

Abstract
Summary
Proteases are enzymes that specifically cleave the peptide backbone of their target proteins. As an important type of irreversible post-translational modification, protein cleavage underlies many key physiological processes. When dysregulated, proteases’ actions are associated with numerous diseases. Many proteases are highly specific, cleaving only those target substrates that present certain particular amino acid sequence patterns. Therefore, tools that successfully identify potential target substrates for proteases may also identify previously unknown, physiologically relevant cleavage sites, thus providing insights into biological processes and guiding hypothesis-driven experiments aimed at verifying protease–substrate interaction. In this work, we present PROSPERous, a tool for rapid in silico prediction of protease-specific cleavage sites in substrate sequences. Our tool is based on logistic regression models and uses different scoring functions and their pairwise combinations to subsequently predict potential cleavage sites. PROSPERous represents a state-of-the-art tool that enables fast, accurate and high-throughput prediction of substrate cleavage sites for 90 proteases.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Proteases are enzymes that specifically cleave the peptide backbone of target proteins (Chou, 1996; Chou et al., 1996; López-Otín and Matrisian, 2007). This cleavage represents an important type of irreversible post-translational modification, and is involved in many key physiological processes (Overall and Blobel, 2007). Dysregulation of proteases has been associated with numerous diseases (Turk, 2006). Many proteases are highly specific, cleaving only the target substrates that present the appropriate combination of structural features and amino acid sequence patterns. Thus, the knowledge of protease-specific substrate cleavage is fundamental for our understanding of the functional mechanisms of proteases. The substrate specificity of proteases can generally be characterized using peptide specificity-profiling (Schilling and Overall, 2008) or high-throughput mass spectrometry techniques (Dix et al., 2008; Mahrus et al., 2008). However, as experimental identification of protease cleavage events is often difficult, expensive and time-consuming, it is highly desirable to develop cost-effective computational methods and tools to complement experimental efforts. In this context, computational methods and tools for identifying potential target substrates of proteases can help guide hypothesis-driven experimental studies of protease–substrate interaction (duVerle and Mamitsuka, 2012; Song et al., 2011). A variety of computational tools have been developed for this purpose, including PeptideCutter (Gasteiger et al., 2003), CaSPredictor (Garay-Malpartida et al., 2005), GraBCas (Backes et al., 2005), PoPS (Boyd et al., 2005), HIVcleave (Shen and Chou, 2008), SitePrediction (Verspurten et al., 2009), Pripper (Piippo et al., 2010), Cascleave (Song et al., 2010; Wang et al., 2014) and PROSPER (Song et al., 2012). Among these methods, HIVcleave is focused on predicting HIV protease cleavage sites in proteins, while GraBCas, CaSPredictor, Cascleave and PROSPER can only predict substrate cleavage sites for a limited number of proteases (e.g. caspases and/or granzyme B), and consequently have only a limited applicability. Meanwhile, two methods were developed to identify proteases and their types (Chou and Shen, 2008; Shen and Chou, 2009).
In the present study, we introduce PROSPERous, a tool for rapid, in silico prediction of protease-specific cleavage sites within substrate sequences. PROSPERous is based on logistic regression (LR) models that integrate various scoring functions based on the local sequence environments of cleavage sites. We evaluated the performance of the models and compared this with three popular tools PoPS, SitePrediction and PROSPER.
2 Materials and methods
In this study, protease-specific substrate data were extracted from the MEROPS database (Rawlings et al., 2016), which is a comprehensive information resource for proteases, their substrates and inhibitors. Importantly, we only collected curated substrate sequences and cleavage sites that had been readily validated experimentally. To assess the performance of the models, we constructed both benchmark and independent test datasets. To avoid potential bias and over-fitting, we performed sequence clustering and homology reduction using the CD-HIT program (Fu et al., 2012) to remove any sequence redundancy at and above a sequence identity of 70% between any two sequences from the extracted dataset. After this procedure, the resulting dataset was further split into two parts, namely the benchmark dataset and the independent test dataset. The performance of different scoring functions was evaluated on the benchmark dataset, while the performance of our and other existing methods was evaluated on the independent test dataset. A complete list of substrate sequences and cleavage sites for each protease can be found on the server website https://prosperous.erc.monash.edu/. Supplementary Table S1 provides a statistical summary of substrate sequences and cleavage sites of different proteases in both benchmark and independent test datasets, with sequence logo representations of P4–P4′ sites shown in Supplementary Figure S1.
The scoring function used for ranking potential protease-specific cleavage sites on the basis of their flanking amino acid sequences is a critical determinant of the prediction performance of the tool. Briefly, several different types of scoring functions to choose from are available in PROSPERous. These include Nearest Neighbor Similarity (NNS), Amino Acid Frequency (AAF), WebLogo-based Sequence conservation (WLS), BLOSUM62 Substitution Index (BSI), as well as combinations of pairs of scoring functions, namely AAF + NNS, WLS + BSI and NNS + WLS. These different scoring functions and their combinations are defined in detail in the Supplementary Material.
An important aspect of PROSPERous is the use of an LR routine that builds upon combinations of individual scoring functions and allows for a more accurate identification of potential cleavage sites. Our extensive benchmark tests showed that the integrated use of all individual scoring functions together in one LR framework enhanced the quality of the predictions considerably (see detailed results in the Supplementary Material). We implemented the LR models with the R package for LR (Everitt and Hothorn, 2010). The individual scoring functions described above, and their combinations, were used as input features to train the LR models. For each protease, the scoring functions used to assess and rank the potential cleavage sites were further used as input features to inform and train each protease-specific LR model. To comprehensively evaluate the prediction performance of these scoring functions and their combinations, we performed 5-fold cross-validation, and independent tests.
The overall flowchart of PROSPERous is shown in Figure 1. Processing a query sequence using the server involves several steps. Firstly, users need to choose a proper cleavage site pattern P4–Pn′ (n = 1, 2, 3 and 4) to score the potential cleavage site. Choosing an optimal window for the cleavage site sequence is most relevant for the prediction, and primarily depends on expert knowledge. However, in the absence of such knowledge, we recommend users to choose the P4–P2′ window to make the prediction, as previous studies have indicated that this window can lead to the overall best performance for predicting cleavage sites for a number of proteases (Song et al., 2012, 2010; Wang et al., 2014). Secondly, users need to choose an appropriate scoring function, or a combination of two such functions. Upon query submission, the submitted sequence will be scanned against the known cleavage site database. The score for each potential P4–Pn′ cleavage site will be calculated based on the selected scoring function, and the top-ranking results will be displayed on the screen. The performance comparison between different scoring functions, including pairwise combinations, and different LR models, as evaluated on two types of independent test datasets [with sequence redundancy removed at 70% and 30% sequence identity (SI), respectively], is presented in Supplementary Tables S2–S5.
Fig. 1.
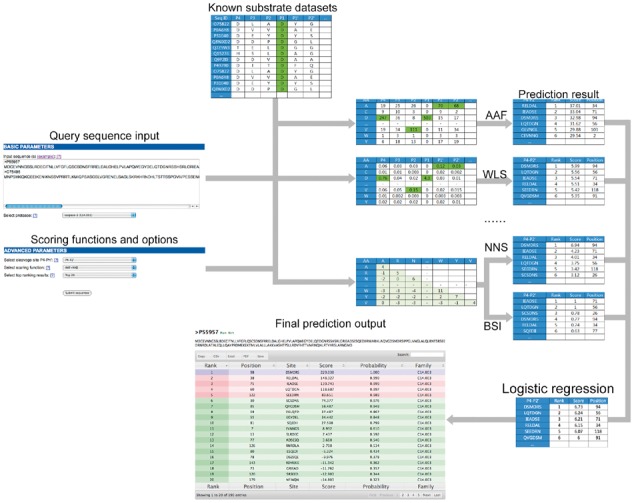
The flowchart of the PROSPERous web server
3 Implementation
A complete description of the PROSPERous web server implementation, including a detailed flowchart, is available in the Supplementary Material. Here, we provide a brief summary. The webserver was implemented in HTML and Perl and configured in the Linux environment on an eight-core server machine with 16GB memory and a 4TB hard disk. In order to submit a job, users need to provide one or more query sequences in the FASTA format. To initiate the prediction, users will also need to select the cleavage site P4–Pn′ (n = 1, 2, 3 and 4) window, individual scoring functions/combinations/LR models, and the number of ranked results (Top-1, −3, −5, −10 or −50). Upon submitting the query sequences, the prediction output by the webserver will be displayed on the screen. The main outputs include ranking, residue position, P4–P2′ cleavage site motif (P1 is the predicted cleavage site), score and protease family that are predicted to cleave the submitted sequence. To demonstrate how to use the server, we show an example sequence in FASTA format in Supplementary Figure S2A, with the corresponding output in Supplementary Figure S2B.
To facilitate high-throughput prediction of potential substrates and cleavage sites, PROSPERous allows users to submit and process up to 1000 sequences per job submission. Users can choose to upload a plain text file in the FASTA format or simply copy and paste the sequence information to perform the high-throughput prediction task. The computational time for completing users’ jobs depends on the number of submitted sequences, the sequence length and also the scoring functions or models used. To name one example, it takes approximately 7 min to complete the Caspase-3 cleavage site predictions of 1000 sequences using the best-performing LR model that was trained based on individual scoring functions and their pairwise combinations.
4 Performance comparison with other methods
To evaluate the performance of PROSPERous for predicting protease-specific substrate cleavage sites, we compared its performance with those of three other popular tools, including PoPS (Boyd et al., 2005), Site-Prediction (Verspurten et al., 2009) and PROSPER (Song et al., 2012). Both PoPS and SitePrediction are based on statistical scoring methods, while PROSPER is based on a learned support vector machine.
The performance comparison results for the independent tests are shown in the Supplementary Material, using all six performance measures AUC, MCC, Accuracy, Sensitivity, Specificity and Precision. Figure 2 and Supplementary Figure S5 show the performance comparison between different tools in terms of the primary measure, the AUC score. We can see that PROSPERous achieved the highest AUC values and clearly outperformed the other three tools for the substrate cleavage site prediction of all the tested proteases. In terms of the ROC curves and other performance measures, PROSPERous also consistently outperformed other tools, as can be seen from Supplementary Tables S4 and S5, and Figures S3 and S4.
Fig. 2.
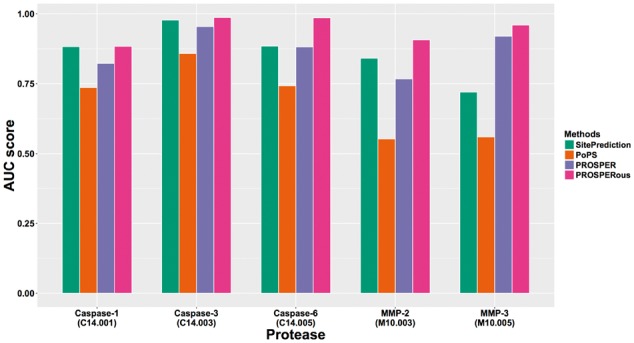
Performance comparison between PROSPERous, PoPS, SitePrediction and PROSPER for cleavage site prediction using independent test datasets with redundancy removed at the 30% SI cutoff
5 Results
We developed the tool PROSPERous to address the need to perform high-throughput prediction and analysis of protease-specific substrate cleavage sites in a cost-effective manner. PROSPERous integrates characteristic scoring functions that describe the local sequence environment of cleavage sites with LR models. Benchmarking experiments indicate that PROSPERous can achieve performances that are generally superior to, the other three existing tools (PoPS, SitePrediction and PROSPER). The webserver provides a straightforward and user-friendly interface for selecting various scoring functions and, accordingly, for predicting the substrate cleavage sites for a particular protease. We believe that PROSPERous is an invaluable tool for assisting cost-effective discoveries of novel target substrates and their cleavage sites and for facilitating community-wide research in the functional characterization of proteases in a high-throughput manner.
Funding
This work was supported by grants from the Australian Research Council (ARC) [LP110200333 and DP120104460] and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health [R01 AI111965].
Conflict of Interest: none declared.
Supplementary Material
References
- Backes C. et al. (2005) GraBCas: a bioinformatics tool for score-based prediction of Caspase- and Granzyme B-cleavage sites in protein sequences. Nucleic Acids Res., 33(Web Server issue), W208–W213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd S.E. et al. (2005) PoPS: a computational tool for modeling and predicting protease specificity. J. Bioinform. Comput. Biol., 3, 551–585. [DOI] [PubMed] [Google Scholar]
- Chou K.C. (1996) Prediction of human immunodeficiency virus protease cleavage sites in proteins. Anal. Biochem., 233, 1–14. [DOI] [PubMed] [Google Scholar]
- Chou K.C., Shen H.B. (2008) ProtIdent: a web server for identifying proteases and their types by fusing functional domain and sequential evolution information. Biochem. Biophys. Res. Commun., 376, 321–325. [DOI] [PubMed] [Google Scholar]
- Chou K.C. et al. (1996) Predicting human immunodeficiency virus protease cleavage sites in proteins by a discriminant function method. Proteins, 24, 51–72. [DOI] [PubMed] [Google Scholar]
- Dix M.M. et al. (2008) Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell, 134, 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- duVerle D.A., Mamitsuka H. (2012) A review of statistical methods for prediction of proteolytic cleavage. Brief Bioinform., 13, 337–349. [DOI] [PubMed] [Google Scholar]
- Everitt B.S., Hothorn T. (2010) A Handbook of Statistical Analyses Using R SECOND EDITION, Preface to First Edition. CRC Press, Boca Raton, Florida, USA.
- Fu L. et al. (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics, 28, 3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garay-Malpartida H.M. et al. (2005) CaSPredictor: a new computer-based tool for caspase substrate prediction. Bioinformatics, 21, i169–i176. [DOI] [PubMed] [Google Scholar]
- Gasteiger E. et al. (2003) ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res., 31, 3784–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C., Matrisian L.M. (2007) Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer, 7, 800–808. [DOI] [PubMed] [Google Scholar]
- Mahrus S. et al. (2008) Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell, 134, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overall C.M., Blobel C.P. (2007) In search of partners: linking extracellular proteases to substrates. Nat. Rev. Mol. Cell Biol., 8, 245–257. [DOI] [PubMed] [Google Scholar]
- Piippo M. et al. (2010) Pripper: prediction of caspase cleavage sites from whole proteomes. BMC Bioinform., 11, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings N.D. et al. (2016) Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res., 44, D343–D350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling O., Overall C.M. (2008) Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat. Biotechnol., 26, 685–694. [DOI] [PubMed] [Google Scholar]
- Shen H.B., Chou K.C. (2008) HIVcleave: a web-server for predicting human immunodeficiency virus protease cleavage sites in proteins. Anal. Biochem., 375, 388–390. [DOI] [PubMed] [Google Scholar]
- Shen H.B., Chou K.C. (2009) Identification of proteases and their types. Anal. Biochem., 385, 153–160. [DOI] [PubMed] [Google Scholar]
- Song J. et al. (2011) Bioinformatic approaches for predicting substrates of proteases. J. Bioinform. Comput. Biol., 9, 149–178. [DOI] [PubMed] [Google Scholar]
- Song J. et al. (2012) PROSPER: an integrated feature-based tool for predicting protease substrate cleavage sites. PLoS One, 7, e50300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J. et al. (2010) Cascleave: towards more accurate prediction of caspase substrate cleavage sites. Bioinformatics, 26, 752–760. [DOI] [PubMed] [Google Scholar]
- Turk B. (2006) Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discov., 5, 785–799. [DOI] [PubMed] [Google Scholar]
- Verspurten J. et al. (2009) SitePredicting the cleavage of proteinase substrates. Trends Biochem. Sci., 34, 319–323. [DOI] [PubMed] [Google Scholar]
- Wang M. et al. (2014) Cascleave 2.0, a new approach for predicting caspase and granzyme cleavage targets. Bioinformatics, 30, 71–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.