

Abstract
Clinical trials have been conducted for the neuronal ceroid lipofuscinoses (NCLs), a group of neurodegenerative lysosomal diseases that primarily affect children. Whereas clinical rating systems will evaluate long-term efficacy, biomarkers to measure short-term response to treatment would be extremely valuable. To identify candidate biomarkers, we analyzed autopsy brain and matching CSF samples from controls and three genetically distinct NCLs due to deficiencies in palmitoyl protein thioesterase 1 (CLN1 disease), tripeptidyl peptidase 1 (CLN2 disease), and CLN3 protein (CLN3 disease). Proteomic and biochemical methods were used to analyze lysosomal proteins, and, in general, we find that changes in protein expression compared with control were most similar between CLN2 disease and CLN3 disease. This is consistent with previous observations of biochemical similarities between these diseases. We also conducted unbiased proteomic analyses of CSF and brain using isobaric labeling/quantitative mass spectrometry. Significant alterations in protein expression were identified in each NCL, including reduced STXBP1 in CLN1 disease brain. Given the confounding variable of post-mortem changes, additional validation is required, but this study provides a useful starting set of candidate NCL biomarkers for further evaluation.
Keywords: neuronal ceroid lipofuscinosis, lysosome, autopsy, brain, cerebrospinal fluid, proteomic
Graphical abstract
Introduction
The neuronal ceroid lipofuscinoses (NCLs) are a group of lysosomal storage diseases (LSDs) with a common distinguishing clinical hallmark, the accumulation of the autofluorescent storage material lipofuscin within neurons and other cells of affected individuals.1 The clinical course of NCLs tends to be similar and reflects neurodegeneration with varying ages of onset and rates of progression. Typical manifestations include seizures, loss of vision and locomotor function, progressive mental decline, and premature death. Whereas these diseases are similar from a clinical perspective, they are genetically diverse, and, to date, mutations in 15 different human genes may result in a diagnosis of NCL (http://www.ucl.ac.uk/ncl/mutation.shtml).2 The most frequently encountered and best characterized of the NCLs were originally clinically defined by the phenotypes of patients with null mutations as the classical infantile (CLN1 disease), late-infantile (CLN2 disease), and juvenile (CLN3 disease) forms, which are caused by defects in the genes encoding palmitoyl protein thioesterase 1 (PPT1), tripeptidyl peptidase 1 (TPP1), and CLN3, respectively. For brevity, we refer to these three diseases as CLN1, CLN2, and CLN3 throughout.
Treatment options for the NCLs are reaching clinical trial3–5 with enzyme replacement therapy for CLN2 now approved,6 and thus there is increasing need for methods to measure disease progression and response to treatment. Clinical rating scales have been developed for the NCLs7–9 and these will play an important role, but given the time-frames associated with decline in these diseases (years to decades), these scales represent long-term options. Methods to measure short-term response to treatment should greatly facilitate clinical studies and this need may be fulfilled by robust, informative and clinically accessible biomarkers. There are no such predictive biomarkers for the NCLs at present although a recent study has identified proteins that are significantly altered in plasma.10
We have analyzed autopsy brain and CSF samples from NCL patients and controls using orthogonal biochemical and proteomic approaches to identify potential biomarkers. In one approach, we specifically interrogate the lysosomal proteome with the rationale that LSDs are frequently associated with multiple changes in lysosomal enzyme activities that are secondary to the primary defect. The causes of such changes are generally unclear, but in many cases, they may be compensatory cellular responses to the accumulation of multiple substrates within the lysosome. There are examples of such secondary alterations in the NCLs: In brain, tripeptidyl peptidase 1 activity,11 lysosomal acid phosphatase,12 and other enzymes13 are reported to be elevated in CLN3; β-glucuronidase is elevated in CLN213 and saposins are a major component of the storage material in CLN1.14 Complementing targeted analysis of lysosomal proteins, we have conducted unbiased quantitative mass spectrometry studies on autopsy brain and CSF samples. We identify significant changes that may provide a basis for useful clinical markers and that may also provide important clues to the molecular mechanisms that underlie pathology in these diseases.
Experimental Procedures
Samples
Research protocols involving human subjects were approved by the Institutional Review Board of Rutgers University. Human cortical brain and matching CSF samples from NCL cases were obtained from The Human Brain and Spinal Fluid Resource Center (Los Angeles, CA) using previously described methods.15 In brief, coronal brain slices were cleaned, digitally imaged, frozen in liquid nitrogen, and stored at −80 °C. Postmortem ventricular CSF was drawn, centrifuged, and stored at −80 °C. Average autolysis time was 14.8 h.
Genotype Analysis
Pathogenic mutations (Table 1) were determined by the Neurogenetics DNA Diagnostic Lab of Massachusetts General Hospital. Genomic DNA of each sample extracted using DNeasy kit (Qiagen, Valencia, CA) was PCR-amplified with a specific primer pair of each exon of the corresponding NCL gene. The PCR amplification was carried out using Taq DNA polymerase (Qiagen) and a standard PCR profile comprising an initial hold of 5 min at 95 °C, 30 cycles of (30 s at 95 °C, 30 s at 60 °C, and 45 s at 72 °C), followed by a final extension of 7 min at 72 °C. PCR products were purified using the QIAquick Multiwell PCR Purification System (Qiagen), and purified products were sequenced bidirectionally on an ABI 3500xl capillary gel electrophoresis system (Applied Biosystems, Foster City, CA). The amplified PCR products included the entire exon and the adjacent intronic sequences. Positive mutations were identified by comparison of bidirectional sequence data against normal reference sequence and were further confirmed by independent reamplification and bidirectional sequencing from the original patient DNA.
Table 1. NCL Patient Samples.
| gene defect | identifier | mutation 1 | mutation 2 | age at death (years) | autolysis time |
|---|---|---|---|---|---|
| CLN1 | HSB 3016 | c.223A>C, p.T75P | c.451C>T, p.R151X | 19 | 14.0 |
| CLN1a | HSB 3616 | c.223A>C, p.T75P | c.451C>T, p.R151X | 21 | 20.0 |
| CLN1 | HSB 3672 | c.619C>T, p.Q207X | c.749G>T, p.G250V | 20 | 3.0 |
| CLN2 | HSB 3235 | 3556G>C | 3670C>T, R208X | 6 | 4.5 |
| CLN2 | HSB 3791 | 3556G>C | not found | 5 | 5.7 |
| CLN2 | HSB 4128 | 3670C>T, R208X | 3990A>T, p.D276V | 8 | 7.2 |
| CLN2 | HSB 4133 | 3670C>T, R208X | 4291 4298del | 8 | 10.8 |
| CLN2 | HSB 4134 | 3670C>T, R208X | 4291 4298del | 6 | 14.7 |
| CLN3 | HSB 2549 | Het 1kb del | c.1247A>G, p.D416G | 34 | 11.0 |
| CLN3 | HSB 3187 | Het 1kb del | c.1056+3A>C | 16 | 54.0 |
| CLN3 | HSB 3685 | Het 1kb del | c.49G>T, p.Glu17X | 21 | 28.0 |
| control | HSB 3540 | 68 | 10.5 | ||
| control | HSB 3545 | 80 | 14.0 | ||
| control | HSB 3565 | 76 | 11.0 | ||
| control | HSB 3589 | 53 | 15.0 | ||
| control | HSB 3504† | 80 | 11.0 | ||
| control | HSB 3543† | 73 | 12.0 | ||
| control | HSB 3558† | 59 | 19.5 |
Note that sample HSB# 3616 was originally listed as CLN2 based on clinical criteria, but enzyme, proteomic, and genetic analyses all indicated a PPT1 deficiency. This case therefore represents CLN1. Brain samples were available for all cases, and CSF was available for all except those indicated by.
Note that survival of CLN1 patients was longer than typically expected (mean survival ∼10 years) due to the presence of hypomorphic mutations T75P and G250 V.
Purification and Analysis of Man6P Glycoproteins
Frozen brain samples were powderized using a Bessman tissue pulverizer and homogenized, and proteins containing Man6P were isolated by affinity purification using bovine soluble cation-independent Man6P receptor (sCI-MPR) and prepared for mass spectrometry analysis as described.16 In brief, cleared homogenates were applied to columns of immobilized sCI-MPR, which were washed, “mock” eluted with glucose 6-phosphate/mannose to determine specificity of protein interaction with sCI-MPR,17 then eluted with Man6P to elute glycoproteins containing Man6P phosphorylated glycans. Five micrograms of purified Man6P glycoprotein preparations was processed for mass spectrometry, and we analyzed an equivalent proportion of the total eluate of the corresponding glucose-6 phosphate/mannose eluates (e.g., if 5 μg represented 10% of the total purified Man6P eluate, then we would also analyze 10% of the corresponding glucose-6 phosphate/mannose eluate). For each purified sample, we performed two independent tryptic digests. Data for each sample were obtained from six individual LC–MS runs, with four runs for the first digest and two runs for the second.
Mass Spectrometry
Samples were routinely digested with trypsin (specificity: Arg and Lys). Preparations of purified Man6P glycoproteins and their corresponding mock eluates were analyzed using a Thermo Orbitrap Velos and analyzed, as previously described in detail.16 iTRAQ 8-plex labeling was conducted using manufacturer's (Thermo Scientific) methods. Samples were fractionated by alkaline RP-HPLC, and ∼20 fractions were analyzed in duplicate by HCD on a Thermo Orbitrap Velos using column and gradient conditions as described.16 For each cycle, one full MS was scanned in the Orbitrap with resolution of 60 000 from 300 to 2000 m/z and the 10 most intense peaks fragmented by HCD using a normalized collision energy of 38% and scanned in the Orbitrap from 110 to 2000 m/z with resolution of 7500.
Generation of Peak Lists
Peak lists were generated using Proteome Discoverer 1.4 with minimum and maximum precursor masses of 350 Da and 10 000, respectively, minimum signal/noise of 1.5, and no constraints with respect to retention time, charge state, or peak count.
Database Search
A local implementation of the Global Proteome Machine18,19 (GPM) Cyclone XE (Beavis Informatics, Winnipeg, Canada) with X!Tandem version CYCLONE (2013.09.11) was used to search mass spectrometry data against the human genome assembly GRCh37.70 (Aug 2012; 56 680 total genes, 20 447 protein coding genes).
For spectral count analysis, the following search parameters were used. For protein identification and spectral counting: fragment mass error, 0.4 Da; parent mass error, 10 ppm; maximum charge, +4; one missed cleavage allowed; cysteine carbamidomethylation was a constant modification and methionine mono-oxidation was a variable modification throughout the search; tryptophan mono- and dioxidation, methionine dioxidation, asparagine deamidation, and glutamine deamidation were variable modifications during model refinement.
For iTRAQ analysis: fragment mass error, 20 ppm; parent mass error, 10 ppm; maximum charge, +4; minimum 15 peaks assigned; one missed cleavage allowed; cysteine carbamidomethylation, iTRAQ8 at lysine and N-terminus were constant modifications, methionine oxidation was a variable modification throughout the search; tryptophan mono- and dioxidation, methionine dioxidation, asparagine deamidation, glutamine deamidation, iTRAQ8 at tyrosine, and minus iTRAQ8 at lysine and N-terminus were variable modifications during model refinement.
MS data files from brain and CSF experiments were analyzed together in a MudPit analysis. Ensembl protein identifiers were converted to associated gene names to collapse multiple protein identifiers that can be assigned to the same gene. Assignments were filtered for a log GPM expectation score of −10 or better and a minimum of two unique peptides. Note that there is a small number of proteins that are labeled as “acceptable gene product assignments” with a single unique peptide. These represent cases in which a protein identifier with a single unique peptide maps, together with other protein identifiers, to a gene identifier that is represented by two or more unique peptides in total. False-positive rates for peptide identification generated by X!Tandem were 0.07 and 0.1% for the iTRAQ and spectral count studies, respectively.
Deposition of MS Data
Raw files, mgf files, GPM result files, Excel workbooks listing protein and peptide assignments, and keys for data interpretation are archived in the MassIVE (http://massive.ucsd.edu) and ProteomeXchange (http://www.proteomexchange.org/) repositories: MSV000081140, spectral count analysis of purified Man6P glycoproteins; MSV000081143, iTRAQ8 analysis of brain extracts and CSF. Sample information is listed in Table S1.
Normalization of iTRAQ Data
Total iTRAQ-8 reporter ion intensities were extracted from mgf files and corrected for crossover using custom scripts (https://github.com/cgermain/IDEAA). Note that using iTRAQ-8plex for the analysis of 14 samples required two separate experiments for each analyte (brain or CSF). With seven samples in each experiment, the remaining unused channel was used for an internal reference comprising a pool derived from all 14 samples for each analyte. Data were normalized in a two-step procedure. First, for each individual spectrum, total intensity for each reporter ion channel was normalized to the average reporter ion intensity for all assigned spectra for each sample for each experiment. This corrects for differences in labeling efficiency or the total amounts of peptide labeled with each iTRAQ reagent. Second, to allow comparison of data between experiments, ion intensities for each spectrum for each individual channel were normalized to the internal reference channel.
Statistical Analysis
Spectral Count Analysis
Protein assignments were filtered for a minimum of two unique peptides, a minimum protein log(e) score of −5, and a minimum average of 10 spectral counts for protein per sample. There were three major considerations in the analysis of spectral count data for purified Man6P glycoproteins. First, there were unequal numbers of samples per group (n = 7 for control samples, n = 3 for CLN1, n = 5 for CLN2, and n = 3 for CLN3). Second, whereas nominally equivalent amounts of each purified Man6P glycoprotein preparation were analyzed by MS, selective losses during digestion and peptide extraction result in variable total spectral counts for each sample. Third, because there are several observations in each group, we need to account for the person-to-person variation within each group as well as the inherent variability in the count data. To account for these factors, we first fitted (for each protein) a binomial (logistic regression) model to the spectral counts, with the total counts across proteins as a denominator and group indicators as a basis for comparison. To adjust for the count-to-count variability, we incorporated into the binomial model an overdispersion parameter.20 We used a base 2 logit link so that the coefficient estimates represent doubling factors of mean counts between comparison groups.
Isobaric Labeling Analysis
The goal of this analysis was to determine biological differences between control and disease samples. To reduce variability arising from sample preparation, in addition to normalizing iTRAQ data as described above, peptides were filtered for fully tryptic cleavage with no missed sites, complete iTRAQ labeling of lysines and amino termini, and a lack of posttranslational modifications deemed to increase variability (asparagine or glutamine deamidation, methionine dioxidation, tryptophan mono- and dioxidation, and isobaric labeling of tyrosine at positions other than the amino terminus). The large number of biological samples (14 NCL cases and controls) were randomly assigned to two separate iTRAQ labeling experiments. Thus for both analytes (brain and CSF), we conducted four individual experiments. For each, we compared data between the two respective experiments using a standard reference sample derived from a pool of all of the samples analyzed in both experiments. To ensure that data were acquired equally for all of the biological samples, peptide data were filtered to include only those proteins that were represented by one or more spectra in both experiments conducted for each analyte. We calculated errors for relative expression of individual proteins in each NCL (Figures 6 and 10 and Figure S3) using a nested analysis that accounted for variability at both the peptide and protein level as follows. For proteins with more than one peptide, a random effects model was fitted for “level” with “peptide” as a random effect and a common variance across all groups. When there was only one peptide, a linear model was used instead of a random effects model. P values were calculated using degrees of freedom based on Satterthwaite's approximation, as implemented in the “lmerTest” R package and corrected for multiple comparisons using the Bonferroni method.
Figure 6.
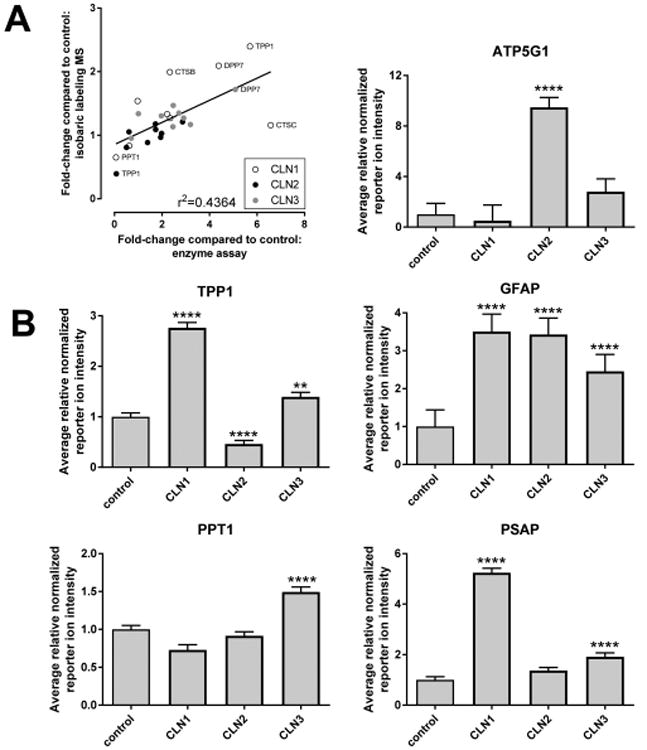
Validation of isobaric-label MS quantitation. (A) Comparison of lysosomal protein expression and activity determined by isobaric labeling/quantitative MS and enzyme assay, respectively. (B) Expression levels of proteins that are known to be altered in NCL diseases. For each NCL, the average relative normalized reporter ion intensity for all samples is shown and error bars indicate standard error. Data are expressed relative to control. Ratios, errors, and significance compared with control were calculated as described in the Experimental Procedures section. *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001.
Figure 10.
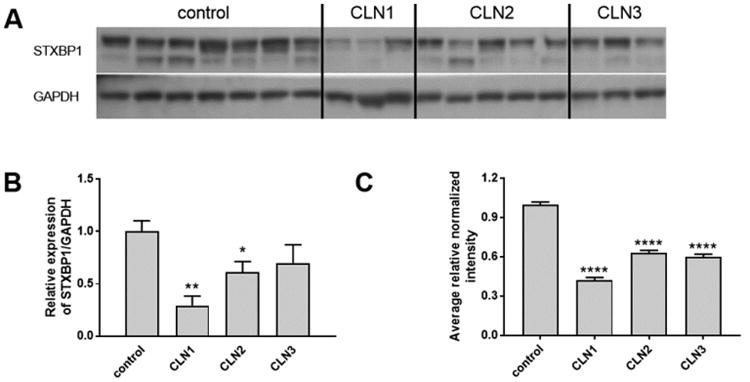
Western blot analysis of STXBP1. (A) Western blot of STXBP1 and GAPDH loading control in brain extracts. (B) Quantitation of Western blot. Data were normalized to GAPDH and are expressed relative to control and error bars show SEM. Significance compared with control was calculated by one-way ANOVA using Dunnett's multiple comparison test. (C) Quantitative MS data for STXBP1. Data are expressed relative to control. Ratios, errors, and significance compared with control were calculated as described in the Experimental Procedures section. *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001.
Enzyme Assays
Enzyme activities were measured as described previously.21–24
Western Blotting
To detect Man6P glycoproteins, protein extracts were fractionated on 10% acrylamide NuPAGE gels using MOPS running buffer (Invitrogen), transferred to nitrocellulose membrane, and probed with 125I-labeled sCI-MPR receptor as described previously.17 Subunit c of mitochondrial ATP synthase (SCMAS) was detected by Western blotting of protein extracts fractionated by electrophoresis on 10% polyacrylamide NuPAGE gels with MES buffer using an affinity-purified rabbit polyclonal antipeptide primary25 and AlexaFluor 488-labeled goat antirabbit secondary antibodies. STXBP1 was detected by Western blotting of brain extracts fractionated by electrophoresis on 10% polyacrylamide NuPAGE gels with MOPS buffer followed by transfer on a PVDF membrane and incubation with rabbit polyclonal STXBP1 primary antibody (ThermoFisher) and AlexaFluor 633-labeled antirabbit secondary antibodies (ThermoFisher).
Gel Quantitation
Radioactive or fluorescent signals in blots were detected by using a Typhoon scanner (GE Healthcare) and quantified by using ImageQuant 5.2 (Molecular Dynamics).
Results and Discussion
Experimental Strategy
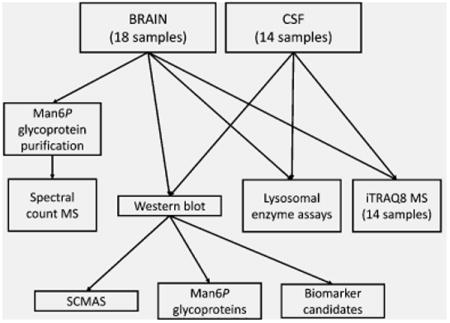
Our overall experimental approach is summarized in Figure 1. We analyzed 14 sets of matched human brain and CSF autopsy specimens from NCL patients and unaffected controls as well as four additional control brain specimens. Note that there is various nomenclature for the NCL diseases including infantile, late-infantile, and juvenile NCL for PPT1, TPP1, and CLN3 deficiencies, respectively. We have chosen to avoid clinical-based nomenclature given the overlap in presentation between the diseases, especially where hypomorphic mutations result in late-onset, slowly progressing phenotypes. Instead, we use CLN1, CLN2, and CLN3 throughout to refer to PPT1, TPP1, and CLN3 deficiencies, respectively. Specimens represented controls and cases diagnosed with NCL based on clinical criteria (Table 1). While information regarding individual clinical progression was not available, note that patients with each CLN2 and CLN3 disease died at ages that were consistent with typical clinical expression rather than late onset/slow progression seen in patients with hypomorphic mutations. The two CLN1 patients studied survived for longer than is typical with this disease (∼10 years), which is consistent with the presence of hypomorphic mutations T75P and G250V in the gene encoding PPT1.26 HSB# 3616 was obtained with a diagnosis of CLN2, but enzyme assay and proteomic analysis of purified Man6P glycoproteins revealed normal levels of TPP1 but deficiencies in PPT1. Genetic analysis revealed mutations in PPT1, and thus this case was reclassified as CLN1 (Table 1). All NCL gene mutations were reported previously. (http://www.ucl.ac.uk/ncl/mutation.shtml), except for the PPT1 Q207X nonsense mutation. Note that there is variability in autolysis time between the samples (Table 1). This is inherent to autopsy samples and cannot be avoided: Moreover, analysis indicates that, other than a marginally significant difference in autolysis time between CLN2 and CLN3 (p = 0.024), we find no significant differences between the groups that could potentially influence data (Figure S1A).
Figure 1.
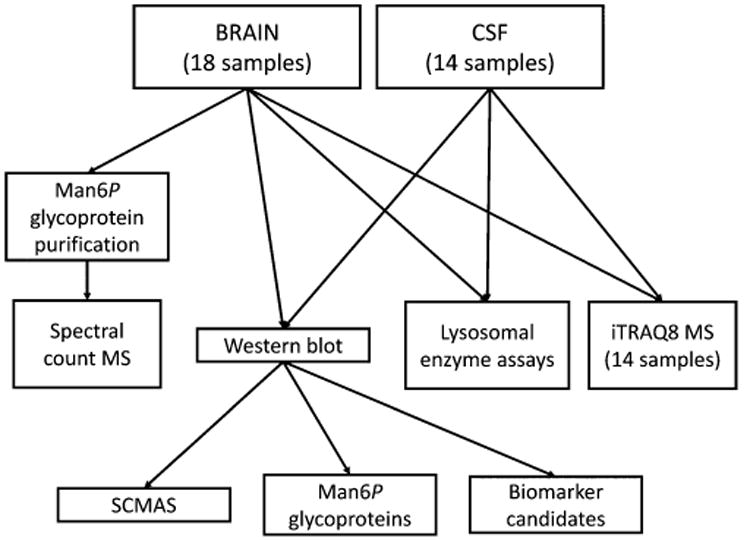
Experimental strategy.
Our goal was to identify pathophysiological changes associated with brain disease that are specific to or shared by three different NCLs as well as to identify potential biomarkers that could be exploited clinically. We initially focused on lysosomal enzymes because these are known to be altered in many LSDs. Lysosomal proteins are normally intracellular but also are secreted into the CSF.27 In addition to assaying a wide range of lysosomal activities, we also measured the spectrum of glycoproteins that contain Man6P, a modification used for targeting soluble lysosomal proteins to the lysosome. For the latter, we conducted SDS-PAGE and blotting for both brain and CSF, and for brain, where there was sufficient material available, we also analyzed affinity-purified Man6P glycoproteins by LC–MS/MS. We also analyzed SCMAS, a known storage material in several NCLs. Finally, we conducted a broad proteomic survey of both brain and CSF using isobaric labeling and multidimensional LC–MS/MS.
Because these are diseases of childhood, it is not possible to obtain autopsy samples from age-matched, healthy control individuals. As a result, there is a significant difference in ages between the controls and NCL cases (Figure S1B). For NCL-specific changes, our inability to age-match controls does not affect conclusions because comparisons are drawn not only against the controls but also against the other NCL diseases. However, when we consider protein expression changes that are detected in all three diseases, we cannot exclude systematic age-related changes. Given that there is a compelling clinical need for biomarkers in these diseases and that there has been no comprehensive proteomic study of the NCLs to date, we decided to proceed using the available control cases. However, this is an important caveat, and as a result, the data set outlined here should be regarded as an initial starting resource for further validation studies in clinical cohorts or animal models.
Lysosomal Enzyme Activities
In an initial analysis of lysosomal changes in the NCLs, we measured the activities of a range of lysosomal enzymes in NCL cases and unaffected controls in brain and CSF (Figure 2). The rationale was that lysosomal alterations observed in brain that are secondary to the primary defect may translate to the CSF.
Figure 2.
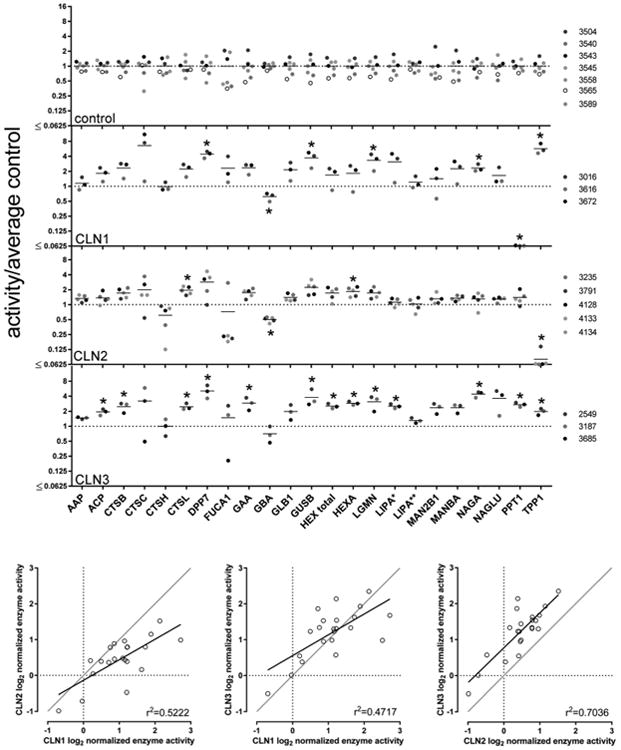
Relative lysosomal activities in NCL brain autopsy samples. Upper panel: Fold change is the average enzyme activity for each NCL normalized to the average control activity. Asterisks indicate significantly different from control (p ≤ (0.05/23), that is, Bonferroni correction for false discovery rate from multiple comparisons). Gene names, enzyme name, and substrate were as follows: AAP, alanine aminopeptidase (a nonlysosomal control activity), Ala-AMC; ACP, total acid phosphatase (ACP2 and ACP5), 4MU-phosphate; CTSB, cathepsin B, Z-Arg-Arg-AMC; CTSC, cathepsin C/dipeptidyl peptidase 1, Gly-Arg-AMC; CTSH, cathepsin H, Arg-AMC; CTSL, cathepsin L, Z-Phe-Arg-AMC-(Z-Phe-Arg-AMC + Z-Phe-Phe-CHN2); DPP7, dipeptidylpeptidase 2, Lys-Ala-AMC; FUCA1, α-fucosidase, 4MU-alpha-fucoside; GAA, α-glucosidase, 4MU-alpha-glucoside; GBA, glucocerebrosidase, 4MU-beta-glucoside; GLB1, β-galactosidase, 4MU-beta-galactoside; GUSB, β-glucuronidase, 4MU-beta-glucuronide; HEX total, hexosaminidase total, 4MU-2-acetamido-2-deoxy-beta-d-glucopiranoside; HEXA, hexosaminidase A, 4MU-2-acetamido-2-deoxy-6-sulfo-beta-d-glucopiranoside; LGMN, legumain, Z-Ala-Ala-Asn-AMC; LIPA, acid lipase, 4MU-oleate* or 4MU-palmitate**; MAN2B1, α-mannosidase, 4MU-alpha-mannoside; MANBA, β-mannosidase, 4MU-beta-mannoside; NAGA, α-N-acetylgalactosaminidase, 4-MU-2-acetamido-2-deoxy-alpha-d-galactopiranoside; NAGLU, α-N-acetylglucosaminidase, 4MU-alpha-N-acetyl-glucosaminide; PPT1, palmitoyl protein thioesterase 1, 4MU-6-thiopalmitoyl-8-glucoside; TPP1, tripeptidylpeptidase 1, Ala-Ala-Phe-AMC. Lower panel: Correlation between average activity in each NCL normalized to the average control activity. Black line, linear regression of log2-transformed enzyme assay data; gray line, x = y. Note that TPP1 and PPT1 are excluded from correlation plots.
In brain, the most marked changes were observed with the CLN3 samples, where 13 of the 23 measured lysosomal enzymes were >1.5 fold higher than average control that was statistically significant (i.e., P ≤ 0.05 with Bonferroni correction for multiple comparisons). These included PPT1 and TPP1, which were elevated 2.7 and 2.0 times, respectively, while DPP7, NAGA, and NAGLU were elevated >3.5-fold. While there were fewer enzymes elevated in CLN1 (five significantly elevated >1.5-fold) compared with CLN3, activities that were increased were elevated to a greater magnitude (on average 3.9-fold compared to control), including TPP1, which was increased 5.7 fold. As expected, PPT1 was extremely low, and GBA was significantly decreased. There were relatively few changes that reached statistical significance in CLN2, but GBA was significantly decreased and TPP1 was absent, as expected, while CTSL and HEXA were both elevated ∼2-fold. AAP (alanine aminopeptidase), a nonlysosomal control, was unaltered in NCL samples compared with controls. Changes in enzyme activities tended to be similar between the different NCLs (Figure 2) with the greatest correlation found between CLN2 and CLN3 (r2 = 0.7036). In general, GBA was consistently decreased, whereas CTSC and DPP7 were elevated.
Some lysosomal activities were detectable in the patient-matched CSF samples (Figure S2), but no significant changes were detected in NCL samples compared with controls. There was considerable sample-to-sample variability in enzyme assay data from CSF (Figure S2), probably reflecting differences in enzyme stability under different collection and processing conditions as well as with the duration and type of storage. Thus we have used additional approaches to measure the amounts rather than activities of proteins in NCL and control samples.
Subunit c of Mitochondrial ATP Synthase (SCMAS)
SCMAS is a small hydrophobic protein that is a major component of the storage material in CLN2, CLN3, and other NCLs, but not CLN1.28,29 We investigated whether SCMAS might provide a useful readout on disease given that its storage can be reduced in brain in an NCL animal model by effective treatment.30,31 As expected, immunoblotting of extracts from the autopsy brain samples (Figure 3) indicated that SCMAS was slightly elevated in CLN3 but markedly and consistently increased in CLN2. In CLN1, SCMAS appeared to be somewhat decreased compared with controls, but this may reflect the age difference between the controls and CLN1 patients (Table 1).
Figure 3.
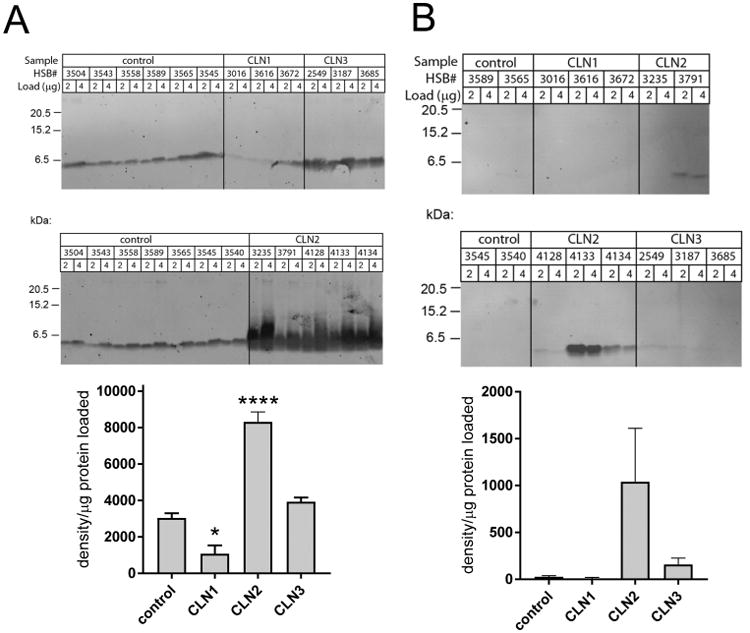
SCMAS in the neuronal ceroid lipofuscinoses. SCMAS detected by Western blotting in brain (A) or CSF (B). In quantitative graphs, the average of duplicate determinations is shown for each sample and error bars indicate SEM. Significance compared with control was measured using one-way ANOVA with p values corrected using Dunnett's multiple comparison test. *, p < 0.05; ****, p < 0.001.
SCMAS is detectable in the urine of NCL patients,32,33 and its presence in this extracellular fluid suggested that it could be similarly detectable in CSF. By Western blotting, SCMAS was not detectable in CSF samples from normal controls or CLN1 (Figure 3) but was clearly detectable in four out of five CLN2 samples. However, there was considerable sample-to-sample variation that did not correspond with the levels that were detected in brain (Figure 3). For example, SCMAS was strikingly elevated in CSF from CLN2 HSB#4133 but undetectable in HSB#3235. Overall, SCMAS may be a good candidate CSF biomarker for follow-up studies in patients.
Man6P Glycoproteins
Most newly synthesized soluble lysosomal hydrolases and accessory proteins contain Man6P, a specific carbohydrate modification that is recognized by Man6P receptors (MPRs) that target these proteins to the lysosome. Purified sCI-MPR can be used for the visualization34 or affinity isolation35,36 of Man6P containing proteins.
We conducted blotting experiments to measure Man6P glycoprotein levels in brain samples from the various NCL samples and controls (Figure 4A). The purpose of this experiment was two-fold. First, this analysis allowed us to determine whether there were any gross alterations in the expression of Man6P glycoproteins and hence lysosomal proteins, associated with disease. Second, this allowed us to determine the relative levels of Man6P glycoproteins in each sample, which provides useful information in the interpretation of proteomic analyses (see below). In an previous study,13 we found characteristic changes in the expression of Man6P glycoproteins in brain samples from various NCL cases.
Figure 4.
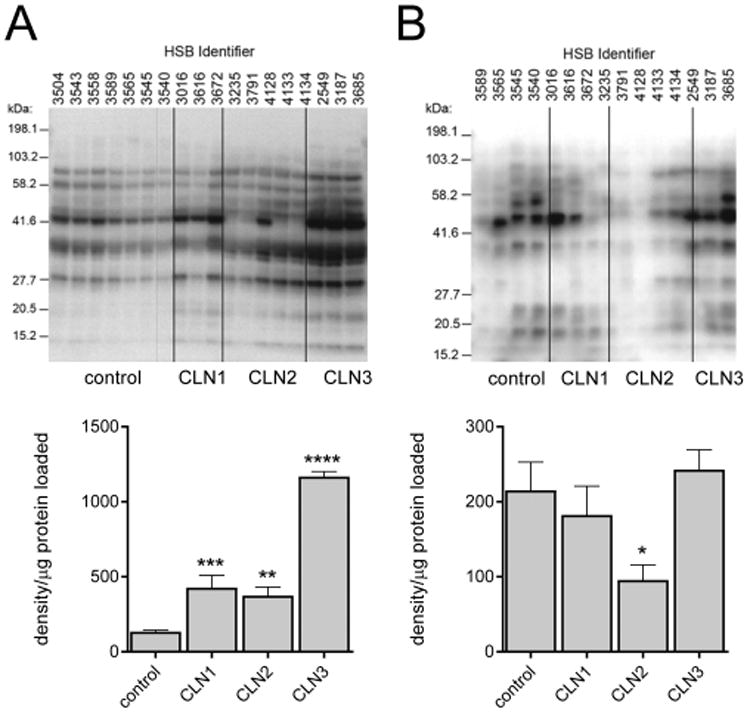
Man6P glycoproteins in the neuronal ceroid lipofuscinoses. Man6P glycoproteins in brain (A) or CSF (B). In quantitative graphs, the average of duplicate determinations is shown for each sample and error bars indicate SEM. Significance compared with control was measured using one-way ANOVA with p values corrected using Dunnett's multiple comparison test. *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001.
In terms of total levels of Man6P glycoproteins, we find a ∼3-fold increase in the CLN1 and CLN2 samples and ∼9-fold increase in the CLN3 samples compared with control (Figure 4A), which is consistent with previous results.13 When the blots are probed in the presence of free Man6P, little signal is detected, confirming that each band represented a Man6P-containing glycoprotein (data not shown13). In most of the CLN2 samples, a prominent band of ∼45 kDa was missing, which corresponds to the mature processed form of TPP1.37,38 In CLN2 case HSB#4128, significant levels of the band corresponding to TPP1 were observed. Presumably, this reflects the expression of stable but inactive protein from the allele containing the missense mutation Asp276Val.
Man6P glycoproteins are present in CSF at low but detectable levels (Figure 4B). There were no significant differences among the control, CLN1, and CLN3 groups. Total levels in the CLN2 samples appeared somewhat diminished, but this likely reflects loss of the prominent band corresponding to TPP1. Overall, in the analysis of either brain or CSF, there were no striking qualitative differences in the patterns of Man6P glycoproteins detected in the NCL samples compared with controls, with the exception of the band corresponding to TPP1 in most CLN2 samples.
Relative Quantitation of Lysosomal Man6P Glycoproteins in NCL Samples
We purified Man6P-containing glycoproteins from the brain samples and determined relative expression levels in NCL and control cases by spectral count analysis of mass spectrometry data. It is worth noting that we analyzed equal amounts of purified Man6P glycoproteins from each sample and thus measured relative rather than absolute amounts. For example, total levels of brain Man6P glycoproteins appear to be elevated in the NCL samples compared with controls (Figure 4A), although we cannot discount the possibility that there may be differences in the affinity of individual proteins for the radiolabeled sCI-MPR probe. We do not attempt to correct for such potential differences; therefore, our approach is conservative and likely underestimates the magnitude of absolute changes. However, the approach should detect proteins that are selectively enriched or depleted in the purified mixture.
Brain extracts were applied to a column of immobilized sCI-MPR, which was then washed, mock-eluted with mannose and glucose 6-phosphate, and then specifically eluted with Man6P. Quantitative comparison of protein levels in the mock versus specific eluate provides a useful measure of the specificity of purification, helping to distinguish true Man6P glycoproteins from contaminants.16,39 This was achieved for each protein by calculating the ratio of counts in the specific versus mock eluates and the upper and lower 95% confidence indices for the ratio (Table S2). In aggregate analysis of all samples, 62 known lysosomal Man6P glycoproteins met our criteria for protein assignment (Table S2), and all were significantly elevated in the specific versus mock eluate, with a minimum enrichment (i.e., a lower 95% confidence index) of ∼3.2 for CTSH. In addition, 128 other proteins were also identified that were significantly elevated in the specific versus mock eluates. Of these, 83 proteins were enriched to the same or greater extent than the lowest observed for a known Man6P lysosomal protein (CTSH). These proteins may represent novel human Man6P glycoproteins that may or may not have lysosomal function. Alternatively, they may be contaminants or other proteins that copurify in association with true Man6P glycoproteins. For example, cytosolic protease inhibitors (e.g., cystatins) may bind to lysosomal proteases during preparation of sample homogenates and specifically copurify with these bona fide Man6P glycoproteins.
Significant alterations in the relative levels of known lysosomal Man6P glycoproteins were observed in the preparations from all three NCL diseases (Table S3, Figure 5A). Most changes were observed in CLN1, with elevated NPC2 and decreased PLD3 being particularly marked. In CLN2, and CLN3, there were fewer alterations, and these were typically of a lower magnitude than observed in CLN1, although there was a large decrease in NAAA (N-acylethanol-amine-hydrolyzing acid amidase) in CLN2. Some changes were observed in more than one NCL (Figure 5B). For example, GUSB was elevated in all, whereas HEXA, ARSA, SGSH, and NAAA were all altered in both CLN2 and CLN3. Other than GUSB, no significant changes found in CLN1 were found in CLN2 or CLN3. (ASAH1 was altered in both CLN1 and CLN2 but elevated in the former and decreased in the latter.) This suggests that there may be similarities between CLN2 and CLN3 diseases at the lysosomal protein level, and this was supported when average changes were compared between the different diseases (Figure 5C). There is little or no correlation between changes in CLN1 and either CLN2 (r2 = 0.0225) or CLN3 (r2 = 0.2110), while changes in lysosomal protein levels were better correlated between CLN2 and CLN3 (r2 = 0.5589).
Figure 5.
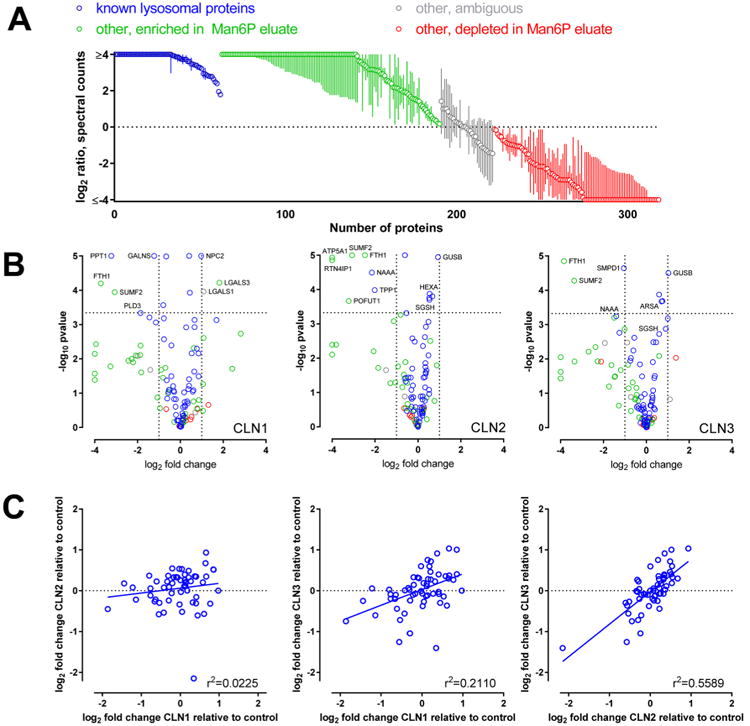
Identification and quantitation of proteins identified in Man6P preparations from human brain. Man6P glycoproteins were isolated from NCL and unaffected control brain samples, analyzed by LC–MS/MS and relative quantitation conducted by spectral counting. (A) Point estimate and 95% confidence limits for ratio of spectral counts in Man6P eluate/mock eluates, combining spectral count data from all samples. Analysis was restricted to proteins with a minimum sum of 10 spectral counts (Table S3). (B) Volcano plots for enriched proteins. Fold change is the ratio of average NCL to average control after normalization for total spectral counts per sample. Dashed line indicates threshold for significance using Bonferroni correction for multiple comparisons (i.e., 0.05/total number of proteins measured). Proteins are filtered for a minimum of 10 spectral counts per sample and contaminants are excluded. (C) correlation between changes in relative levels of known lysosomal Man6P glycoproteins between the three NCLs.
We compared quantitative data for lysosomal proteins measured by enzyme assay with spectral counting in purified Man6P glycoprotein preparations (Figure S3). Changes compared with controls tended to be of a lower magnitude when measured by enzyme assay, but there was some correlation between data obtained using the different methods, with r2 values of 0.319, 0.225, and 0.353 for CLN1, CLN2, and CLN3 respectively. It is not clear why the correlations between activity and protein measurements in the three NCLs are so modest, but one possibility is that this may reflect changes in cellularity in the NCL brain, where gliosis accompanies neurodegeneration. Note that in most cell types other than neurons, the Man6P marker is rapidly removed from lysosomal proteins by acid phosphatase 540 after targeting to the lysosome, so total activity of a given lysosomal protein may not necessarily correlate with the amount of the Man6P-containing form, even if both forms have the same specific activity.
In addition to lysosomal proteins and other known Man6P glycoproteins, the affinity purified preparations contain numerous other proteins that have not been reported to contain Man6P that are specifically eluted from the affinity column with Man6P. In CLN2, ATP5A1 and RTN4IP1 are greatly decreased or absent compared with control. However, when we measure ATP5A1 in homogenates from human brain samples, we do not detect any significant decrease in total amount of this protein (Figure S4). It is possible that ATP5A1 or RTN4IP1 copurify in association with TPP1 and hence their absence in the Man6P glycoprotein preparations from the CLN2 samples. SUMF2 (sulfatase-modifying factor 2) and FTH1 (ferritin) were decreased in Man6P glycoprotein preparations from all three NCLs. SUMF2 was previously shown to contain Man6P.41
Validation of Quantitative Proteomic Analysis of Brain Extracts and CSF
Complementing the focused analysis of lysosomal proteins isolated from brain samples, we also analyzed total proteins in brain and CSF samples using isobaric labeling, followed by 2D LC–MS/MS. The complete data set is shown in Table S4. Results were validated in two ways.
First, we compared quantitative data obtained from enzyme assay with protein measurements obtained by isobaric labeling using proteins that were common to both data sets. We find correlation (r2 = 0.4364) with determinations made using the two different methods (Figure 6A). However, it is worth noting that the magnitude of isobaric labeling measurements (increase or decrease) was less than observed with enzyme assay. While activity does not always reflect protein levels (e.g., an enzymatically inactive mutant protein may be stable and detectable by MS, for example, see comments regarding TPP1 in sample HSB# 4128), the lower magnitude of change measured from the MS data likely arises from ratio compression associated with iTRAQ and other isobaric labeling approaches.42 Given the complexity of both samples examined here (whole brain extracts and CSF), we investigated the degree of ratio compression by including a bacterial standard (DrR57) added at different amounts to each sample (Figure S5). For both brain and CSF, there is a strong linear relationship for measured intensities, but there is clearly some ratio compression (note the divergence of linear regression plots for brain and CSF from the ideal). Taken together, this indicates that isobaric-label MS can detect changes in protein abundance, but the magnitudes of change (increase or decrease) are likely to be underestimates.
Second, in addition to the individual defective proteins in each NCL (PPT1, TPP1, and CLN3 in CLN1, CLN2, and CLN3, respectively), there are several proteins that previous studies have demonstrated to be elevated or decreased in brain. As a further validation of the quantitative MS, levels of each of these proteins have been evaluated individually in the control and NCL samples. For TPP1 (Figure 6B), levels were reduced in the CLN2 samples as expected. However, it is worth noting that in one sample (HSB# 4128) levels of TPP1 were not diminished to the same extent as the other samples. This is consistent with blotting (Figure 4A) where a Man6P glycoprotein corresponding to TPP1 was retained in this case. TPP1 was elevated in CLN3 (Figure 6B), as previously reported in a mouse model.11 PPT1 was reduced in CLN1 (Figure 6B) but only to ∼70% of levels detected in control samples. This again may reflect the synthesis of an inactive but stable protein from one of the pathogenic alleles (T75P). As previously demonstrated (Figure 3), as a major component of storage material, ATP5G1 was highly elevated in CLN2 and also in CLN3 albeit to a lesser degree (Figure 6B). PSAP is a known component of the storage material in CLN114 and is highly elevated compared with control samples when quantified by MS (Figure 6B). Although not a component of storage material but instead reflecting glial activation, GFAP is elevated in CLN243 and other NCLs, and, consistent with this, quantitative MS analysis indicates that levels in the NCL samples are elevated compared with controls. Taken together, the strong correlation between quantitative MS and functional assay data and the detection of specific changes in brain that are predicted from previous studies provide strong validation of the MS-based approach and provide confidence in the interpretation of previously unrecognized protein expression changes in NCL samples.
Disease Specificity of Altered Protein Expression
In Figure 7, protein expression in brain and CSF samples was analyzed using volcano plots, a scatter plot to compare magnitude of change with probability of significance. It is interesting to note that in each disease compared with controls, levels of known lysosomal proteins are not particularly elevated compared to other proteins, with the exception of PSAP in CLN1. For both brain and CSF, there is a strong correlation between changes in protein levels relative to controls for each of the three NCLs (Figure 8).
Figure 7.
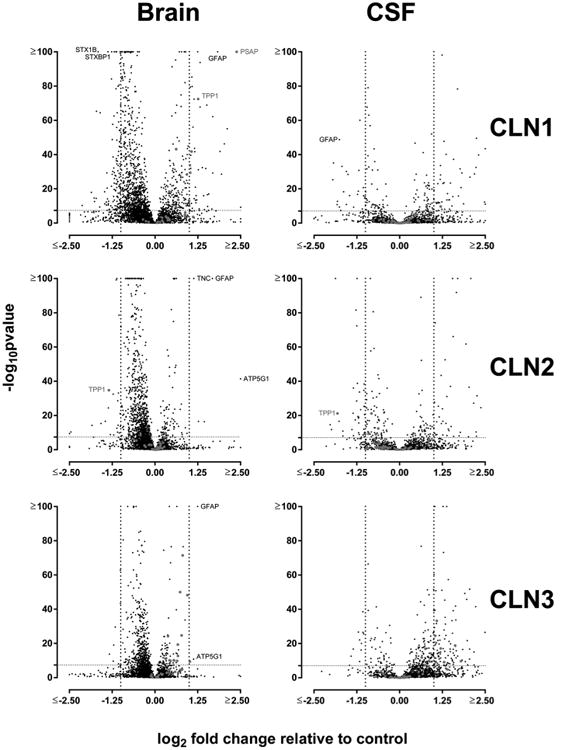
Visualization of protein expression levels compared with controls. Dashed lines on the x axis indicate limits of arbitrary two-fold difference and on y axis indicate statistical significance when accounting for false discovery using Bonferroni correction. Known lysosomal proteins are indicated in red.
Figure 8.
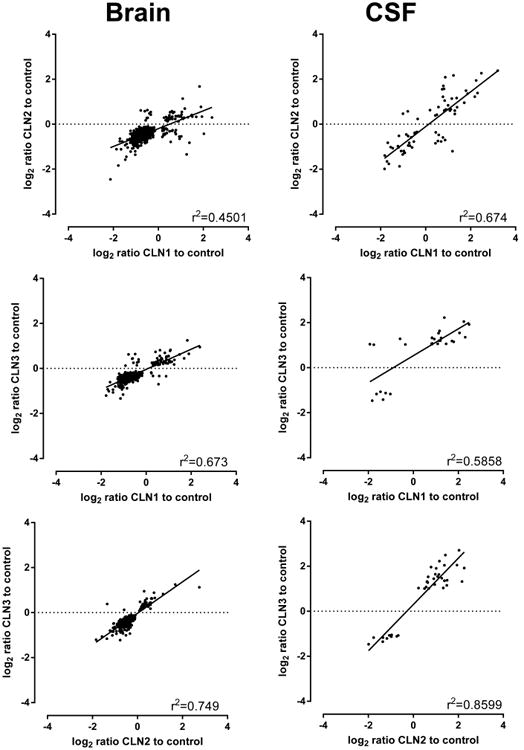
Correlation of significant changes in brain and CSF. For proteins that are significantly altered in both NCLs in pairwise combinations, correlation coefficients are determined when magnitude of changes are compared.
Proteins That Are Significantly Altered in All Three NCL Diseases
When proteins that are significantly altered are compared in the three NCL diseases (Figure 9A), we found significant overlap in brain, and, to a lesser degree, CSF. In brain, out of a total of 2553 significantly altered proteins in one or more of the three diseases, 392 (∼15%) were altered in all three NCLs. In CSF, out of a total of 469 significantly altered proteins in any of the three diseases, 18 (∼4%) were altered in all three NCLs. To investigate whether this overlap was biologically significant or whether it simply represented a stochastic intersection of the data sets reflecting the large number of proteins analyzed, we compared the direction and magnitude of changes between the three NCLs in CSF (Figure 9B). We found that the magnitude of changes for the subset of proteins that were altered in all three NCL diseases was extremely well correlated in both brain and CSF: When the magnitude of change in CSF is examined for individual proteins, all except one of the 18 common proteins are altered in a similar direction (i.e., increased or decreased) and to a similar magnitude. These data are consistent with the overall correlations detected between the NCLs (Figure 8) and indicate that proteins that are altered in all three NCLs likely reflect a uniform response to neurodegeneration, which may be NCL-specific or may more broadly reflect neurological disease. As previously discussed, it is also possible that changes observed in all three NCLs compared with controls reflect the inability to age-match control samples. Most (17/18) of such proteins were not identified in previous studies of age-dependent changes in the CSF proteome.44,45 The one protein that was reported to show age-related changes in CSF expression (KLK6) was actually decreased with normal aging, while our data indicate that it is higher in the older controls than younger NCL cases.
Figure 9.
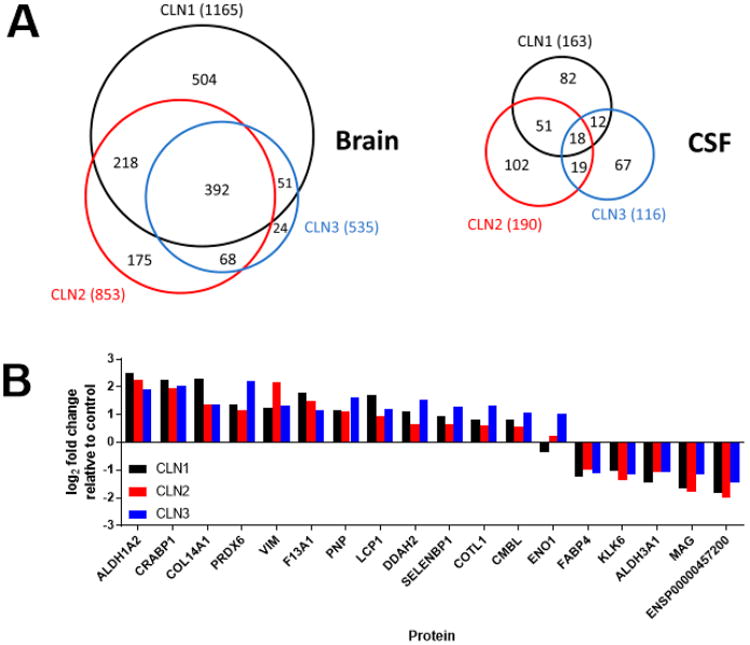
Significant changes that are common to all three NCL diseases. (A) Venn diagram showing overlap in specific changes found in brain and CSF. Note that a number of proteins were significantly altered in all three NCLs in either brain or CSF. (B) Magnitude of change for individual proteins that were significantly altered in CSF in all three NCLs.
Alterations in Brain
In brain, we detected a large number of significant changes, but most were of modest magnitude. The greatest number of significant changes of more than 2-fold elevation or decrease compared with control samples were detected in CLN1, where out of 3678 proteins quantified, 1165 were significantly altered and 174 were altered by 2-fold or more. For CLN2, similar analyses indicated 853 significantly altered with 26 ≥ 2-fold, and for CLN3, 535 significantly altered with 10 ≥ 2-fold.
For CLN1, two proteins were markedly elevated compared with controls, and these were consistent with previous studies: PSAP (elevated 4.6-fold) is a well-characterized component of the INCL storage material,14 and levels of GFAP (elevated 3.6-fold) also increase in INCL.46 Detection of elevated PSAP and GFAP in INCL was expected, but these observations provide a further validation of our analytical approaches (see above). Many proteins were decreased in CLN1 brain, but two of particular interest are STXBP1 and STX1B, which were both decreased to ∼0.4-fold of control levels, which is likely an underestimate of the actual decrease (see earlier comments above regarding reporter ion ratio compression). A recent report47 indicates that haploinsufficiency for STX1B is associated with myoclonic epilepsy, while mutations in STXBP1 are associated with various neurodevelopmental disorders including epilepsy and intellectual disability.48 The clinical phenotypes associated with STX1B and STXBP1 bear some similarity with NCL diseases, raising the intriguing possibility that, for CLN1 at least, disease may partly reflect decreased expression of these two proteins. Western blot analysis (Figure 10) confirms reduced levels of STXBP1 in CLN1 brain (see below).
Alterations in CSF
One of the major goals of this study was to identify potential biomarkers in CSF that may reflect NCL disease. Tables 2–4 summarize proteins that are altered by >2.5-fold compared with controls in CSF samples from the three NCL diseases.
Table 2. Potential CSF Biomarkers in CLN1a.
| gene name | ENSEMBL protein ID | CSF ratio compared with control | brain ratio compared with control |
|---|---|---|---|
| MB | ENSP00000380489 | 9.19 | |
| REG3A | ENSP00000386630 | 7.11 | |
| ALDH1A2* | ENSP00000249750 | 5.58 | |
| COL14A1* | ENSP00000311809 | 4.95 | ns |
| CRABP1* | ENSP00000299529 | 4.74 | ns |
| APCS | ENSP00000255040 | 4.71 | |
| CA3 | ENSP00000285381 | 4.55 | |
| DCN | ENSP00000052754 | 4.16 | ns |
| S100A11 | ENSP00000271638 | 3.65 | 1.90 |
| CD163 | ENSP00000352071 | 3.53 | |
| F13A1* | ENSP00000264870 | 3.47 | 1.86 |
| CD14 | ENSP00000304236 | 3.40 | |
| LCP1* | ENSP00000381581 | 3.29 | 2.21 |
| FGA | ENSP00000306361 | 3.23 | ns |
| GC | ENSP00000273951 | 2.98 | ns |
| ENSP00000459226 | ENSP00000459226 | 2.94 | ns |
| CHIT1 | ENSP00000356198 | 2.88 | |
| BGN | ENSP00000327336 | 2.80 | ns |
| AMBP | ENSP00000265132 | 2.80 | |
| GALM | ENSP00000272252 | 2.75 | ns |
| LYVE1 | ENSP00000256178 | 2.75 | |
| LUM | ENSP00000266718 | 2.71 | ns |
| ANXA2 | ENSP00000379342 | 2.64 | 3.23 |
| PRDX6* | ENSP00000342026 | 2.60 | 2.49 |
| F2 | ENSP00000308541 | 2.56 | |
| FUCA2 | ENSP00000002165 | 0.39 | |
| ALDH3A1* | ENSP00000225740 | 0.36 | |
| TARS | ENSP00000424387 | 0.35 | 1.15 |
| ANXA4* | ENSP00000386756 | 0.34 | 1.81 |
| PRDX5 | ENSP00000265462 | 0.34 | ns |
| MAG* | ENSP00000376048 | 0.32 | ns |
| CLIC6* | ENSP00000290332 | 0.31 | |
| HAPLN2 | ENSP00000255039 | 0.30 | 0.52 |
| GFAP | ENSP00000253408 | 0.30 | 3.55 |
| NEFM | ENSP00000221166 | 0.30 | 0.50 |
| TPPP | ENSP00000353785 | 0.28 | 0.50 |
| ENSP00000457200* | ENSP00000457200 | 0.28 | 0.68 |
| CKB | ENSP00000299198 | 0.26 | 0.87 |
| CAPS | ENSP00000465883 | 0.26 | 4.07 |
Proteins that are significantly elevated or reduced in CSF by more than 2.5-fold are shown, together with respective brain fold-changes (blank indicates not found in brain; ns, no significant difference between NCL and control). Data are expressed as fold-change compared with average control. Data in bold indicate proteins for which changes in expression between brain and CSF are not consistent.
Asterisks indicate proteins that are significantly altered in all three NCLs in the same direction.
Table 4. Potential CSF Biomarkers in CLN3a.
| gene name | ENSEMBL protein ID | CSF ratio compared with control | brain ratio compared with control |
|---|---|---|---|
| CALB1 | ENSP00000265431 | 6.55 | ns |
| HIBCH | ENSP00000352706 | 6.47 | ns |
| DCLK1 | ENSP00000353846 | 6.12 | ns |
| ACOT1 | ENSP00000311224 | 5.64 | ns |
| PRDX6* | ENSP00000342026 | 4.68 | ns |
| CRYGS | ENSP00000376287 | 4.53 | |
| PGAM2 | ENSP00000297283 | 4.51 | 1.54 |
| CRABP1* | ENSP00000299529 | 4.14 | 0.63 |
| HSPD1 | ENSP00000373620 | 4.13 | ns |
| INPP1 | ENSP00000325423 | 4.12 | ns |
| PEA15 | ENSP00000353660 | 3.97 | ns |
| LHPP | ENSP00000357835 | 3.94 | ns |
| ALDH1A1 | ENSP00000297785 | 3.90 | ns |
| ALDH1A2* | ENSP00000249750 | 3.78 | |
| SORD | ENSP00000267814 | 3.75 | ns |
| HRSP12 | ENSP00000254878 | 3.59 | ns |
| CYB5R3 | ENSP00000338461 | 3.56 | ns |
| GAA | ENSP00000374665 | 3.47 | ns |
| BBOX1 | ENSP00000435781 | 3.25 | ns |
| JAM2 | ENSP00000383376 | 3.15 | ns |
| PIR | ENSP00000369785 | 3.14 | ns |
| FABP7 | ENSP00000357429 | 3.13 | ns |
| PNP* | ENSP00000354532 | 3.10 | ns |
| ABAT | ENSP00000268251 | 3.10 | 0.81 |
| ARSA | ENSP00000348406 | 3.08 | ns |
| GPD1 | ENSP00000301149 | 3.02 | ns |
| TSPAN3 | ENSP00000267970 | 2.99 | |
| HADH | ENSP00000385638 | 2.97 | ns |
| GPT | ENSP00000433586 | 2.97 | ns |
| MDH2 | ENSP00000327070 | 2.97 | 0.72 |
| HNMT | ENSP00000386940 | 2.97 | ns |
| DCN | ENSP00000052754 | 2.92 | ns |
| SHISA4 | ENSP00000355064 | 2.92 | 0.74 |
| ENSP00000352980 | ENSP00000352980 | 2.91 | 1.32 |
| DDAH2* | ENSP00000389552 | 2.89 | ns |
| PPP1R7 | ENSP00000385022 | 2.85 | 0.87 |
| ALDH2 | ENSP00000261733 | 2.81 | 0.68 |
| BCAM | ENSP00000270233 | 2.79 | ns |
| PLCD1 | ENSP00000335600 | 2.79 | 1.23 |
| H3F3A | ENSP00000355779 | 2.76 | |
| DBI | ENSP00000348116 | 2.73 | |
| ACO2 | ENSP00000379769 | 2.69 | |
| CSRP1 | ENSP00000356275 | 2.68 | |
| ASRGL1 | ENSP00000400057 | 2.67 | |
| TMEM189-UBE2V1 | ENSP00000344166 | 2.64 | |
| KCTD12 | ENSP00000366694 | 2.60 | |
| PSAT1 | ENSP00000365773 | 2.60 | |
| NUDT1 | ENSP00000349148 | 2.58 | |
| COL14A1* | ENSP00000311809 | 2.55 | |
| DPYSL3 | ENSP00000343690 | 2.54 | |
| GLUD1 | ENSP00000277865 | 2.53 | |
| ACYP2 | ENSP00000378161 | 2.52 | |
| PYGM | ENSP00000164139 | 2.50 | |
| NELL2 | ENSP00000390680 | 0.39 | |
| FUCA2 | ENSP00000002165 | 0.37 | |
| ENSP00000457200* | ENSP00000457200 | 0.36 |
Proteins that are significantly elevated or reduced in CSF by more than 2.5-fold are shown, together with respective brain fold-changes (blank indicates not found in brain; ns, no significant difference between NCL and control). Data are expressed as fold-change compared with average control. Data in bold indicate proteins for which changes in expression between brain and CSF are not consistent.
Asterisks indicate proteins that are significantly altered in all three NCLs in the same direction.
As discussed above, there were a number of proteins that were significantly altered in all three NCLs that may be generally related to neurodegeneration (Figure 9B). MB (myoglobin) was highly elevated in CLN1 and CLN2 (∼9 and 5-fold, respectively) and in CLN3 to a lesser degree that did not reach significance (1.7 fold). The significance of MB in the CSF and its elevation in the NCLs is unclear. One possibility is that, as a consequence of muscle wasting in the bed-bound NCL patients, serum MB levels may be elevated, and this is reflected in the CSF. Another protein that was markedly elevated in the three NCLs (∼3- to 4-fold) was cellular retinoic acid binding protein 1 (CRABP1). CRABP1 is elevated in CSF in the progressive cerebrovascular disorder, Moyamoya disease,49,50 and is expressed within neurons.51 Other proteins that were markedly elevated in CSF from NCL cases include ALDH1A2 and COL14A1, which were, on average, ∼5 and ∼3 times higher in the NCLs compared with controls. Several number of proteins were consistently downregulated in NCL CSF, including myelin-associated glycoprotein (MAG). Changes in MAG expression are potentially very relevant given the central role played by MAG in axonal regeneration and are worth further investigation in the NCLs. Carnosine dipeptidase 1 (ENSP00000457200), which is reduced in several progressive neurological disorders,52 reviewed in ref 53, was also consistently lowered in NCL CSF samples.
Other changes appeared to be NCL-specific. One of the most highly elevated proteins in CLN1 CSF was REG3A, which was increased ∼7-fold compared with controls. REG3A was previously shown to be transcriptionally regulated after peripheral nerve inflammation or injury,54,55 which is consistent with elevated levels in CLN1. STXBP1 was reduced in CLN1 CSF, as observed in brain (see above). A particularly interesting observation in CLN2 and CLN3 was that calbindin1 (CALB1) was highly elevated (∼7- and 4-fold, respectively). CALB1, a calcium-binding protein that is relatively highly expressed in the cerebellum, has long been proposed as a potential biomarker for diseases with cerebellar involvement.56 CALB1 is highly elevated in CSF from Niemann-Pick type C1 disease patients and can be lowered by effective treatment.57 Our data suggest that it may also be of value in following disease progression in CLN2 and CLN3.
Concluding Remarks
The current lack of methods to evaluate short-term efficacy of treatment is a significant problem in the design and interpretation of clinical studies for the NCLs. In this study, we have used both biochemical and proteomic methods to identify candidate biomarkers in the three most frequently encountered of the NCL diseases. As a general conclusion, it is worth noting that we consistently observed a greater correlation between biochemical and proteomic changes in CLN2 and CLN3 versus either compared with CLN1. This is consistent with the common accumulation of SCMAS in both diseases and may be suggestive of common pathological pathways. Moreover, whereas the NCLs are generally considered as a group of related lysosomal diseases with common clinical and pathological features, the relative lack of concordance between CLN1 and the other two NCLs strongly suggests that at the cellular level these diseases may be quite dissimilar.
For each NCL, we compared the magnitude of change for proteins that were significantly altered in both brain and CSF. Overall, the degree of correlation was not high between the two analytes (Figure 11), indicating that disease-related changes in brain are not necessarily reflected in the CSF. For example, GFAP was highly elevated in all three NCL brain samples, but in CSF, GFAP was detected at levels that were similar to or lower than the control samples. Other examples where proteins are altered significantly but in different directions are highlighted in Tables 2–4. The overall lack of concordance between NCL-related changes in brain and CSF may reflect the fact that most of the proteins found within the CSF are derived from the plasma, with only ∼20% being derived from the brain itself.58 In addition, while the brain analysis was conducted on a homogenate representing all cell types, brain-derived CSF proteins are thought to be secreted from the cells of the choroid plexus and also originate from parenchymal interstitial fluid, and thus they may not proportionately represent all cells of the organ. It is interesting to note that in nearly all cases where the direction of change was different between brain and CSF, elevated levels in brain were associated with decreased levels in the NCL CSF samples. For PSAP in CLN1 and possibly other proteins, this could reflect intracellular sequestration within insoluble storage material. Regardless, despite the overall lack of correlation between brain and CSF, there are proteins that are elevated similarly in both samples, suggesting that they are brain- rather than plasma-derived. Whereas this provides some rationale for continued evaluation of brain for discovery-based studies applicable to the CSF, the ability to distinguish the CSF proteome of brain-secreted versus plasma-derived proteins would provide a useful tool in evaluating future studies. This may be achieved by concomitant analysis of both CSF and blood samples.
Figure 11.
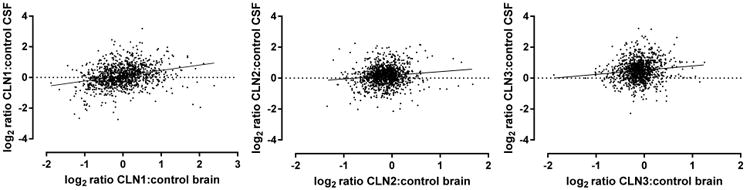
Correlation between magnitude of change of proteins in brain and CSF for each NCL disease.
Given the rarity of the NCLs, the intrusive nature of sample collection, and the fact that these diseases primarily affect children, CSF from living NCL patients and healthy, age-matched controls are not available, necessitating our use of post-mortem samples. There are considerations that must be borne in mind when analyzing data obtained from autopsy CSF (reviewed in ref 59), particularly with respect to protein composition and concentration that can change after death.60–63 For example, the measured CSF protein concentrations were outside the range normally found in living subjects (Figure S6).64 There are two main areas of concern. First, blood contamination is a general concern with ante and post-mortem CSF given that minor levels of contamination can have dramatic effects given the low protein concentration of CSF compared with blood. We have examined the CSF samples used in this study for established blood markers (HBB, PRDX2, and CAT65) (Figure S7), and whereas there is evidence for blood contamination in two samples (CLN1 HSB#3672 and CLN2#3791), most samples appear to have relatively similar levels of blood markers. Moreover, there is no correlation between disease status and blood contamination. Thus blood contamination may contribute to intrasample variability but does not appear to be a source of systematic changes. Second, post-mortem cell lysis within the CNS could potentially introduce intracellular proteins into the CSF that would not normally be observed in a living subject. Whereas there is little evidence of systematic differences in autolysis time (Figure S1A), we investigated the levels of three proteins that are elevated in post-mortem CSF, presumably reflecting cell lysis (GSTP1, YWHAB, and CAPS62) (Figure S8). Levels of GSTP1 and YWHAB were relatively similar in all brain samples but were elevated in some CSF samples, notably controls #3565 and #3589 and CLN2 #3235. Similar to GSTP1 and YWHAB, CAPS was elevated in control #3589 and CLN2 #3235 CSF samples but was similar in most brain samples but elevated notably in CLN1 #3616. From these data, we conclude that post-mortem release of cellular proteins is not NCL-related therefore does not exert any systemic influence on the data. Moreover, it is also worth noting that there is no correspondence between levels of SCMAS in CSF (Figure 3) and markers of cell lysis: CLN2 #3235 had high levels of the three cell lysis markers but had no detectable SCMAS. Third, it should be borne in mind that our studies on autopsy samples have been conducted at a single time point, the conclusion of the disease process. Proteomic changes of interest may thus simply reflect disease and as such, depending on treatment approach, may not necessarily be useful biomarkers for following disease progression and response to treatment. Longitudinal studies will therefore be required to evaluate the value of any candidate biomarkers.
Brain tissue is obviously not a suitable biological sample for following progression of disease and response to treatment. However, in the future, CSF will be available from NCL patients with the advent of enzyme replacement and gene therapy trials and will be free of the potential caveats associated with autopsy samples. This is a relatively accessible biological sample that will be an excellent resource for more extensive discovery-based analysis as well as further investigation of candidates identified here, although obtaining age-matched control CSF will continue to be difficult or impossible for these pediatric diseases. While validation in humans will be necessary, the use of animal models for the NCLs could circumvent such issues and will also allow for time-course studies to monitor the response of potential biomarkers to disease progression and to effective therapy.
Supplementary Material
Figure S1. Analysis of clinical variables for NCL cases and controls.Figure S2. Relative lysosomal activities in NCL CSF samples. Figure S3. Quantitative comparison of lysosomal proteins measured by enzyme assay and spectral count analysis of purified Man6P glycoprotein preparations. Figure S4. Relative abundance of ATP5A1 and RTN4IP1. Figure S5. Measured versus observed levels of DrR57 bacterial internal standard. Figure S6. CSF protein concentrations. Figure S7. Blood markers in CSF. Figure S8. Cell-lysis markers in CSF. Figure S8. Cell-lysis markers in CSF. (PDF)
Table S1. Key for iTRAQ analysis. Table S2. Statistical analysis of relative protein abundance in specific and mock eluates. Table S3. Spectral count analysis of purified Man6P glycoprotein preparations. Table S4. iTRAQ analyses of brain and CSF. (XLSX)
Table 3. Potential CSF Biomarkers in CLN2a.
| gene name | ENSEMBL protein ID | CSF ratio compared with control | brain ratio compared with control |
|---|---|---|---|
| MB | ENSP00000380489 | 5.19 | |
| ALDH1A2* | ENSP00000249750 | 4.80 | |
| HBG2 | ENSP00000369609 | 4.75 | 0.59 |
| FBP1 | ENSP00000364475 | 4.71 | |
| VIM* | ENSP00000224237 | 4.49 | 1.49 |
| HBB* | ENSP00000333994 | 4.24 | 0.81 |
| CALB1 | ENSP00000265431 | 4.09 | 1.30 |
| CRABP2 | ENSP00000357205 | 4.03 | |
| CRABP1* | ENSP00000299529 | 3.84 | ns |
| FLNC | ENSP00000327145 | 3.42 | 1.62 |
| PON1 | ENSP00000222381 | 3.34 | |
| APOB* | ENSP00000233242 | 3.25 | ns |
| CA1 | ENSP00000430656 | 3.17 | 0.70 |
| EEA1 | ENSP00000317955 | 2.99 | 0.82 |
| HBD | ENSP00000369654 | 2.96 | ns |
| HBA2 | ENSP00000251595 | 2.93 | 0.84 |
| KCTD12 | ENSP00000366694 | 2.84 | ns |
| F13A1* | ENSP00000264870 | 2.81 | ns |
| ANXA1 | ENSP00000257497 | 2.72 | ns |
| ACOT1 | ENSP00000311224 | 2.68 | ns |
| HSPD1 | ENSP00000373620 | 2.61 | 0.82 |
| COL14A1* | ENSP00000311809 | 2.61 | ns |
| S100A11 | ENSP00000271638 | 2.56 | ns |
| TIMP2 | ENSP00000262768 | 0.39 | |
| KLK6* | ENSP00000366047 | 0.39 | |
| NELL2 | ENSP00000390680 | 0.39 | ns |
| TPPP | ENSP00000353785 | 0.38 | 0.59 |
| HAPLN2 | ENSP00000255039 | 0.32 | 0.55 |
| CBLN3 | ENSP00000267406 | 0.32 | |
| MAG* | ENSP00000376048 | 0.29 | 1.45 |
| TPP1 | ENSP00000299427 | 0.28 | 0.39 |
| CNDP1* | ENSP00000351682 | 0.27 | ns |
| ENSP00000457200* | ENSP00000457200 | 0.25 | 1.12 |
| PMP2 | ENSP00000256103 | 0.25 | 0.61 |
Proteins that are significantly elevated or reduced in CSF by more than 2.5-fold are shown, together with respective brain fold-changes (blank indicates not found in brain; ns, no significant difference between NCL and control). Data are expressed as fold-change compared with average control. Data in bold indicate proteins for which changes in expression between brain and CSF are not consistent.
Asterisks indicate proteins that are significantly altered in all three NCLs in the same direction.
Acknowledgments
Tissue/fluid specimens were obtained from the Human Brain and Spinal Resource Center, VA West Los Angeles Healthcare Center, Los Angeles, CA 90073, which is sponsored by NINDS/NIMH, the National Multiple Sclerosis Society, and Department of Veterans Affairs. Mass spectrometry was conducted by the Biological Mass Spectrometry Facility of Robert Wood Johnson Medical School and Rutgers University, supported in part by NIH P30NS046593 and S10RR024584. This project was supported by NIH grants R01NS37918 (P.L.), R21NS088786 (D.E.S.), P30NS046593 (Hong Li and P.L.), and P30CA072720 (D.F.M.).
Footnotes
Associated Content: Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.7b00460.
Orcid: David E. Seat 0000-0002-5159-9928
Notes: The authors declare no competing financial interest. Raw files, mgf files, GPM result files, Excel workbooks listing protein and peptide assignments, and keys for data interpretation are archived in the MassIVE (http://massive.ucsd.edu) and ProteomeXchange (http://www.proteomexchange.org/) repositories: MSV000081140, spectral count analysis of purified Man6P glycoproteins; MSV000081143, iTRAQ8 analysis of brain extracts and CSF.
References
- 1.Goebel HH, Mole SE, Lake BD. The Neuronal Ceroid Lipofuscinoses (Batten Disease) IOS Press; Washington, DC: 1999. [Google Scholar]
- 2.Warrier V, Vieira M, Mole SE. Genetic basis and phenotypic correlations of the neuronal ceroid lipofusinoses. Biochim Biophys Acta. 2013;1832:1827–1830. doi: 10.1016/j.bbadis.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 3.Selden NR, Al-Uzri A, Huhn SL, Koch TK, Sikora DM, Nguyen-Driver MD, Guillaume DJ, Koh JL, Gultekin SH, Anderson JC, Vogel H, Sutcliffe TL, Jacobs Y, Steiner RD. Central nervous system stem cell transplantation for children with neuronal ceroid lipofuscinosis. J Neurosurg Pediatr. 2013;11:643–652. doi: 10.3171/2013.3.PEDS12397. [DOI] [PubMed] [Google Scholar]
- 4.Crystal RG, Sondhi D, Hackett NR, Kaminsky SM, Worgall S, Stieg P, Souweidane M, Hosain S, Heier L, Ballon D, Dinner M, Wisniewski K, Kaplitt M, Greenwald BM, Howell JD, Strybing K, Dyke J, Voss H. Clinical protocol. Administration of a replication-deficient adeno-associated virus gene transfer vector expressing the human CLN2 cDNA to the brain of children with late infantile neuronal ceroid lipofuscinosis. Hum Gene Ther. 2004;15:1131–1154. doi: 10.1089/hum.2004.15.1131. [DOI] [PubMed] [Google Scholar]
- 5.Schulz A, Specchio N, Gissen P, de los Reyes E, Williams R, Cahan H, Genter F, Jacoby D. Intracerebroventricular Cerliponase Alfa (BMN 190) in children with CLN2 disease: Interim results from a Phase 1/2, Open-Label, dose-escalation study. Neuropediatrics. 2016;47 47-FV02-06. [Google Scholar]
- 6.Markham A. Cerliponase Alfa: First Global Approval. Drugs. 2017;77:1247–1249. doi: 10.1007/s40265-017-0771-8. [DOI] [PubMed] [Google Scholar]
- 7.Marshall FJ, de Blieck EA, Mnk JW, Dure L, Adams H, Messing S, Rothberg PG, Levy E, McDonough T, DeYoung J, Wang M, Ramirez-Montealegre D, Kwon JM, Pearce DA. A clinical rating scale for Batten disease: reliable and relevant for clinical trials. Neurology. 2005;65:275–279. doi: 10.1212/01.wnl.0000169019.41332.8a. [DOI] [PubMed] [Google Scholar]
- 8.Steinfeld R, Heim P, von Gregory H, Meyer K, Ullrich K, Goebel HH, Kohlschutter A. Late infantile neuronal ceroid lipofuscinosis: quantitative description of the clinical course in patients with CLN2 mutations. Am J Med Genet. 2002;112:347–354. doi: 10.1002/ajmg.10660. [DOI] [PubMed] [Google Scholar]
- 9.Worgall S, Kekatpure MV, Heier L, Ballon D, Dyke JP, Shungu D, Mao X, Kosofsky B, Kaplitt MG, Souweidane MM, Sondhi D, Hackett NR, Hollmann C, Crystal RG. Neurological deterioration in late infantile neuronal ceroid lipofuscinosis. Neurology. 2007;69:521–535. doi: 10.1212/01.wnl.0000267885.47092.40. [DOI] [PubMed] [Google Scholar]
- 10.Hersrud SL, Geraets RD, Weber KL, Chan CH, Pearce DA. Plasma Biomarkers for Neuronal Ceroid Lipofuscinosis. FEBS J. 2016;283:459–471. doi: 10.1111/febs.13593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitchison HM, Bernard DJ, Greene ND, Cooper JD, Junaid MA, Pullarkat RK, de Vos N, Breuning MH, Owens JW, Mobley WC, Gardiner RM, Lake BD, Taschner PE, Nussbaum RL. Targeted disruption of the Cln3 gene provides a mouse model for Batten disease. The Batten Mouse Model Consortium [corrected] Neurobiol Dis. 1999;6:321–334. doi: 10.1006/nbdi.1999.0267. [DOI] [PubMed] [Google Scholar]
- 12.Pohl S, Mitchison HM, Kohlschutter A, van Diggelen O, Braulke T, Storch S. Increased expression of lysosomal acid phosphatase in CLN3-defective cells and mouse brain tissue. J Neurochem. 2007;103:2177–2188. doi: 10.1111/j.1471-4159.2007.04920.x. [DOI] [PubMed] [Google Scholar]
- 13.Sleat DE, Sohar I, Pullarkat PS, Lobel P, Pullarkat RK. Specific alterations in levels of mannose 6-phosphorylated glycoproteins in different neuronal ceroid lipofuscinoses. Biochem J. 1998;334(Pt 3):547–551. doi: 10.1042/bj3340547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tyynela J, Palmer DN, Baumann M, Haltia M. Storage of saposins A and D in infantile neuronal ceroid-lipofuscinosis. FEBS Lett. 1993;330:8–12. doi: 10.1016/0014-5793(93)80908-d. [DOI] [PubMed] [Google Scholar]
- 15.Tourtellotte WW, Nagra RM, Atkinson R, Baumhefner RW, Riehl J, Guntrip D. Banking Human Neurospecimens. Encyclopedia of Neuroscience. 2003:1–19. [Google Scholar]
- 16.Sleat DE, Sun P, Wiseman JA, Huang L, El-Banna M, Zheng H, Moore DF, Lobel P. Extending the mannose 6-phosphate glycoproteome by high resolution/accuracy mass spectrometry analysis of control and acid phosphatase 5-deficient mice. Mol Cell Proteomics. 2013;12:1806–1817. doi: 10.1074/mcp.M112.026179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sleat DE, Della Valle MC, Zheng H, Moore DF, Lobel P. The mannose 6-phosphate glycoprotein proteome. J Proteome Res. 2008;7:3010–3021. doi: 10.1021/pr800135v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Craig R, Cortens JP, Beavis RC. Open source system for analyzing, validating, and storing protein identification data. J Proteome Res. 2004;3:1234–1242. doi: 10.1021/pr049882h. [DOI] [PubMed] [Google Scholar]
- 19.Beavis RC. Using the global proteome machine for protein identification. Methods Mol Biol. 2006;328:217–228. doi: 10.1385/1-59745-026-X:217. [DOI] [PubMed] [Google Scholar]
- 20.McCullagh P, Nelder JA. Generalized Linear Model. 2nd. CRC Press; 1990. [Google Scholar]
- 21.Sleat DE, Sohar I, Lackland H, Majercak J, Lobel P. Rat brain contains high levels of mannose-6-phosphorylated glycoproteins including lysosomal enzymes and palmitoyl-protein thioesterase, an enzyme implicated in infantile neuronal lipofuscinosis. J Biol Chem. 1996;271:19191–19198. doi: 10.1074/jbc.271.32.19191. [DOI] [PubMed] [Google Scholar]
- 22.Kim KH, Pham CT, Sleat DE, Lobel P. Dipeptidyl-peptidase I does not functionally compensate for the loss of tripeptidyl-peptidase I in the neurodegenerative disease late-infantile neuronal ceroid lipofuscinosis. Biochem J. 2008;415:225–232. doi: 10.1042/BJ20080411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dando PM, Fortunato M, Smith L, Knight CG, McKendrick JE, Barrett AJ. Pig kidney legumain: an asparaginyl endopeptidase with restricted specificity. Biochem J. 1999;339(Pt 3):743–749. [PMC free article] [PubMed] [Google Scholar]
- 24.Sleat DE, Wiseman JA, Sohar I, El-Banna M, Zheng H, Moore DF, Lobel P. Proteomic analysis of mouse models of Niemann-Pick C disease reveals alterations in the steady-state levels of lysosomal proteins within the brain. Proteomics. 2012;12:3499–3509. doi: 10.1002/pmic.201200205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elleder M, Sokolova J, Hrebicek M. Follow-up study of subunit c of mitochondrial ATP synthase (SCMAS) in Batten disease and in unrelated lysosomal disorders. Acta Neuropathol. 1997;93:379–390. doi: 10.1007/s004010050629. [DOI] [PubMed] [Google Scholar]
- 26.Das AK, Becerra CH, Yi W, Lu JY, Siakotos AN, Wisniewski KE, Hofmann SL. Molecular genetics of palmitoyl-protein thioesterase deficiency in the U.S. J Clin Invest. 1998;102:361–370. doi: 10.1172/JCI3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balducci C, Pierguidi L, Persichetti E, Parnetti L, Sbaragli M, Tassi C, Orlacchio A, Calabresi P, Beccari T, Rossi A. Lysosomal hydrolases in cerebrospinal fluid from subjects with Parkinson's disease. Mov Disord. 2007;22:1481–1484. doi: 10.1002/mds.21399. [DOI] [PubMed] [Google Scholar]
- 28.Fearnley IM, Walker JE, Martinus RD, Jolly RD, Kirkland KB, Shaw GJ, Palmer DN. The sequence of the major protein stored in ovine ceroid lipofuscinosis is identical with that of the dicyclohexylcarbodiimide-reactive proteolipid of mitochondrial ATP synthase. Biochem J. 1990;268:751–758. doi: 10.1042/bj2680751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmer DN, Martinus RD, Cooper SM, Midwinter GG, Reid JC, Jolly RD. Ovine ceroid lipofuscinosis. The major lipopigment protein and the lipid-binding subunit of mitochondrial ATP synthase have the same NH2-terminal sequence. J Biol Chem. 1989;264:5736–5740. [PubMed] [Google Scholar]
- 30.Xu S, Wang L, El-Banna M, Sohar I, Sleat DE, Lobel P. Large-volume intrathecal enzyme delivery increases survival of a mouse model of late infantile neuronal ceroid lipofuscinosis. Molecular therapy Mol Ther. 2011;19:1842–1848. doi: 10.1038/mt.2011.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meng Y, Sohar I, Sleat DE, Richardson JR, Reuhl KR, Jenkins RB, Sarkar G, Lobel P. Effective intravenous therapy for neurodegenerative disease with a therapeutic enzyme and a peptide that mediates delivery to the brain. Mol Ther. 2014;22:547–553. doi: 10.1038/mt.2013.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wisniewski KE, Kaczmarski W, Golabek AA, Kida E. Rapid detection of subunit c of mitochondrial ATP synthase in urine as a diagnostic screening method for neuronal ceroid-lipofuscinoses. Am J Med Genet. 1995;57:246–249. doi: 10.1002/ajmg.1320570227. [DOI] [PubMed] [Google Scholar]
- 33.Wisniewski KE, Golabek AA, Kida E. Increased urine concentration of subunit c of mitochondrial ATP synthase in neuronal ceroid lipofuscinoses patients. J Inherited Metab Dis. 1994;17:205–210. doi: 10.1007/BF00711619. [DOI] [PubMed] [Google Scholar]
- 34.Valenzano KJ, Kallay LM, Lobel P. An assay to detect glycoproteins that contain mannose 6-phosphate. Anal Biochem. 1993;209:156–162. doi: 10.1006/abio.1993.1096. [DOI] [PubMed] [Google Scholar]
- 35.Lubke T, Lobel P, Sleat DE. Proteomics of the lysosome. Biochim Biophys Acta. 2009;1793:625–635. doi: 10.1016/j.bbamcr.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sleat DE, Jadot M, Lobel P. Lysosomal proteomics and disease. Proteomics: Clin Appl. 2007;1:1134–1146. doi: 10.1002/prca.200700250. [DOI] [PubMed] [Google Scholar]
- 37.Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, Lobel P. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. 1997;277:1802–1805. doi: 10.1126/science.277.5333.1802. [DOI] [PubMed] [Google Scholar]
- 38.Liu CG, Sleat DE, Donnelly RJ, Lobel P. Structural organization and sequence of CLN2, the defective gene in classical late infantile neuronal ceroid lipofuscinosis. Genomics. 1998;50:206–212. doi: 10.1006/geno.1998.5328. [DOI] [PubMed] [Google Scholar]
- 39.Sleat DE, Lackland H, Wang Y, Sohar I, Xiao G, Li H, Lobel P. The human brain mannose 6-phosphate glycoproteome: a complex mixture composed of multiple isoforms of many soluble lysosomal proteins. Proteomics. 2005;5:1520–1532. doi: 10.1002/pmic.200401054. [DOI] [PubMed] [Google Scholar]
- 40.Sun P, Sleat DE, Lecocq M, Hayman AR, Jadot M, Lobel P. Acid phosphatase 5 is responsible for removing the mannose 6-phosphate recognition marker from lysosomal proteins. Proc Natl Acad Sci U S A. 2008;105:16590–16595. doi: 10.1073/pnas.0807472105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sleat DE, Zheng H, Qian M, Lobel P. Identification of sites of mannose 6-phosphorylation on lysosomal proteins. Mol Cell Proteomics. 2006;5:686–701. doi: 10.1074/mcp.M500343-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Karp NA, Huber W, Sadowski PG, Charles PD, Hester SV, Lilley KS. Addressing accuracy and precision issues in iTRAQ quantitation. Mol Cell Proteomics. 2010;9:1885–1897. doi: 10.1074/mcp.M900628-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu S, Sleat DE, Jadot M, Lobel P. Glial fibrillary acidic protein is elevated in the lysosomal storage disease classical late-infantile neuronal ceroid lipofuscinosis, but is not a component of the storage material. Biochem J. 2010;428:355–362. doi: 10.1042/BJ20100128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baird GS, Nelson SK, Keeney TR, Stewart A, Williams S, Kraemer S, Peskind ER, Montine TJ. Age-dependent changes in the cerebrospinal fluid proteome by slow off-rate modified aptamer array. Am J Pathol. 2012;180:446–456. doi: 10.1016/j.ajpath.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J, Goodlett DR, Peskind ER, Quinn JF, Zhou Y, Wang Q, Pan C, Yi E, Eng J, Aebersold RH, Montine TJ. Quantitative proteomic analysis of age-related changes in human cerebrospinal fluid. Neurobiol Aging. 2005;26:207–227. doi: 10.1016/j.neurobiolaging.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 46.Paetau A, Elovaara I, Paasivuo R, Virtanen I, Palo J, Haltia M. Glial filaments are a major brain fraction in infantile neuronal ceroid-lipofuscinosis. Acta Neuropathol. 1985;65:190–194. doi: 10.1007/BF00686997. [DOI] [PubMed] [Google Scholar]
- 47.Vlaskamp DR, Rump P, Callenbach PM, Vos YJ, Sikkema-Raddatz B, van Ravenswaaij-Arts CM, Brouwer OF. Haploinsufficiency of the STX1B gene is associated with myoclonic astatic epilepsy. Eur J Paediatr Neurol. 2016;20:489–492. doi: 10.1016/j.ejpn.2015.12.014. [DOI] [PubMed] [Google Scholar]
- 48.Stamberger H, Nikanorova M, Willemsen MH, Accorsi P, Angriman M, Baier H, Benkel-Herrenbrueck I, Benoit V, Budetta M, Caliebe A, Cantalupo G, Capovilla G, Casara G, Courage C, Deprez M, Destree A, Dilena R, Erasmus CE, Fannemel M, Fjaer R, Giordano L, Helbig KL, Heyne HO, Klepper J, Kluger GJ, Lederer D, Lodi M, Maier O, Merkenschlager A, Michelberger N, Minetti C, Muhle H, Phalin J, Ramsey K, Romeo A, Schallner J, Schanze I, Shinawi M, Sleegers K, Sterbova K, Syrbe S, Traverso M, Tzschach A, Uldall P, Van Coster R, Verhelst H, Viri M, Winter S, Wolff M, Zenker M, Zoccante L, De Jonghe P, Helbig I, Striano P, Lemke JR, Moller RS, Weckhuysen S. STXBP1 encephalopathy: A neurodevelopmental disorder including epilepsy. Neurology. 2016;86:954–962. doi: 10.1212/WNL.0000000000002457. [DOI] [PubMed] [Google Scholar]
- 49.Jeon JS, Ahn JH, Moon YJ, Cho WS, Son YJ, Kim SK, Wang KC, Bang JS, Kang HS, Kim JE, Oh CW. Expression of cellular retinoic acid-binding protein-I (CRABP-I) in the cerebrospinal fluid of adult onset moyamoya disease and its association with clinical presentation and postoperative haemodynamic change. J Neurol, Neurosurg Psychiatry. 2014;85:726–731. doi: 10.1136/jnnp-2013-305953. [DOI] [PubMed] [Google Scholar]
- 50.Kim SK, Yoo JI, Cho BK, Hong SJ, Kim YK, Moon JA, Kim JH, Chung YN, Wang KC. Elevation of CRABP-I in the cerebrospinal fluid of patients with Moyamoya disease. Stroke. 2003;34:2835–2841. doi: 10.1161/01.STR.0000100159.43123.D7. [DOI] [PubMed] [Google Scholar]
- 51.Zhou FC, Wei LN. Expression of cellular retinoic acid-binding protein I is specific to neurons in adult transgenic mouse brain. Brain Res Gene Expr Pattern. 2001;1:67–72. doi: 10.1016/s1567-133x(01)00010-2. [DOI] [PubMed] [Google Scholar]
- 52.Hu Y, Hosseini A, Kauwe JS, Gross J, Cairns NJ, Goate AM, Fagan AM, Townsend RR, Holtzman DM. Identification and validation of novel CSF biomarkers for early stages of Alzheimer's disease. Proteomics: Clin Appl. 2007;1:1373–1384. doi: 10.1002/prca.200600999. [DOI] [PubMed] [Google Scholar]
- 53.Bellia F, Vecchio G, Rizzarelli E. Carnosinases, their substrates and diseases. Molecules. 2014;19:2299–2329. doi: 10.3390/molecules19022299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kalinski AL, Sachdeva R, Gomes C, Lee SJ, Shah Z, Houle JD, Twiss JL. mRNAs and Protein Synthetic Machinery Localize into Regenerating Spinal Cord Axons When They Are Provided a Substrate That Supports Growth. J Neurosci. 2015;35:10357–10370. doi: 10.1523/JNEUROSCI.1249-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He SQ, Yao JR, Zhang FX, Wang Q, Bao L, Zhang X. Inflammation and nerve injury induce expression of pancreatitis-associated protein-II in primary sensory neurons. Mol Pain. 2010;6:23. doi: 10.1186/1744-8069-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kiyosawa K, Mokuno K, Murakami N, Yasuda T, Kume A, Hashizume Y, Takahashi A, Kato K. Cerebrospinal fluid 28-kDa calbindin-D as a possible marker for Purkinje cell damage. J Neurol Sci. 1993;118:29–33. doi: 10.1016/0022-510x(93)90241-p. [DOI] [PubMed] [Google Scholar]
- 57.Bradbury A, Bagel J, Sampson M, Farhat N, Ding W, Swain G, Prociuk M, O'Donnell P, Drobatz K, Gurda B, Wassif C, Remaley A, Porter F, Vite C. Cerebrospinal Fluid Calbindin D Concentration as a Biomarker of Cerebellar Disease Progression in Niemann-Pick Type C1 Disease. J Pharmacol Exp Ther. 2016;358:254–261. doi: 10.1124/jpet.116.232975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thompson EJ. Proteins of the Cerebrospinal Fluid: Analysis and Interpretation in the Diagnosis and Treatment of Neurological Disease. Academic Press; 2005. [Google Scholar]
- 59.Choi YS, Choe LH, Lee KH. Recent cerebrospinal fluid biomarker studies of Alzheimer's disease. Expert Rev Proteomics. 2010;7:919–929. doi: 10.1586/epr.10.75. [DOI] [PubMed] [Google Scholar]
- 60.Finehout EJ, Franck Z, Relkin N, Lee KH. Proteomic analysis of cerebrospinal fluid changes related to postmortem interval. Clin Chem. 2006;52:1906–1913. doi: 10.1373/clinchem.2006.070508. [DOI] [PubMed] [Google Scholar]
- 61.Paulson GW, Stickney D. Cerebrospinal fluid after death. Confin Neurol. 1971;33:149–162. doi: 10.1159/000103129. [DOI] [PubMed] [Google Scholar]
- 62.Burgess JA, Lescuyer P, Hainard A, Burkhard PR, Turck N, Michel P, Rossier JS, Reymond F, Hochstrasser DF, Sanchez JC. Identification of brain cell death associated proteins in human post-mortem cerebrospinal fluid. J Proteome Res. 2006;5:1674–1681. doi: 10.1021/pr060160v. [DOI] [PubMed] [Google Scholar]
- 63.Lescuyer P, Allard L, Zimmermann-Ivol CG, Burgess JA, Hughes-Frutiger S, Burkhard PR, Sanchez JC, Hochstrasser DF. Identification of post-mortem cerebrospinal fluid proteins as potential biomarkers of ischemia and neurodegeneration. Proteomics. 2004;4:2234–2241. doi: 10.1002/pmic.200300822. [DOI] [PubMed] [Google Scholar]
- 64.Lentner C, Lentner C, Wink A. eigy Scientific Tables. 8th. CIBA-Geigy; West Caldwell, NJ: 1981. [Google Scholar]
- 65.You JS, Gelfanova V, Knierman MD, Witzmann FA, Wang M, Hale JE. The impact of blood contamination on the proteome of cerebrospinal fluid. Proteomics. 2005;5:290–296. doi: 10.1002/pmic.200400889. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Analysis of clinical variables for NCL cases and controls.Figure S2. Relative lysosomal activities in NCL CSF samples. Figure S3. Quantitative comparison of lysosomal proteins measured by enzyme assay and spectral count analysis of purified Man6P glycoprotein preparations. Figure S4. Relative abundance of ATP5A1 and RTN4IP1. Figure S5. Measured versus observed levels of DrR57 bacterial internal standard. Figure S6. CSF protein concentrations. Figure S7. Blood markers in CSF. Figure S8. Cell-lysis markers in CSF. Figure S8. Cell-lysis markers in CSF. (PDF)
Table S1. Key for iTRAQ analysis. Table S2. Statistical analysis of relative protein abundance in specific and mock eluates. Table S3. Spectral count analysis of purified Man6P glycoprotein preparations. Table S4. iTRAQ analyses of brain and CSF. (XLSX)