.K. (2012) Ranking differential hubs in gene co-expression networks. J. Bioinform. Comput. Biol., 10, 1240002.. [DOI] [PubMed] [Google Scholar]
- Uppal K. et al. (2016) Computational Metabolomics: A Framework for the Million Metabolome. Chem Res Toxicol, 29, 1956–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. et al. (2016) A comparative analysis of community detection algorithms on artificial networks. Sci. Rep., 6, 30750.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanichthanarak K. et al. (2015) Genomic, proteomic, and metabolomic data integration strategies. Biomark Insights, 10, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.