

Abstract
De novo design provides an attractive approach to test the mechanism by which metalloproteins define the geometry and reactivity of their metal ion cofactors. While there has been considerable progress in designing proteins that bind transition metal ions including iron-sulphur clusters, the design of tetranuclear clusters with oxygen-rich environments has not been accomplished. Here, we describe the design of tetranuclear clusters, consisting of four Zn2+ and four carboxylate oxygens situated at the vertices of a distorted cube-like structure. The tetra-Zn2+ clusters are bound at a buried site within a four-helix bundle, with each helix donating a single carboxylate (Glu or Asp) and imidazole (His) ligand, as well as second- and third-shell ligands. Overall, the designed site consists of four Zn2+ and 16 polar sidechains in a fully connected hydrogen-bonded network. The designed proteins have apolar cores at the top and bottom of the bundle, which drive the assembly of the liganding residues near the center of the bundle. The steric bulk of the apolar residues surrounding the binding site was varied to determine how subtle changes in helix-helix packing affect the binding site. The crystal structures of two of four proteins synthesized were in good agreement with the overall design; both formed a distorted cuboidal site stabilized by flanking second and third-shell interactions that stabilize the primary ligands. A third structure bound a single Zn2+ in an unanticipated geometry and the fourth bound multiple Zn2+ at multiple sites at partial occupancy. The metal-binding and conformational properties of the helical bundles in solution, probed by circular dichroism spectroscopy, analytical ultracentrifugation and NMR, were consistent with the crystal structures.
Keywords: de novo design, protein design, multinuclear transition metal ion clusters, helix bundles, coiled coils
TOC image
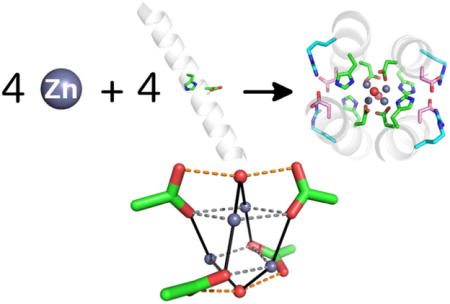
Introduction
De novo protein design is an excellent approach to test and refine our understanding of protein structure and function.1–4 Here, we use this approach to probe the features governing the assembly of metalloproteins that incorporate covalently bridged multinuclear metal clusters.5 Natural metalloproteins generally include direct interactions between sidechains and the metal ions (first-shell interactions) as well as second- and third-shell hydrogen-bonded interactions that create hydrogen-bonded networks important for function.6,7 Thus, it has been important to include hydrogen-bond networks in the de novo design of metalloproteins.8–12 Recently, Rosetta has been used to design hydrogen-bonded networks into helical bundles,13 which has been heralded as a major accomplishment in de novo protein design.2 Although these proteins designed using Rosetta lacked any function, the desired structures were confirmed by X-ray crystallography. Also, Woolfson and coworkers have implanted a hydrogen-bond in a heptameric helical bundle, which is crucial for hydrolytic function14 but the use of extensive hydrogen-bonded networks to reinforce binding has not been reported, outside of early work on the DF class of proteins.8–12 In the present work we reinforce the binding of an abiological cofactor by designing significantly more extensive networks, than in previously designed proteins, including the bundles described by Baker and coworkers13 or our previously designed metalloproteins.8–12
Natural enzymes use these bridged multinuclear centers to perform diverse transformations, including some of the more difficult chemical conversions: Oxygen is reductively activated in multi-copper laccases,15,16 bridged diiron proteins17,18 and type 3 copper proteins.15,19 Di-Mn(II/III) catalases catalyze the disproportionation of hydrogen peroxide.20 Cytochrome c oxidase (CcO) catalyzes the four-electron reduction of O221–23 while water is oxidized to O2 at the Mn4CaO5 oxygen evolving complex (OEC) of photosystem II.24,25 NO and N2O are reduced at the FeB site of nitric oxide reductase (NOR) and at the CuZ site of nitrous oxide reductase, respectively.26–28 Electron and metal transfer reaction are mediated by metallothioneins (MT)29 and FeS proteins.30 Finally, small molecule fixation occurs at hybrid FeS clusters, as in sulfite reductase,31 CO dehydrogenase (CODH),32,33 the [FeFe] and [NiFe] hydrogenases34 and FeMo nitrogenase.35,36
Structural and functional studies of these multinuclear inorganic cofactors have been hampered by the intrinsic complexity. Common strategies in the design of small synthetic ligands have led to significant results in the stabilization of these bridged multinuclear clusters,5,37,38 but the unique geometries are particularly demanding in terms of the relative positioning of metals and ligands. Inherent synthetic difficulties may be overcome in principle by self-assembly, since a thermodynamic driving force has to be expected in the spontaneous formation of some of these clusters.37,39 However, ungoverned oligomerization may represent a serious issue. Therefore, several groups have undertaken the challenge to design tailored metallopeptides and proteins recapitulating the structural and hopefully the catalytic features of their natural counterparts.40–42 Progress has been made in both introducing novel sites into natural proteins43 as well as de novo design. De novo approaches show the following advantages: (i) a high degree of confidence in the design of secondary and tertiary structures can be reached thanks to the modern computational techniques of protein design; (ii) the intrinsic modularity of these compounds allows one to design the geometry and identity of first and second interactions surrounding the metal ion; (iii) the solvent accessibility of the site can be varied to assess the role of the aqueous solvent and substrate trafficking.
There has been significant progress in the design of proteins with stable metal ion clusters,40, 44–46 including many examples of metal-sulphur and metal-Cys complexes. Several groups have adopted the well-known Cys-Xaa-Xbb-Cys (CXXC) binding motif, which is frequently found in several unrelated proteins.47 Nanda, Noy and co-workers have designed a symmetrical four-helix bundle that encapsulates a Fe4S4 cluster.48 Dutton and co-workers inserted the CXXCXXC ferredoxin loop to bind the Fe4S4 cluster in heme-binding helical bundles,49 and Laplaza and Holm used a related approach to bind a {Ni2+-(μ2-S•Cys)-[Fe4S4]2+} cluster.50 Tanaka and co-workers designed a protein harboring a purple copper site similar to the CuA site of the CcO.51 Multi-copper clusters have been also obtained by Ogawa and co-workers, who reported the Cu(I) binding properties of a designed four-helix bundle,52,53 with spectroscopic features similar to cysteine rich metallothioneins. More recently, the same group has reported the design of a three-helix bundle binding a tetra-Cd2+ cluster54 featuring a tetrahedral adamantane like cluster [Cd4(μ2-S•Cys)6(O2C•Glu)3(H2O)] stabilized by a CXXCE motif. Of particular significance, the structure was determined by X-ray crystallography, which has not always been possible for many of the above-mentioned examples.
There has been significantly less diversity in the design of multinuclear centers with oxygen-rich carboxylate and water ligands, despite of their importance in many enzymes involved in oxygen reduction or water oxidation. Our groups have designed the DF (Due Ferri) series of di-metal carboxylate bridged clusters stabilized helical bundles,55,56 originally designed as mimics of diiron proteins. A number of versions of these proteins were prepared in which the four-helix bundle was assembled from four helical peptides, two helix-loop-helix peptides or a recombinantly expressed single-chain protein.55,56 In each case, the crystallographic and NMR studies showed the proteins folded into their designed conformations, which recapitulated the main structural features of much larger and more complex diiron proteins.8–10,57 A highly symmetrical starting protein was used in the initial design, but this symmetry was eliminated in subsequent functional studies in which the coordination environment of each metal ion was systematically varied.10–12,57–59 The DF scaffold was ultimately exploited to examine the structural requirements for two-electron and four-electron reduction of dioxygen with concomitant substrate oxidation.10–12,57,59 Futhermore, a di-Zn2+ form of a DF protein was also found to stabilize the radical semiquinone form of catechols in aqueous solution at room temperature for weeks.11
In this paper we address two design challenges in the area of de novo protein design and metalloprotein engineering. The first challenge was to design tetranuclear clusters that assembled within a protein without the aid of elemental sulphur or Cys as bridging ligands. The ability to design such clusters represents an important step towards the ultimate construction of mimics of the oxygen-evolving complex and other complex multi-nuclear clusters. A second challenge is to design extensive hydrogen-bonded networks that stabilize the primary ligands that surrounding the metal ions, as discussed above. Here, we explore solutions to these goals through the design of a tetra-Zn2+cluster, whose geometry is not found in biological proteins or small molecule complexes as defined by a search in the Cambridge Structural Database (CSD).60 The metal ion clusters are assembled in a vast hydrogen-bonded network of 16 polar amino acids, all involved in predetermined metal ion-ligand and hydrogen-bonding interactions.
Results
Protein design
We designed a series of D2-symmetric tetra-Zn2+-binding proteins, starting from the backbone geometry of DF1 protein,9 a dimeric antiparallel four-helix bundle with precise C2 symmetry and quasi-D2 symmetry (Figure 1A). The two-fold axis, which is orthogonal to the main axis of the bundle, bisects the center of mass of the two metal ions (Figure 1A). The four helices are arranged with approximate D2 symmetry, each donating a single Glu to the site. Two additional His ligands reduce the D2 symmetry of the structure to C2 (in DF proteins, the ligation sites trans to the two His residues are often left vacant to facilitate interactions with O2 and substrates). The two His residues pull the metal ions away from the central axis of the bundle by 1 to 2 Å.61 This displacement from the central bundle axis provides an opportunity to build a binding site for two additional metal ions, creating a tetranuclear complex (Figure 1B).
Figure 1.
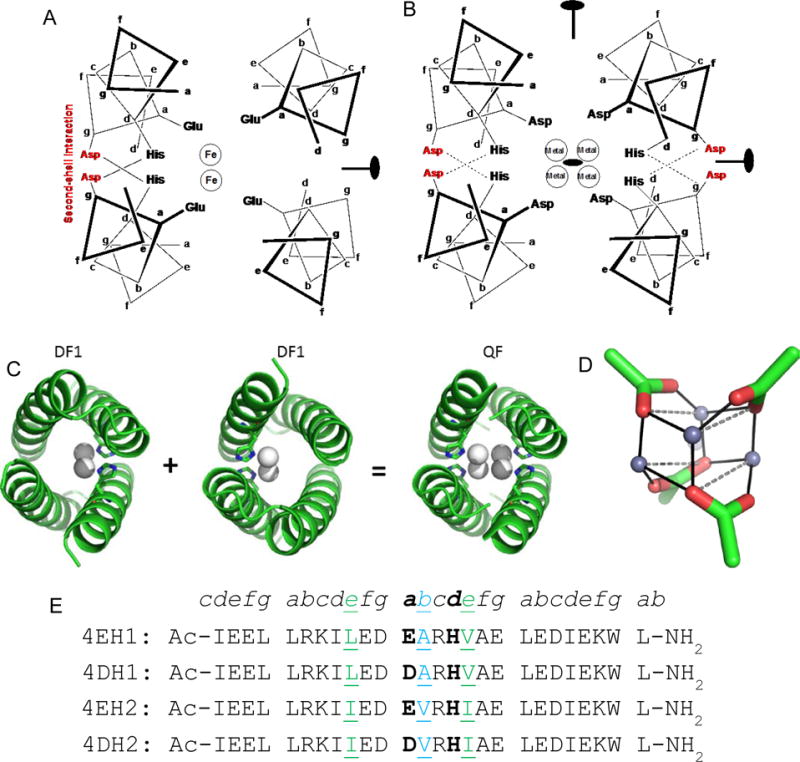
Design of tetranuclear clusters in helix bundles. (A) Previously designed diiron protein DF1 has 4 Glu and 2 His residues with 2 metal ions bound positioned off of the superhelical axis. The positions of second-shell Asp and the C2 symmetry axis are also indicated. (B) D2-symmetrical DF1 has four Asp and four His residues coordinating 4 ions at the core. The positions of the three orthogonal two-fold axes are indicated. Second-shell Asp residues at an interfacial g position (shown in red) are also included to interact with the first-shell Asp ligands. (C) Crystal structure of dimeric DF1, showing the displacement of the two metal ions away from the central bundle axis towards the His ligands. Two orientations of DF1 can be conceptually combined to create a structure that binds four metal ions. (D) Designed tetranuclear binding site, showing the positions of the carboxylates and Zn2+ ions. (E) Designed peptide sequences are shown with heptad repeat designation. The peptides are N-acetylated and C-amidated. The primary ligands are in bold. Residues at positions b and e, which were varied to probe the effect of interhelical packing on the structure of the metal-binding site are shown in green.
To generate a tetranuclear complex, we built an antiparallel, four-stranded coiled coil using the Crick parameterization62,63 to create an idealized D2-symmetric backbone9 (Figure 1C). Each helix was 26 residues long, with the N- and C-termini acetylated and amidated, respectively. With the capping groups, this length corresponds to 4-heptads, which can facilitate crystallization by end-to-end packing of the helices via hydrogen-bond formation, as previously described.64,65 Asp and His ligands were added to each of the four helices at the a and d positions of the middle helptad of the coiled coil. This placement positions four metal ions in close juxtaposition at four corners of a tetrahedron (Figure 1D); the Asp carboxylate ligands bridge the metal ions and each His serves as a monodentate ligand.
The designed tetranuclear-binding site differs in two significant aspects from DF1. First, the EXXH motif of DF1 was converted to a DXXH motif. Modeling suggested that replacing Glu with the smaller Asp sidechain would better accommodate four rather than two metal ions. Peptides with Glu as the primary carboxylate ligand were also synthesized to verify this hypothesis. Secondly, the number of His ligands per bundle was increased from two, as in DF1, to four in the present design.
The remainder of the peptide sequence was designed using a combination of structural bioinformatics and de novo computational methods. A database search of the crystallographic structures using the program MASTER66 revealed that the idealized backbone of the designed structure was nearly identical to tetrameric antiparallel coiled coils in the PDB, with Leu and Ile occupying each a and d position, respectively.67 Therefore, we placed these residues in the hydrophobic core, except those a and d positions occupied by the metal ligands. An Asp residue from a neighbouring helix (at a g position of the heptad repeat) was included to form a second-shell hydrogen-bond to the His ligand, stabilizing the +8 charge associated with 4 Zn2+ ions. Thus, considering these second-shell Asp residues together and the four Asp/Glu primary ligands, the charge on the four metal ions would be entirely neutralized. An Arg sidechain was included to salt bridge to this second-shell Asp, forming a third-shell interaction.
All other positions of the sequence were designed using the Rosetta fixed-backbone symmetry design module,68 with the exception of a Trp that was manually placed at a C-terminal g position, for peptide concentration measurement. The sequences were designated 4DH1 and 4EH1 (Figure 1E), in which the integer 4 reflects the 4-helix design and the letters define the ligands in single-letter code. In addition to the primary ligand exploration as either Glu or Asp, we also varied the interfacial residues at positions b and e. It was expected that these positions could indirectly affect the binding site by modulating the inter-helical distance and solvent access to the binding site. Thus, the residues at these positions (Leu9, Ala13 and Val16 in 4DH1 and 4EH1) were substituted to all beta-branched amino acids, Ile9, Val13, Ile16 in two additional peptides designated 4DH2 and 4EH2.
The crystallographic structures of 4EH1, 4DH1, 4EH2 and 4DH2 reveal closely related 4-helix bundles, stacked into an infinite coiled coil in the crystal
Despite the differences in their sequences, the four peptides crystallized as antiparallel tetrameric helical bundles in the P 31 2 1 space group (Table 1, Figure S1). The structures were solved using molecular replacement and the locations of the zinc ions were unambiguously determined by anomalous scattering (Figure S2). As in the design, exposed C-terminal amides hydrogen-bond with neighbouring helices to create infinite coiled coils (Figure 2B). Each helical bundle makes one quarter turn of a super-helix, such that four 4-helix bundles comprise a single turn of the super-helix, with a pitch of about 130 Å. The crystal structures match very well the design model with Cα RMSDs less than 1 Å (Table S1).
Table 1.
Statistics of data collection and structure refinement
| Dataset | 4EH1 (PDB: 5WLJ) |
4EH2 (PDB: 5WLK) |
4DH1*** (PDB: 5WLL) |
4DH2 (PDB: 5WLM) |
|---|---|---|---|---|
| Data Collection: | ||||
| Software | HKL2000 | HKL2000 | XDS | XDS |
| Wavelength (Å) | 1.11587 | 1.11587 | 1.11587 | 1.11587 |
| Resolution (Å)* | 80.9–1.60 (1.66–1.60) |
80.9–1.80 (1.86–1.80) |
80.8–1.90 (2.02–1.90) |
81.2–1.95 (2.06–1.95) |
| Cell constants | ||||
| a, b, c (Å) | 80.828, 80.828, 64.388 |
80.995, 80.995, 65.308 |
80.782, 80.782, 64.923 |
81.156, 81.156, 66.331 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 |
| Space group | P3(1)21 | P3(1)21 | P3(1)21 | P3(1)21 |
| Unique reflections | 32467 (3210) | 23242 (2285) | 19614 (3086) | 17914 (2867) |
| Completeness (%) | 99.9 (100.0) | 99.6 (100.0) | 99.2 (96.2) | 99.8 (99.2) |
| Redundancy | 10.8 (10.6) | 3.7 (3.7) | 10.4 (10.0) | 7.5 (7.4) |
| I/σ(I) | 25.9 (2.9) | 14.6 (4.4) | 11.8 (2.2) | 7.0 (1.9) |
| Rmerge (%) | 8.6 (76.3) | 12.7 (74.4) | 9.7 (84.4) | 17.9 (84.8) |
| Refinement: | ||||
| Resolution (Å) | 70.0–1.60 (1.64–1.60) |
70.1–1.80 (1.85–1.80) |
70.0–1.90 (1.95–1.90) |
48.2–1.95 (2.00–1.95) |
| R factor (%)** | 14.8 | 16.9 | 22.0 | 17.2 |
| Rfree factor (%) | 16.7 | 17.8 | 23.8 | 18.7 |
| RMSD from standards | ||||
| Bonds (Å) | 0.008 | 0.006 | 0.009 | 0.010 |
| Angles (°) | 1.1 | 0.9 | 1.2 | 1.1 |
| Ramachandran plot | ||||
| Favored (%) | 100.0 | 100.0 | 100.0 | 100.0 |
Values in parentheses are for the outmost resolution shell.
Rfree factor value was calculated for R factor using a subset of reflections data (5%, randomly chosen). For dataset 4DH2, an overall 2.5% randomly chosen data, which corresponded to 5% of data free of twin pairs, were used for Rfree factor calculation.
This dataset was subjected to anisotropic scaling.
Figure 2.
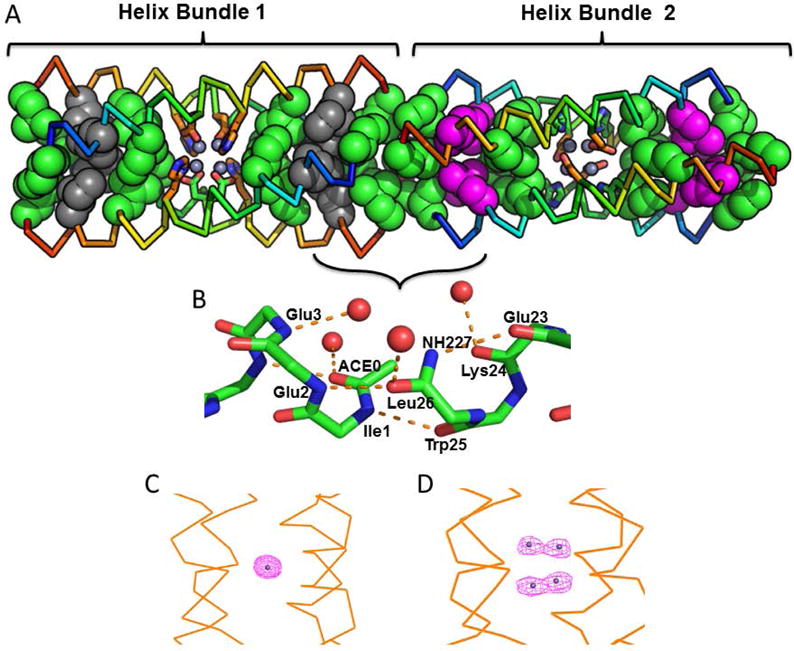
Packing of the hydrophobic cores and hydrogen-bonding interactions connecting individual four-helix bundles within the unit cell. (A) Each protein has two hydrophobic cores at the top and bottom of the bundle, which enforce the geometry of the central metal-binding site. The hydrophobic Leu and Ile residues pack in layers, alternately coloured in grey and green in bundle 1, and magenta and green in the neighbouring bundle 2 in the asymmetric unit. (B) Hydrogen-bonded interactions that extend the superhelix running through the crystal. These interactions include direct hydrogen-bonds between amides from the N-terminus of one helix and the C-terminus of the neighbour. Additionally, water-mediated hydrogen-bonds are observed at this interface. (C) – (D) Map computed from the Zn2+ anomalous scattering observed in 4EH2 and 4DH2, respectively.
The overall structures of the four-helix bundles observed for the four proteins, 4DH1, 4EH1, 4DH2 and 4EH2 are very closely related (Cα RMSD ranges from 0.24 to 0.59 Å, Table S2). For each protein, the unit cells are built from two nearly identical (Cα RMSD ranges from 0.35 to 0.80 Å) four-helix bundles, which have precise crystallographic two-fold symmetry and deviate only slightly from D2 symmetry. In the crystallographic lattice one of the two four-helix bundles is surrounded by water channels and has systematically higher Debye-Waller crystallographic b-factors (Figure S3). Thus, the following analysis focuses on the helical bundles with the lower b-factors and more clearly defined electron density maps.
Each of the proteins has two well-packed hydrophobic cores at opposite ends of the bundle, flanking the metal-binding site near the central region of the coiled coil. In precise agreement with the design, the hydrophobic residues within these cores pack in three layers, each layer comprised of two Leu and two Ile residues from the a and d positions of the heptad repeats, respectively (Figure 2A). These well-packed cores provide structural and thermodynamic stability and help position the metal-binding residues Glu/Asp12 and His15 within the interior of the central portion of the helical bundle. The helical bundles all display a-d packing, as assessed by the global axial helix rotation69,70 (Table S3).
4DH1 and 4DH2 form tetranuclear clusters
4DH1 and 4DH2 both bind four Zn2+ in a distorted cuboidal geometry. The cluster is formed by the four Zn2+ ions and four carboxylate oxygens from the Asp12 sidechains (Figures 3A – 3D). The site has nearly precise D2 symmetry, matching the symmetry of the bundle. Each Zn2+ is strongly coordinated by three ligands (metal-ligand distance < 2.5 Å; bold line, Figures 3A and 3B): one carboxylate oxygen of Asp12, the Nδ of His15 and a bridging water. One or two weaker carboxylate-Zn2+ interactions (< 3 Å distance, dashed lines, Figures 3A and 3B) complete the first coordination sphere. Two bridging solvent molecules were assigned as water, based on its shape and ability to donate two hydrogen-bonds with a carboxylate in the 4DH1 complex (Figure 3A). Although not explicitly included in the design, they appear to help stabilize the designed coordination geometry of the metal site, and are well positioned to participate in proton-coupled electron transfer reactions (in future extensions of this work with redox-active metal ions). The designed Asp11 second-shell interaction to His15 Nε is clearly observed, as is the third-shell Arg interaction.
Figure 3.
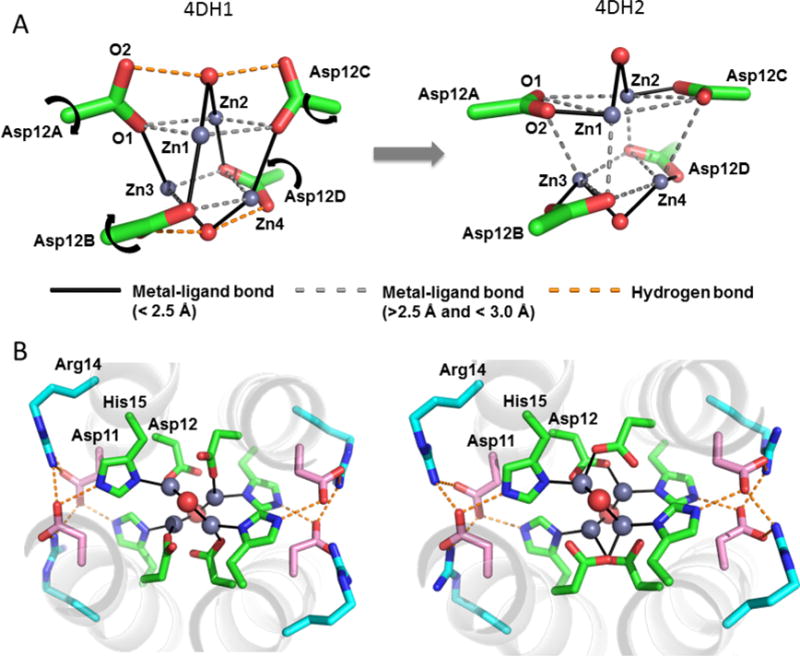
Crystallographic structure of the metal-binding site in 4DH1 and 4DH2. (A) The tetranuclear cluster formed in 4DH1 and 4DH2; for clarity, only the Asp carboxylates, waters and Zn2+ ions are shown (carbon atoms are shown in green; oxygen in red; zinc in grey and interactions are drawn with dashed and solid lines as indicated). A carboxylate shift leads to a difference in the primary coordination shell and hydrogen-bonding to a bound water molecule. (B) Overall axial view of the 4DH1 and 4DH2 sites, including the first-shell ligand His16, the second-shell Asp11 and the third-shell Arg14. Carbon atoms of the secondary Asp11 and tertiary Arg14 ligands are in pink and cyan, respectively.
There are no significant differences in the binding-site geometries of 4DH1 in the two distinct tetramers seen in the asymmetric unit. On the other hand, there are significant differences in the geometries of tetranuclear clusters seen for 4DH1 versus 4DH2 (Figure 3), which we ascribe to differences in the protein sequence rather than crystal packing. In the 4DH1 cluster one oxygen of each of the Asp12 ligands binds to Zn2+ ions (Figure 3A), while the other oxygen forms a hydrogen-bond with the bound water. However, in 4DH2 the hydrogen-bonds to water are broken, and each Asp forms a primary liganding interaction with a different Zn2+ ion (to the left or right of it as viewed in Figure 3C). The other carboxylate oxygen of Asp12 forms weaker electrostatic interactions with the cluster. This same arrangement was seen in both helical bundles in the asymmetric unit of 4DH2 (Figure 3D), supporting the conclusion that the geometric differences between the tetranuclear sites in 4DH1 and 4DH2 are due to differences in their primary sequences rather than packing within the crystal lattice.
The shift in geometry between the two proteins is a result of a rotation of approximately 40° about the Cβ-Cγ bond (χ2 torsional angle) of the Asp sidechain, along with small shifts in the helical bundle geometry discussed below. Small changes in the second-and third-shell ligands in 4DH2 (relative to 4DH1) accompany these changes in geometry. These findings show how small geometric changes in a helical bundle can influence the detailed geometry of their bound metal ion cofactors.
Comparison of tetranuclear Zn2+ clusters from 4DH1 and 4DH2 with small molecule complexes
Proteins often enforce somewhat unusual geometries onto metal ions or metal ion clusters, which are not easily achieved in small molecule complexes.71,72 We therefore searched the Cambridge Crystallographic Database, to probe the novelty of the tetranuclear stabilized in the designed structures. We discovered one tetranuclear Zn2+ cuboidal site.73 The ligands in this cuboidal site are methoxides and t-butoxides, and hence would not be stable in water. A second 4-Zn2+ site is organized around a central O2−, but, again, this site is not stable in water.74 Also, the metal-metal distances in both complexes are significantly closer (3.1-3.3 Å), due to 1,1 bridging interactions. Other tetranuclear Zn2+ sites appeared to be embedded in larger complexes that contain up to more than eight Zn2+ ions.75,76
4EH2 binds a single Zn2+ at the center of an extended network of hydrogen-bonded sidechains
The Glu sidechain ligands are accommodated in the core of 4EH1 and 4EH2, relative to the corresponding 4DH1 and 4DH2, without a significant accompanying change in the radius of the helical bundle. The longer Glu sidechains are therefore more compressed in a cavity of fixed size, which has a large influence on the metal-binding sites for these proteins. In 4EH2, the single Zn2+ is tetrahedrally coordinated by one Glu13 carboxylate oxygen in each chain (Figure 4A). This primary ligand is stabilized by a hydrogen-bond network of second-, third- and fourth-shell interactions. His15 serves as a second-shell ligand, rather than binding directly to the metal as in 4DH1 and 4DH2. His15 appears protonated based on the fact that both basic nitrogens make close approaches to the primary Glu13 ligand and a third-shell ligand, Asp12. Additionally, Cε-H of His15 also makes a very close approach (3.1 Å) to the Oδ of Asp12 from a neighboring chain, suggesting that it might provide additional stabilization by “C-H–O hydrogen-bonds”.77 Finally, the hydrogen-bonded network surrounding the primary ligand extends to Arg14, which serves as a fourth-shell ligand by interacting with Asp12. In summary, the structure of mono-Zn2+ 4EH2 shows a remarkable network of concentric shells of interacting sidechains, each shell alternating in charge in the order of Zn2+ (+2), four Glu13 residues (−4), four His37 (+4), four Asp12 (−4), and finally four Arg14 (+4) (Figure 4B).
Figure 4.
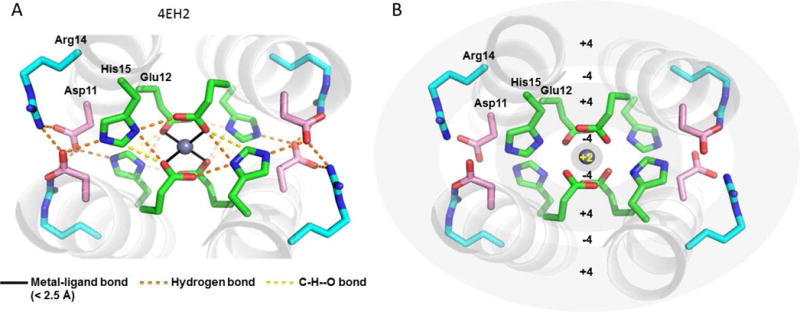
Crystal structure of 4EH2. (A) A single Zn2+ is bound, surrounded by four monodentate Glu ligands. (B) The primary ligand sphere is surrounded by concentric rings of oppositely charged residues, which form an extensive hydrogen-bonded network.
To confirm that His15 was positively charged in solution, we examined its NMR spectrum in the absence and presence of a single equivalent of Zn2+ per tetramer, matching the stoichiometry seen for this peptide in the crystal structure. The chemical shifts of the His residue clearly indicated that this residue was protonated under both conditions (Figure S4 and Table S4 for peak assignment).
4EH1 binds multiple Zn2+ at partial occupancy
4EH1 binds multiple metal ions at a number of partially occupied sites (Figure 5A). In 4EH1 five Zn2+ ions are seen near the center of the structure: four form a 4-Zn2+ cuboidal structure similar to that seen in 4DH1 and 4DH2 (Figures 5B and 5C); additional density is seen at a position similar to the monovalent site observed in 4EH2. It is not sterically feasible to accommodate both the 4-Zn2+ and 1-Zn2+ structures within the same bundle, which is consistent with the partial occupancy seen for the two overlapping sites. Thus, the crystal lattice contains a mixture of the two new ion complexes. One final interesting feature of this structure is the presence of yet additional Zn2+ ions that intercalate between His15 and Asp12, in a doubly coordinated imidazolate geometry (Figures 5D and 5E), similar to that recently been seen in a designed beta-sheet forming peptide.78
Figure 5.
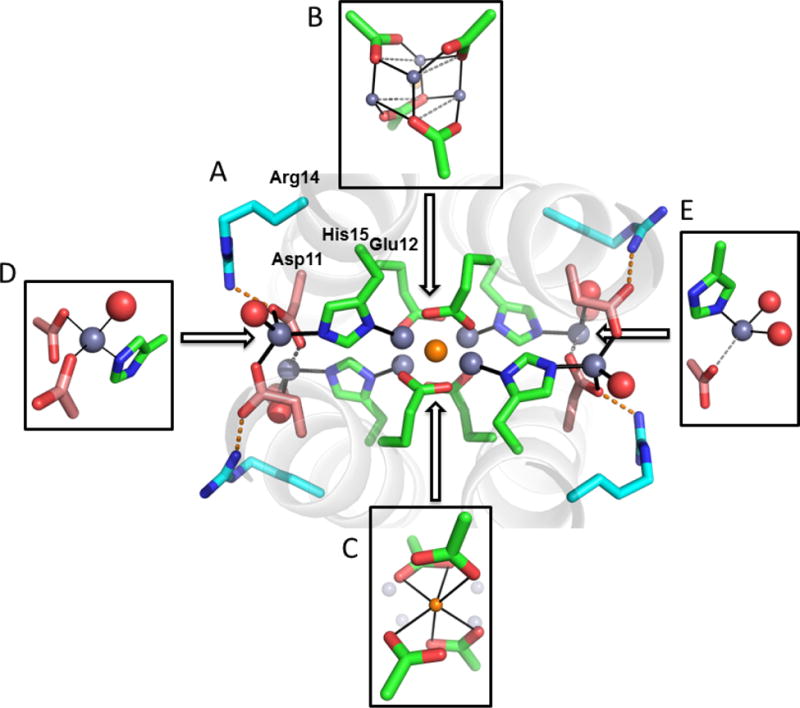
Crystal structure of 4EH1. (A) A large number of Zn2+ ions are observed, all at partial occupancy. (B) and (C) depict in grey and orange, respectively, two alternatively occupied Zn2+ complexes observed near the center of the bundle. They resemble the tetranuclear and mononuclear clusters shown in Figures 3 and 4, respectively. The same arrangement was observed in the other crystallographically distinct 4-helix bundle in the structure of 4EH1. (D) and (E) depict partially occupied Zn2+ ions, observed at the periphery of the bundle.
Concerted helical rotations and translations accompany changes in Zn2+ coordination geometry
The four peptides studied here vary in terms of the bulk of the hydrophobic residues at the b and e heptad positions surrounding the metal-binding site, as well as the nature of the carboxylate ligand (Glu versus Asp). While these changes had little effect on the overall structure of the helical bundles or their packing within the lattice, they led to deep-seated changes in the geometry and stoichiometry of the bound metal ions. Thus, we sought to identify more fine-tuned variations in the backbone geometries of the helical bundles that underlie these differences. The geometric features that mostly contribute to the variation between the structures (Figures 6A – 6C, Table S5) consist of a rotation of the helices about their own axes by up to 10° (Figure 6C) and a shift of the antiparallel helices relative to one another by up to 0.8 Å (Figure 6B). By contrast, the radius remains constant at 7.41 ± 0.08 Å, and the super-helical phase remains constant at 104.4° ± 0.4°. The small helical rotation allows the bulkier residues of 4EH2 and 4DH2 to be displaced towards the protein surface (Figure 6D); in parallel the helices slide relative to one another, thereby maintaining good packing along the b/e interface. These correlated rotations and translations, along with more local carboxylate shifts (associated with changes in the χ angles of Asp and Glu) define a continuum of designable geometries that might be used in future designs of helical bundles with pre-programmed structures and dynamics.
Figure 6.
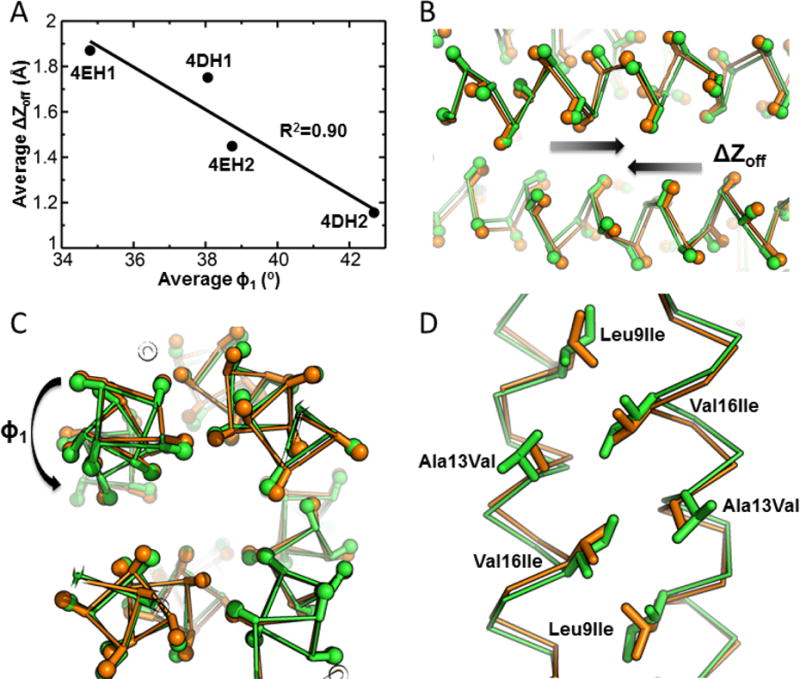
Structural differences between the crystal structures of 4EH1, 4EH2, 4DH1 and 4DH2. The backbones surrounding the metal-binding sites (11 residues/helix) were analysed using a mathematical parameterization of the coiled coil, resulting in an excellent fit to the experimental structures (0.2 to 0.4 Å Cα RMSD, Table S5). (A) Plot of the parameters that mostly contributed to the structural variability: (B) shift of the helices relative to one another, ΔZoff and (C) their axial rotation, ϕ1. (D) Slight helix rotation and sliding that avoid steric overlap as the bulkiness of the varied residues (A13V, V16I) and the degree of β branching (A13V, V16I, L9I) increase. These two motions appear to be correlated, as shown in (A): the values plotted represent the average over the two crystallographically distinct 4-helix bundles observed for each crystal structure.
Association of the peptides into helical bundles in solution
The association state of each peptide was next examined using sedimentation equilibrium ultracentrifugation. The apparent MW of 4EH2, 4DH2, and 4EH1 were slightly lower than expected for a tetramer (Table 2), but the computed MW increased to that expected for a tetramer in the presence of a 1.5-fold excess of Zn2+ (6 equivalents per tetramer). Thus, binding of Zn2+ appears to stabilize the 4-helical conformation observed in the crystal structures. By contrast, 4EH1 formed a dimer in the absence of metal ions, which was converted to a tetramer only in the presence of a large excess of Zn2+. At lower Zn2+, this peptide formed higher-order aggregates. Thus, the tetrameric form of this peptide is less stable and conformationally exploratory than the other three bundles. Finally, far UV CD spectroscopy confirmed that all peptides had substantial helical content, both in the presence and absence of metal ions (Figure S6).
Table 2.
Sedimentation equilibrium results.
| Protein | Monomer M.W. (Dalton) | AUC Mass (Apo) (Dalton) | AUC Mass (Holo) (Dalton) |
|---|---|---|---|
| 4EH1 | 3232 | 10.08 K – 11.91 K (3.38)* |
14.82 K – 17.51 K (+1.5 eq Zn) (4.97)* |
| 4DH1 | 3218 | 5.184 K – 6.119 K (1.74)* |
23.37 K – 27.58 K (+1.5 eq Zn) (7.86)* 13.67 K – 16.14 K (+5.0 eq Zn) (4.60)* |
| 4EH2 | 3274 | 11.63 K – 13.77 K (3.85)* |
13.34 K – 15.80 K (+1.5 eq Zn) (4.42)* |
| 4DH2 | 3260 | 10.01 K – 11.84 K (3.33)* |
13.91 K – 16.45 K (+1.5 eq Zn) (4.62)* |
Conclusions
In this work, we report the structural characterization of a tetranuclear zinc site stabilized by a de novo designed four-helix bundle scaffold. The design procedure started by the parametric generation of the backbone coordinates resulting in a perfect D2 symmetric anti-parallel coiled coil. The protein core was then stabilized by strong hydrophobic packing on both ends of the bundle, thus allowing the insertion of the metal cofactor in the middle of the protein. The key factor for the stabilization of the hydrophilic metal binding residues has been the rational design of the second up to the third-shell H-bond interactions, and even fourth-shell interactions were observed in 4EH2 mono-Zn2+ complex. Recently, Baker and coworkers13 demonstrated the incorporation of hydrogen-bonded networks into the structures of helical bundles by introducing up to nine hydrogen-bonds in helical bundles, although these proteins lacked any binding or other function. Previously, we had designed complex hydrogen-bonded networks to tune the properties of diiron-binding proteins,8–12 which help stabilize the protein in the metal-free state,8 and additionally tune the electronic properties in the metal-coordinated state.10–12,59 Here, we use even more extensive networks to reinforce the binding of an abiological cofactor building networks of sixteen sidechains and four metal ions. Moreover, we have shown that the fine modulation of the metal binding geometry could be achieved by changing the size of the hydrophobic packing interactions, resulting in slightly different metal complexes differing in the relative ligand topology and/or in the metal content.
This new class of metalloproteins should serve as an attractive starting point to design more complex redox-active multinuclear clusters. In our previous studies with dinuclear centers, we began with a simple, highly symmetrical di-Zn2+ protein, which served as a versatile scaffold for varying both the metal ions and the protein sequence. Ultimately, it was possible to generate fully asymmetric proteins capable of two and four-electron reduction of O2 with concomitant oxidation and hydroxylation of substrates.10,12,59 Here, we have engineered a more complex cofactor with four transition metal ions, which might similarly serve as a starting point for the design of demanding O2-dependent functions. The tetranuclear cluster in 4DH1 and 4DH2 includes bridging water molecules – well positioned to serve as proton donors for future studies of proton-coupled electron transfer in mimics of the OEC of PSII. Such future designs might also incorporate proton-pathways79 and additional light and redox-active cofactors, which have previously been built in 4-helix bundle architectures.50,80,81 Thus, this work provides a minimal system for studying the physical principles underlying water oxidation, oxygen reduction, with the ultimate goal of building devices for energy conversion and storage. Finally, as described recently by Suzuki, Tezcan and coworkers, the addition of metal-binding sites onto the surface of natural proteins can yield supramolecular lattices with remarkable auxetic material properties.82 The ability to expand the pallet to fully designed metalloproteins should provide additional attractive possibilities for the design of responsive soft materials.
Methods
Protein design
The starting backbone was generated by fitting a coiled coil to the DF1 protein (PDB: 1EC5)9 using the program CCCP.63 The identity of liganding residues Asp12 and His14, the second-shell residue Asp11, and the third-shell residue Arg15 were based on their abilities to form the desired interactions in low-energy rotamers. We used the TERMS method66 to define the hydrophobic core of the structure. In the course of search for the fragment, we found the backbone fitted well to an existing antiparallel homotetrameric 4-helix bundle (PDB: 2CCN)67 with a Cα RMSD of 0.86 Å. The hydrophobic packing motif of 2CCN, Leu on position a and Ile on position d, was therefore adopted for the designed scaffolds. Peripheral residues are computationally picked by the fixbb module in Rosetta,68 and residues on positions b and e are systematically varied to β-branched amino acids in the second-generation designs.
Peptide Synthesis
Peptides were solid-state synthesis at 0.1 mmole scale on a Biotage Initiator+ microwave synthesizer with Rink Amide Chemmatrix resin. Double coupling was conducted on each residue by 5 eq. Fmoc amino acid, 4.98 eq. HCTU and 10 eq. DIPEA in DMF for 5 minutes at 75°C (30 min at room temperature for His residue). Decoupling is performed by 20% 4-methylpiperidine and 0.1 M HOBt in DMF for 5 minutes at 70°C. Cleavage was done in a TFA cocktail with TFA:TIPS:H2O at a ratio 95:2.5:2.5. Purification was performed by reverse-phase HPLC with water and acetonitrile as eluants. Purity of the peptides was verified by analytical reverse-phase HPLC.
Crystallography
The peptides were dissolved at 5 mg/ml in water with addition of 1 equivalent of ZnCl2. The hanging-drop vapor-diffusion method at room temperature was used for crystallization. The crystallization condition for 4EH1 is 0.2 M NaCl, 6% (v/v) isopropanol and 0.1 M HEPES pH 7.5, for 4DH1 is 2.0% PEG1000, 1.0 M AmSO4 and 0.1 M HEPES pH 7.5, for 4EH2 is 0.2 M Li2SO4, 1.0 M potassium/sodium tartrate and 0.1 M Tris pH 7.0, and for 4DH2 is 0.01 M NiCl2, 1.0 M Li2SO4 and 0.1 M Tris pH 8.5. Crystals were flash frozen with the cryoprotectant 25%-30% (v/v) glycerol, and diffraction data were collected at the Advanced Light Source, Lawrence Berkeley National Laboratory in Berkeley CA at the Beamline 8.3.1 on an ADSC Q315 CCD Detector with X-ray wavelength of 1.1158 A. Data collection temperature was 100 K.
Data were processed with HKL200083 and/or XDS84 packages. Statistics for data processing and structural refinement were shown in Table 1. The 4EH1 structure was first solved by molecular replacement with Phaser85 using the designed 4EH1 single helix as searching model. For the other 3 structures, a single helix of 4EH1 served as search models. In general, Refmac86 was used for the most of refinements. For helical bundles, rigid-body refinement appeared to be particularly important and was carried out at any time whenever large displacement observed and fixed. TLS domains were set down to single helices, with binding metal ions technically grouped into a tightly interacted helix. To remove initial model biases, simulated-annealing refinement was carried out with Phenix.87 Metal ion sites were determined based on the anomalous maps phased from metal ion-free models after molecular repacement. The occupancy refinement for metal sites, as well as for partially occupied site chains, was performed also in Phenix. Protein model rebuilding and adjustment were done in Coot.88 It is also noteworthy that 4DH2 dataset had pseudo-hexagonal symmetry and the refinement was largely improved with twinning operators applied.
NMR
The 1D spectra were recorded at 298K on a Bruker Avance II 900 MHz spectrometer equipped with a cryogenic probe with the pulse sequence zgesgp with tmax = 151 ms, 256 scans. The 1H carrier frequency was set at 4.75 ppm. All spectra were processed and analyzed using the program topspin.
Analytical Ultracentrifugation
Sedimentation equilibrium studies were performed with the peptides at concentrations of 80-100 μM in the buffer 10 mM MOPS, pH 7.0, 100 mM NaCl, at speeds of 25 K, 30 K, 35 K, 40 K and 45 K r.p.m. in a Beckman XL-I ultracentrifuge at 25°C. Data collected on UV absorbance at 280 nm for each sample from the five speeds were fit globally to single-species of equilibrium sedimentation by a non-linear least squares method using Igor Pro.
Supplementary Material
Acknowledgments
The authors thank Dr. Yibing Wu for recording and analyzing NMR data. This work was primarily supported by NIH grant R35GM122603 to W.F.D., with additional support from the NSF (CHE1413295) for the MRSEC program to the LRSM at the University of Pennsylvania, Research Department of Campania Region (STRAIN Project, POR FSE 2007/2013, grant number B25B0900000000 for a postdoctoral fellowship to M. C.), and University of Napoli “Federico II” for mobility grants to W.F.D. and M.C.
Beamline 8.3.1 at the Advanced Light Source is operated by the University of California Office of the President, Multicampus Research Programs and Initiatives grant MR-15-328599, the National Institutes of Health (R01 GM124149 and P30 GM124169), Plexxikon Inc. and the Integrated Diffraction Analysis Technologies program of the US Department of Energy Office of Biological and Environmental Research. The Advanced Light Source (Berkeley, CA) is a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the US Department of Energy under contract number DE-AC02-05CH11231, Office of Basic Energy Sciences.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Crystal structure characterization figures and tables, 1H NMR spectra; chemical shift statistics; sedimentation equilibrium ultracentrifugation, circular dichroism characterization (PDF). Atomic coordinates and structure factors have been deposited in the Protein Data Bank, with the following accession numbers: 5WLJ (for the protein 4EH1); 5WLK (for the protein 4EH2); 5WLL (for the protein 4DH1); 5WLM (for the protein 4DH2).
ORCID
Shao-Qing Zhang: 0000-0001-5853-592X
Marco Chino: 0000-0002-0436-3293
Lijun Liu: 0000-0003-0514-281X
William F. DeGrado: 0000-0003-4745-263X
Angela Lombardi: 0000-0002-2013-3009
References
- 1.DeGrado WF, Summa CM, Pavone V, Nastri F, Lombardi A. Ann Rev Biochem. 1999;68(1):779–819. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 2.Huang PS, Boyken SE, Baker D. Nature. 2016;537(7620):320–327. doi: 10.1038/nature19946. [DOI] [PubMed] [Google Scholar]
- 3.Woolfson DN, Bartlett GJ, Burton AJ, Heal JW, Niitsu A, Thomson AR, Wood CW. Curr Opin Struct Biol. 2015;33:16–26. doi: 10.1016/j.sbi.2015.05.009. [DOI] [PubMed] [Google Scholar]
- 4.Cheng RP. Curr Opin Struct Biol. 2004;14(4):512–520. doi: 10.1016/j.sbi.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Holm RH. Pure Appl Chem. 1995;67:217–224. [Google Scholar]
- 6.Christianson DW, Cox JD. Annu Rev Biochem. 1999;68:33–57. doi: 10.1146/annurev.biochem.68.1.33. [DOI] [PubMed] [Google Scholar]
- 7.Migliore A, Polizzi NF, Therien MJ, Beratan DN. Chem Rev. 2014;114(7):3381–465. doi: 10.1021/cr4006654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maglio O, Nastri F, Pavone V, Lombardi A, DeGrado WF. Proc Natl Acad Sci US A. 2003;100(7):3772–3777. doi: 10.1073/pnas.0730771100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lombardi A, Summa CM, Geremia S, Randaccio L, Pavone V, DeGrado WF. Proc Natl Acad Sci U S A. 2000;97:6298–6305. doi: 10.1073/pnas.97.12.6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reig AJ, Pires MM, Snyder RA, Wu Y, Jo H, Kulp DW, Butch SE, Calhoun JR, Szyperski T, Solomon EI, DeGrado WF. Nat Chem. 2012;4:900–906. doi: 10.1038/nchem.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ulas G, Lemmin T, Wu YB, Gassner GT, DeGrado WF. Nat Chem. 2016;8(4):354–359. doi: 10.1038/nchem.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chino M, Leone L, Maglio O, D’Alonzo D, Pirro F, Pavone V, Nastri F, Lombardi A. Angew Chem Int Ed. 2017 doi: 10.1002/anie.201707637. [DOI] [PubMed] [Google Scholar]
- 13.Boyken SE, Chen Z, Groves B, Langan RA, Oberdorfer G, Ford A, Gilmore JM, Xu C, DiMaio F, Pereira JH, Sankaran B, Seelig G, Zwart PH, Baker D. Science. 2016;352(6286):680–687. doi: 10.1126/science.aad8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burton AJ, Thomson AR, Dawson WM, Brady RL, Woolfson DN. Nat Chem. 2016;8(9):837. doi: 10.1038/nchem.2555. [DOI] [PubMed] [Google Scholar]
- 15.Solomon EI, Sundaram UM, Machonkin TE. Chem Rev. 1996;96:2563–2606. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 16.Giardina P, Sannia G. Cell Mol Life Sci. 2015;72:855–856. doi: 10.1007/s00018-014-1821-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sazinsky MH, Lippard SJ. Methane Monooxygenase: Functionalizing Methane at Iron and Copper. In: Kroneck PMH, Torres MES, editors. Sustaining Life on Planet Earth: Metalloenzymes Mastering Dioxygen and Other Chewy Gases. Springer International Publishing; Cham, Switzerland: 2015. pp. 205–256. [Google Scholar]
- 18.Acheson JF, Bailey LJ, Brunold TC, Fox BG. Nature. 2017;544:191–195. doi: 10.1038/nature21681. [DOI] [PubMed] [Google Scholar]
- 19.Kanteev M, Goldfeder M, Fishman A. Protein Sci. 2015;24:1360–1369. doi: 10.1002/pro.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barynin VV, Whittaker MM, Antonyuk SV, Lamzin VS, Harrison PM, Artymiuk PJ, Whittaker JW. Structure. 2001;9:725–738. doi: 10.1016/s0969-2126(01)00628-1. [DOI] [PubMed] [Google Scholar]
- 21.Tsukihara T, Shimokata K, Katayama Y, Shimada H, Muramoto K, Aoyama H, Mochizuki M, Shinzawa-Itoh K, Yamashita E, Yao M, Ishimura Y, Yoshikawa S. Proc Natl Acad Sci U S A. 2003;100:15304–15309. doi: 10.1073/pnas.2635097100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaila VRI, Verkhovsky MI, Wikström M. Chem Rev. 2010;110:7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- 23.Ferguson-Miller S, Babcock GT. Chem Rev. 1996;96:2889–2908. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 24.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 25.Young ID, Ibrahim M, Chatterjee R, Gul S, Fuller FD, Koroidov S, Brewster AS, Tran R, Alonso-Mori R, Kroll T, Michels-Clark T, Laksmono H, Sierra RG, Stan CA, Hussein R, Zhang M, Douthit L, Kubin M, de Lichtenberg C, Vo Pham L, Nilsson H, Cheah MH, Shevela D, Saracini C, Bean MA, Seuffert I, Sokaras D, Weng T-C, Pastor E, Weninger C, Fransson T, Lassalle L, Bräuer P, Aller P, Docker PT, Andi B, Orville AM, Glownia JM, Nelson S, Sikorski M, Zhu D, Hunter MS, Lane TJ, Aquila A, Koglin JE, Robinson J, Liang M, Boutet S, Lyubimov AY, Uervirojnangkoorn M, Moriarty NW, Liebschner D, Afonine PV, Waterman DG, Evans G, Wernet P, Dobbek H, Weis WI, Brunger AT, Zwart PH, Adams PD, Zouni A, Messinger J, Bergmann U, Sauter NK, Kern J, Yachandra VK, Yano J. Nature. 2016;540:453–457. doi: 10.1038/nature20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown K, Tegoni M, Prudêncio M, Pereira AS, Besson S, Moura JJ, Moura I, Cambillau C. Nat Struct Mol Biol. 2000;7:191–195. doi: 10.1038/73288. [DOI] [PubMed] [Google Scholar]
- 27.Hino T, Matsumoto Y, Nagano S, Sugimoto H, Fukumori Y, Murata T, Iwata S, Shiro Y. Science. 2010;330:1666–1670. doi: 10.1126/science.1195591. [DOI] [PubMed] [Google Scholar]
- 28.Matsumoto Y, Tosha T, Pisliakov AV, Hino T, Sugimoto H, Nagano S, Sugita Y, Shiro Y. Nat Struct Mol Biol. 2012;19:238–245. doi: 10.1038/nsmb.2213. [DOI] [PubMed] [Google Scholar]
- 29.Calderone V, Dolderer B, Hartmann H-J, Echner H, Luchinat C, Bianco CD, Mangani S, Weser U. Proc Natl Acad Sci U S A. 2005;102:51–56. doi: 10.1073/pnas.0408254101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Chakraborty S, Hosseinzadeh P, Yu Y, Tian S, Petrik I, Bhagi A, Lu Y. Chem Rev. 2014;114:4366–4469. doi: 10.1021/cr400479b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crane BR, Siegel LM, Getzoff ED. Science. 1995;270:59–67. doi: 10.1126/science.270.5233.59. [DOI] [PubMed] [Google Scholar]
- 32.Dobbek H, Svetlitchnyi V, Gremer L, Huber R, Meyer O. Science. 2001;293:1281–1285. doi: 10.1126/science.1061500. [DOI] [PubMed] [Google Scholar]
- 33.Doukov TI, Iverson TM, Seravalli J, Ragsdale SW, Drennan CL. Science. 2002;298:567–572. doi: 10.1126/science.1075843. [DOI] [PubMed] [Google Scholar]
- 34.Caserta G, Roy S, Atta M, Artero V, Fontecave M. Curr Opin Chem Biol. 2015;25:36–47. doi: 10.1016/j.cbpa.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 35.Einsle O, Tezcan FA, Andrade SLA, Schmid B, Yoshida M, Howard JB, Rees DC. Science. 2002;297(5587):1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 36.Spatzal T, Aksoyoglu M, Zhang L, Andrade SLA, Schleicher E, Weber S, Rees DC, Einsle O. Science. 2011;334:940–940. doi: 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venkateswara Rao P, Holm RH. Chem Rev. 2004;104:527–560. doi: 10.1021/cr020615+. [DOI] [PubMed] [Google Scholar]
- 38.Lee SC, Holm RH. Chem Rev. 2004;104:1135–1158. doi: 10.1021/cr0206216. [DOI] [PubMed] [Google Scholar]
- 39.Citek C, Lyons CT, Wasinger EC, Stack TD. Nat Chem. 2012;4(4):317–322. doi: 10.1038/nchem.1284. [DOI] [PubMed] [Google Scholar]
- 40.Yu F, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, Mocny CS, Ruckthong L, Qayyum H, Pecoraro VL. Chem Rev. 2014;114(7):3495–3578. doi: 10.1021/cr400458x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peacock AF. Curr Opin Chem Biol. 2016;31:160–165. doi: 10.1016/j.cbpa.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 42.Nastri F, Chino M, Maglio O, Bhagi-Damodaran A, Lu Y, Lombardi A. Chem Soc Rev. 2016;45:5020–5054. doi: 10.1039/c5cs00923e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu Y, Yeung N, Sieracki N, Marshall NM. Nature. 2009;460(7257):855–62. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zastrow ML, Peacock AFA, Stuckey JA, Pecoraro VL. Nat Chem. 2012;4:118–123. doi: 10.1038/nchem.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Der BS, Machius M, Miley MJ, Mills JL, Szyperski T, Kuhlman B. J Am Chem Soc. 2012;134:375–385. doi: 10.1021/ja208015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghadiri MR, Case MA. Angew Chem Int Ed. 1993;32:1594–1597. [Google Scholar]
- 47.Lombardi A, Marasco D, Maglio O, Di Costanzo L, Nastri F, Pavone V. Proc Natl Acad Sci U S A. 2000;97:11922–11927. doi: 10.1073/pnas.97.22.11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grzyb J, Xu F, Weiner L, Reijerse EJ, Lubitz W, Nanda V, Noy D. Biochim et Biophys Acta, Bioenerg. 2010;1797:406–413. doi: 10.1016/j.bbabio.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 49.Gibney BR, Mulholland SE, Rabanal F, Dutton PL. Proc Natl Acad Sci U S A. 1996;93:15041–15046. doi: 10.1073/pnas.93.26.15041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laplaza CE, Holm RH. J Am Chem Soc. 2001;123:10255–10264. doi: 10.1021/ja010851m. [DOI] [PubMed] [Google Scholar]
- 51.Shiga D, Funahashi Y, Masuda H, Kikuchi A, Noda M, Uchiyama S, Fukui K, Kanaori K, Tajima K, Takano Y, Nakamura H, Kamei M, Tanaka T. Biochemistry. 2012;51:7901–7907. doi: 10.1021/bi3007884. [DOI] [PubMed] [Google Scholar]
- 52.Kharenko OA, Kennedy DC, Demeler B, Maroney MJ, Ogawa MY. J Am Chem Soc. 2005;127:7678–7679. doi: 10.1021/ja042757m. [DOI] [PubMed] [Google Scholar]
- 53.Xie F, Sutherland DEK, Stillman MJ, Ogawa MY. J Inorg Biochem. 2010;104:261–267. doi: 10.1016/j.jinorgbio.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 54.Zaytsev DV, Morozov VA, Fan J, Zhu X, Mukherjee M, Ni S, Kennedy MA, Ogawa MY. J Inorg Biochem. 2013;119:1–9. doi: 10.1016/j.jinorgbio.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 55.Chino M, Maglio O, Nastri F, Pavone V, DeGrado WF, Lombardi A. Eur J Inorg Chem. 2015;(21):3371–3390. doi: 10.1002/ejic.201500470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maglio O, Nastri F, Martin de Rosales RT, Faiella M, Pavone V, DeGrado WF, Lombardi A. C R Chimie. 2007;10(8):703–720. [Google Scholar]
- 57.Faiella M, Andreozzi C, de Rosales RTM, Pavone V, Maglio O, Nastri F, DeGrado WF, Lombardi A. Nat Chem Biol. 2009;5(12):882–884. doi: 10.1038/nchembio.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chino M, Leone L, Maglio O, Lombardi A. Methods Enzymol. 2016;580:471–499. doi: 10.1016/bs.mie.2016.05.036. [DOI] [PubMed] [Google Scholar]
- 59.Snyder RA, Butch SE, Reig AJ, DeGrado WF, Solomon EI. J Am Chem Soc. 2015;137(29):9302–14. doi: 10.1021/jacs.5b03524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Groom CR, Bruno IJ, Lightfoot MP, Ward SC. Acta Crystallogr, Sect B: Struct Sci, Cryst Eng Mater. 2016;72:171–179. doi: 10.1107/S2052520616003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Geremia S, Di Costanzo L, Randaccio L, Engel DE, Lombardi A, Nastri F, DeGrado WF. J Am Chem Soc. 2005;127(49):17266–17276. doi: 10.1021/ja054199x. [DOI] [PubMed] [Google Scholar]
- 62.Crick FH. Acta Crystallogr. 1953;6:685–689. [Google Scholar]
- 63.Grigoryan G, DeGrado WF. J Mol Biol. 2011;405(4):1079–1100. doi: 10.1016/j.jmb.2010.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ogihara NL, Weiss MS, Degrado WF, Eisenberg D. Protein Sci. 1997;6(1):80–88. doi: 10.1002/pro.5560060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lanci CJ, MacDermaid CM, Kang SG, Acharya R, North B, Yang X, Qiu XJ, DeGrado WF, Saven JG. Proc Natl Acad Sci U S A. 2012;109(19):7304–7309. doi: 10.1073/pnas.1112595109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou J, Grigoryan G. Protein Sci. 2015;24(4):508–524. doi: 10.1002/pro.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yadav MK, Leman LJ, Price DJ, Brooks CL, Stout CD, Ghadiri MR. Biochemistry. 2006;45(14):4463–4473. doi: 10.1021/bi060092q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.DiMaio F, Leaver-Fay A, Bradley P, Baker D, Andre I. Plos One. 2011;6(6):e20450. doi: 10.1371/journal.pone.0020450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Szczepaniak K, Lach G, Bujnicki JM, Dunin-Horkawicz S. J Struct Biol. 2014;188(2):123–133. doi: 10.1016/j.jsb.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 70.Dunin-Horkawicz S, Lupas AN. J Struct Biol. 2010;170(2):226–235. doi: 10.1016/j.jsb.2010.01.023. [DOI] [PubMed] [Google Scholar]
- 71.Fraústo da Silva JJR, Williams RJP. The biological chemistry of the elements: the inorganic chemistry of life. 2nd. Oxford University Press; New York: p. 2001. [Google Scholar]
- 72.Lippard SJ, Berg JM. Principles of bioinorganic chemistry. University Science Books; Mill Valley, California: 1994. [Google Scholar]
- 73.Bond AD, Linton DJ, Wheatley AEH. Acta Crystallogr, Sect E: Struct Rep Online. 2001;57(7):m298–m300. [Google Scholar]
- 74.Kubisiak M, Zelga K, Bury W, Justyniak I, Budny-Godlewski K, Ochal Z, Lewiński J. Chem Sci. 2015;6(5):3102–3108. doi: 10.1039/c5sc00600g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lewiński J, Suwała K, Kubisiak M, Ochal Z, Justyniak I, Lipkowski J. Angewandte Chemie International Edition. 2008;47(41):7888–7891. doi: 10.1002/anie.200803254. [DOI] [PubMed] [Google Scholar]
- 76.Lalioti N, Raptopoulou CP, Terzis A, Aliev AE, Gerothanassis IP, Manessi-Zoupa E, Perlepes SP. Angewandte Chemie International Edition. 2001;40(17):3211–3214. doi: 10.1002/1521-3773(20010903)40:17<3211::AID-ANIE3211>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 77.Nanda V, Schmiedekamp A. Proteins. 2008;70(2):489–497. doi: 10.1002/prot.21537. [DOI] [PubMed] [Google Scholar]
- 78.Lee M, Wang T, Makhlynets OV, Wu Y, Polizzi NF, Wu H, Gosavi PM, Stohr J, Korendovych IV, DeGrado WF, Hong M. Proc Natl Acad Sci U S A. 2017;114(24):6191–6196. doi: 10.1073/pnas.1706179114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Joh NH, Wang T, Bhate MP, Acharya R, Wu YB, Grabe M, Hong M, Grigoryan G, DeGrado WF. Science. 2014;346(6216):1520–1524. doi: 10.1126/science.1261172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fry HC, Lehmann A, Sinks LE, Asselberghs I, Tronin A, Krishnan V, Blasie JK, Clays K, DeGrado WF, Saven JG, Therien MJ. J Am Chem Soc. 2013;135(37):13914–13926. doi: 10.1021/ja4067404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Polizzi NF, Wu Y, Lemmin T, Maxwell AM, Zhang SQ, Rawson J, Beratan DN, Therien MJ, DeGrado WF. Nat Chem. 2017 doi: 10.1038/nchem.2846. 10.1038/nchem.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Suzuki Y, Cardone G, Restrepo D, Zavattieri PD, Baker TS, Tezcan FA. Nature. 2016;533(7603):369–373. doi: 10.1038/nature17633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Otwinowski Z, Minor W. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 84.Kabsch W. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Crystallogr. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Emsley P, Lohkamp B, Scott WG, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.