

Abstract
Chronic myeloid leukemia (CML) is defined by the presence of t(9;22)(q34;q11.2)/BCR-ABL1. Additional chromosomal abnormalities confer an adverse prognosis and are particularly common in the blast phase of CML (CML-BP). CBFB rearrangement, particularly CBFB-MYH11 fusion resulting from inv(16)(p13.1q22) or t(16;16)(p13.1;q22), is an acute myeloid leukemia (AML)-defining alteration that is associated with a favorable outcome. The co-occurrence of BCR-ABL1 and CBFB rearrangement is extremely rare, and the significance of this finding remains unclear. We identified 10 patients with myeloid neoplasms harboring BCR-ABL1 and CBFB rearrangement. The study group included six men and four women with a median age of 51 years (range, 20–71 years). The sequence of molecular alterations could be determined in nine cases: BCR-ABL1 preceded CBFB rearrangement in seven, CBFB rearrangement preceded BCR-ABL1 in one, and both alterations were discovered simultaneously in one patient. BCR-ABL1 encoded for p210 kD in all cases in which BCR-ABL1 preceded CBFB rearrangement; a p190 kD was identified in the other three cases. Two patients were treated with the FLAG-IDA regimen (fludarabine, cytarabine, idarubicin, and G-CSF) and tyrosine kinase inhibitors (TKI); seven with other cytarabine-based regimens and TKIs, and one with ponatinib alone. At last follow up (median, 16 months; range 2–85), 7 of 10 patients had died. The co-existence of BCR-ABL1 and CBFB rearrangement is associated with poor outcome and a clinical course similar to that of CML-BP, and unlike de novo AML with CBFB rearrangement, suggesting that high-intensity chemotherapy with TKI should be considered in these patients.
1 | INTRODUCTION
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm that arises from a clonal pluripotent bone marrow (BM) stem cell. CML is defined by the presence of BCR-ABL1 fusion resulting from a reciprocal translocation between chromosomes 9 and 22, t(9;22)(q34;q11.2) that creates a minute derivative chromosome 22, also known as the Philadelphia (Ph) chromosome.1 The translocation is also detected in a subset of B-cell lymphoblastic leukemia (B-ALL) and less commonly in de novo AML.2,3 The most common form of BCR-ABL1 fusion (b2a2 or b3a2) in CML results in a 210 kDa product, whereas in B-ALL the main fusion form (e1a2) results in the 190 kDa product.4 The BCR-ABL1 fusion protein is a constitutively activated receptor tyrosine kinase that results in dysregulated growth and cell replication through the activation of downstream effectors such as RAS, RAF, MYC, and JAK/STAT.4
CML is further divided into three phases: chronic phase (CP), accelerated phase (AP), and blast phase (BP) based on the presence of persistent or increasing WBC (>10×109/L), splenomegaly, thrombocytosis or thrombocytopenia; clonal cytogenetic evolution; 20% or more basophils in the peripheral blood; the number of myeloblasts in the BM or extramedullary tissues; and response to tyrosine receptor kinase inhibitors (TKI).1,4 The 10-year survival of patients with CML has increased dramatically in the era of targeted therapy, approaching 80%–90%.5
The occurrence of additional cytogenetic alterations other than t (9;22) is observed in up to 80% of cases of CML-BP.6–12 The most common additional cytogenetic abnormalities include trisomy 8, an extra copy of the Ph chromosome, 3q26 rearrangements, monosomy 7/del(7q), i(17)(q10), trisomy 21, minus Y, and trisomy 19.6,7
CBFB rearrangement, particularly CBFB-MYH11 fusion, resulting from inv(16)(p13.1q22) or less commonly t(16;16)(p13.1;q22), is an acute myeloid leukemia (AML)-defining alteration that is associated with a favorable outcome.13–15 CBFB is a member of the core binding factor (CBF) family of transcription factors and stabilizes the interaction of the α subunits RUNX1, RUNX2, and RUNX3 with DNA. RUNX1 regulates hematopoietic stem cell self-renewal, survival, and differentiation of B-cells, T-cells, and megakaryocytes. The fusion product encodes the protein CBFB-SMMHC which is thought to be necessary but insufficient for the development of AML. The fusion protein induces defective hematopoietic differentiation; however, usually additional genetic alterations, mostly mutations, are needed for fully developed leukemogenesis.16 CBFB-SMMHC induces a dominant negative effect on wild-type CBFB via its more potent binding ability to RUNX, thereby repressing RUNX1. More recently, it has been suggested that the CBFB-SMMHC fusion protein cooperates with RUNX1 to act as a transcription activator and induce differential gene expression.16 Because of the variability of the genomic breakpoints in CBFB and MYH11 over 10 fusion products of different sizes have been described. The most common form is type A, occurring in more than 85% of cases; type D and E are seen in up to 5%–10% of cases and other fusion forms have been reported in isolated cases.17
The co-occurrence of BCR-ABL1 fusion and CBFB rearrangement is extremely rare and its clinical significance remains largely unknown.18–21 Since therapeutic approaches to neoplasms harboring these potent oncogenic fusion products are different, the co-occurrence of BCR-ABL1 fusion and CBFB rearrangement might pose a clinical management challenge. Herein, we describe a series of patients with myeloid neoplasms harboring BCR-ABL1 fusion and CBFB rearrangement and provide detailed clinicopathologic details, genotype-phenotype correlation, and outcome data.
2 | METHODS
2.1 | Patients and study design
We identified retrospectively 10 patients with AML carrying both BCR-ABL1 and CBFB rearrangement seen and treated at The University of Texas MD Anderson Cancer Center (UTMDACC). These patients included a subset with a well-documented antecedent CML in chronic phase and another group that harbored both alterations at the time of initial diagnosis. Clinical and laboratory data were obtained by electronic chart review. This study was approved by the Institutional Review Board of UTMDACC and was conducted in accordance with the declaration of Helsinki.
2.2 | Morphologic evaluation
All diagnostic BM samples were reviewed. BM cellularity was assessed relative to age according to the EUMNET criteria.22 BM blast, eosinophil, and monocyte percentages were enumerated by a 500-cell count using Wright-Giemsa-stained aspirate smears and/or touch imprints.
2.3 | Flow cytometry immunophenotyping
Multiparameter flow cytometry was performed on BM samples using a standard stain-lyse-wash procedure with ammonium chloride lysis and the FACSCanto II cytometer and FACSDiva software (BD Biosciences). Data were analyzed using FCS Express (De Novo Software, Glendale, CA). The following antibodies were used in various combinations: CD2, sCD3, cytoCD3, CD4, CD5, CD7, CD13, CD14, CD15, CD19, CD22, CD25, CD33, CD34, CD36, CD38, CD41, CD45, CD56, CD64, CD117, CD123, HLA-DR, MPO, and TDT.
2.4 | Cytogenetic analysis
Conventional cytogenetic analysis was performed on unstimulated cultured BM aspirate specimens using standard GTG-banding as described previously.23 At least 20 metaphases were analyzed. Results were reported using the 2013 International System for Human Cytogenetic Nomenclature (ISCN).24
2.5 | Fluorescence in situ hybridization for BCR-ABL1 and CBFB rearrangement
Fluorescence in situ hybridization (FISH) analysis for BCR-ABL1 and CBFB rearrangement was performed on cultured BM cells or G-banded slides with LSI BCR-ABL1 ES fusion probes or LSI CBFB dual-color breakapart probes (Abbott Molecular/Vysis, Des Plaines, IL) using previously described methods.18 A total of 200 interphases were analyzed. The positive cutoff value of 2.0% for BCR/ABL1 rearrangement and 4.2% for CBFB rearrangement has been established in our laboratory.
2.6 | Quantitative reverse transcription PCR for BCR-ABL1 and CBFB-MYH11
Reverse transcription quantitative polymerase chain reaction (Q-PCR) for detection of BCR-ABL1 and CBFB-MYH11 was performed using RNA extracted from BM or PB samples according to methods described previously.25,26 Briefly, the BCR-ABL1 Q-PCR is a multiplex assay designed to detect the e1a2, e13a2 (b2a2), and e14a2 (b3a2) transcripts in a single tube. BCR-ABL1 and ABL1 transcript levels were detected simultaneously and quantitative results were expressed as the percent ratio of BCR-ABL1 to ABL1. The specific fusion transcripts were distinguished using capillary electrophoretic separation of the fluorochrome-labeled products. The CBFB-MYH11 assay is designed to detect type A CBFB-MYH11 fusion transcript.26 The CBFB-MYH11 was also normalized to ABL1 transcript levels for quantification. The sensitivity of detection of BCR-ABL1 and CBFB-MYH11 transcripts was between 1 in 10,000 and 1 in 100,000.
2.7 | Mutation analysis
Mutation analysis was performed using DNA extracted from BM aspirate samples in a subset of patients using the following techniques: Next-generation sequencing-based mutation analysis of exonic regions of ABL1, EGFR, GATA2, IKZF2, MDM2, NOTCH1, RUNX1, ASXL1, EZH2, HRAS, JAK2, MLL, NPM1, TET2, BRAF, IDH1, KIT, NRAS, TP53, DNMT3A, GATA1, IDH2, KRAS, MYD88, PTPN11, and WT1 was performed using the Illumina MiSeq (Illumina, San Diego, CA) sequencer as described previously.27 FLT3 (internal tandem duplication and D835) and NPM1 (exon12, codons 956–971) mutations were assessed by polymerase chain reaction (PCR) followed by capillary electrophoresis on a Genetic Analyzer (Applied Biosystems, Foster City, CA), as described previously.28 In some cases, mutations in NRAS and KRAS (codons 12, 13, and 61), and KIT exon 17 (codons 778 to 838) were analyzed using pyrosequencing (Biotage, Uppsala, Sweden). PCR-based cDNA sequencing of BCR-ABL1 was performed to detect mutations in codons 221 to 500 of the ABL1 kinase domain, including codon 315.
3 | RESULTS
3.1 | Clinical and laboratory findings
The salient clinical features for the 10 patients are summarized in Table 1. There were six (60%) men and four (40%) women with a median age of 51 years (range, 20–71 years) at diagnosis. Patients were classified into three groups based on the sequence of genetic alterations: those who presented with BCR-ABL1 first and later acquired CBFB rearrangement (n =7); those who presented with simultaneous BCR-ABL1 and CBFB rearrangement (n =1); and those who initially presented with CBFB rearrangement and later acquired BCR-ABL1 (n =1). For one patient, the sequence of events is unknown. Accordingly, the first group represented patients who had CML with progression to blast phase at the time of acquiring CBFB rearrangement, whereas the other two groups represented patients who, by definition, had AML. The median interval from CML diagnosis to acquiring CBFB rearrangement was 11 months (range, 5–43) in patients who originally presented with CML in chronic phase.
TABLE 1.
Summary of clinical features
| Patient | Age at initial dx | Sex | Initial dx | Sequence of CG alterationsa | Time from initial dx to CBFB rearrangement (months) | Therapy | Response to therapy | Status at last FU | Interval from CBFB rearrangement to relapse | Length of FU from initial diagnosis (months) | Length of FU from t(9:22) and CBFB rearrangement (months) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 71 | M | AML | A | 0 | FLAG-Ida+ dasatinib | Remission | Alive | NA | 21 | 21 |
| 2 | 66 | M | AML | C | 0 | 3 +7 and consolidation with high-dose cytarabine; MEK inhibitor, GSK1120212; single agent decitabine | Relapse | Dead | 8 | 24 | 24 |
| 3 | 55 | F | AML | U | 0 | High dose cytarabine and idarubicin; imatinib+clofarabine and cytarabine; dasatinib | Relapse/persistent | Alive | 7c | 11 | 2 |
| 4 | 20 | F | CML-CP | B | 8 | Interferon alpha and leukophoresisb; imatinib and Hydrea; fludarabine and cytarabine and IT idarubicin and cytarabine; Allo-SCT | Relapse/persistent | Dead | 8 | 27 | 18 |
| 5 (3) | 61 | F | CML-BP | B | 5 | Hydrea; imatinib; idarubicin, and cytarabine; cytarabine+ imatinib | Relapse/persistent | Dead | 4 | 11 | 6 |
| 6 (1) | 43 | F | CML-CP | B | 11 | Hydrea +; imatinib; cytarabine+fludarabine | Relapse | Dead | 3 | 20 | 9 |
| 7 (2) | 48 | M | CML-AP | B | 30 | Hydrea +leukophoresis; imatinib; cytarabine+ idarubicin+etoposide | Persistent | Dead | 0 | 115 | 85 |
| 8 (4) | 47 | M | CML-CP | B | 6 | Interferon alpha+ cytarabine+ATRA; allo-SCT | Remission | Deadd | NA | 20 | 14 |
| 9 | 54 | M | CML-CP | B | 26 | FLAG-IA+ponatinib; allo-SCT | Remission | Alive | NA | 41 | 27 |
| 10 | 40 | M | CML-CP | B | 43 | Ponatinib | Persistent | Dead | 0 | 47 | 4 |
Abbreviations: Allo-SCT, allogeneic stem cell transplant; AML, acute myeloid leukemia; CG, cytogenetic; CML-BP, chronic myelogenous leukemia-blast phase; CML-CP, chronic myelogenous leukemia-chronic phase; dx, diagnosis; F, female; FU, follow up; IT, intrathecal.
Sequence of CG alterations: A: simultaneous BCR-ABL1 and CBFB rearrangement at initial presentation; B: BCR-ABL1 first CBFB rearrangement later; C: CBFB rearrangement first BCR-ABL1 later; U: unknown.
The patient was pregnant at this time.
Time from initial diagnosis.
Because of graft versus host disease.
At presentation, all patients had anemia (range, 7–13.8 g/dL; normal range, 12–16 g/dL for women and 14–18 g/dL for men), five patients had leukocytosis (range, 21.3–362.7×103/ul; reference range, 4.0–11.0 ×103/ul), and four patients had leukopenia (range, 1.9–3.1 ×103/ul). Eight patients had thrombocytopenia (range, 12–73 ×103/ul; normal range, 140–440× 103/ul) at presentation.
3.2 | Morphologic features
The median BM cellularity was 90% (range, 30–100%). The median BM blast and eosinophil percentages were 26% (range, 20–87%) and 8% (range, 2–30%), respectively, at the time of BCR-ABL1 and CBFB rearrangement co-occurrence. The median BM monocyte percentage was 3% (range, 0–26%). Morphologically abnormal eosinophils and eosinophilic precursors with immature eosinophilic granules were identifiable in 6 of 10 cases. (Figures 1 and 2)
FIGURE 1.
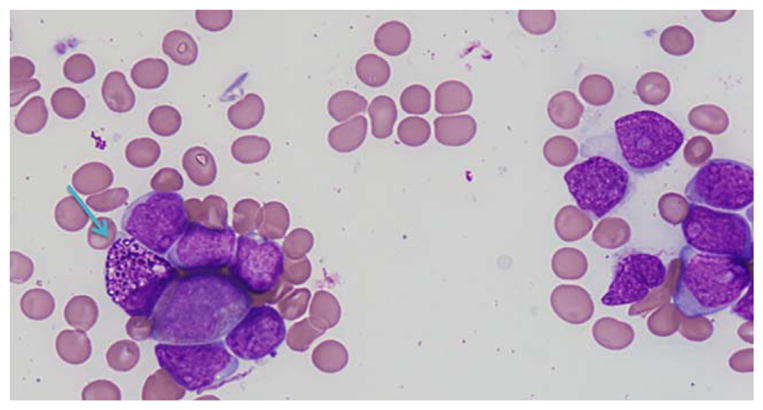
Bone marrow aspirate smear from patient #2 showing increased blasts and an abnormal eosinophilic precursor (blue arrow). (Giemsa stain, 600×) This patient presented with de novo AML harboring concurrent BCR-ABL1 (e1a2) and CBFB rearrangement [Color figure can be viewed at wileyonlinelibrary.com]
FIGURE 2.
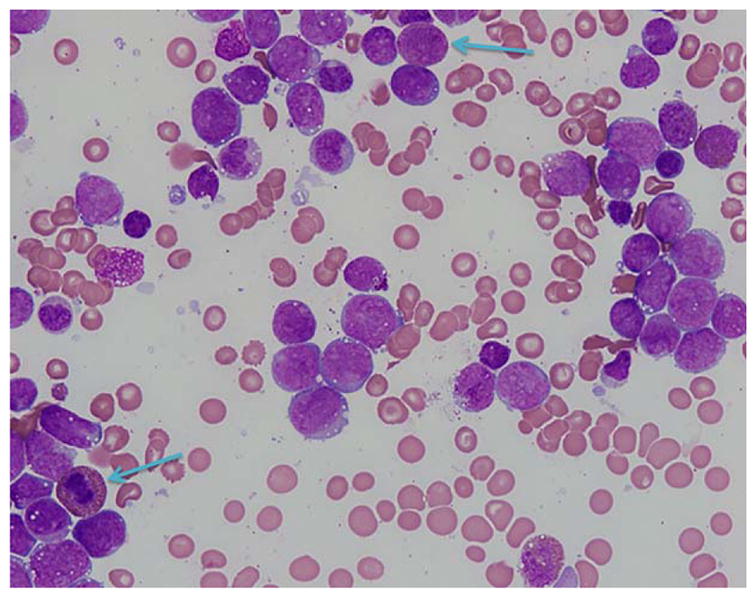
Bone marrow aspirate smear from patient #4 showing increased blasts with monocytic features and occasional eosinophilic precursors (blue arrows). (Giemsa stain, 600×) This patient presented with CML blast phase with BCR-ABL1 (b3a2 and b2a2) and later acquired CBFB rearrangement. The photograph depicts the sample with concurrent BCR-ABL1 and CBFB rearrangement [Color figure can be viewed at wileyonlinelibrary.com]
3.3 | Flow cytometry immunophenotyping results
Aberrant myeloid blasts were identified in all cases (n =9) with available data. The most common alterations included increased expression of CD13 and decreased expression of CD33, CD38, and HLA-DR. Flow cytometric immunophenotyping details for each case are provided in Table 2.
TABLE 2.
Summary of clinical features and immunophenotypic features at the time of BCR-ABL1 and CBFB rearrangement co-occurrence
| Case | BM blasts% | Blast immunophenotype | BM eosinophil % | BM monocyte % | Abnormal eosinophils in BM |
|---|---|---|---|---|---|
| 1 | 20 | CD2dim+, sCD3−, CytoCD3−, CD4partial+, CD5−, CD7−, CD10−, CD13+/increased, CD14−, CD15partial +, CD19−, CD22−, CD33+/decreased, CD34+, CD36−, CD38+/decreased, CD41−, CD45dim+, CD56−, CD64 partial+, CD117+, CD123+/increased, HLA-DR+/decreased, MPO+, TDT dim+. | 10 | 26 | Yes |
| 2 | 62 | CD2−, sCD3−, CytoCD3−, CD5−, CD7−, CD10−, CD13+, CD14−, CD15partial +, CD19−, CD33+/decreased, CD34+, CD38+/decreased, CD41−, CD45dim+, CD56−, CD64 −, CD117+, HLA-DR+/decreased, MPO+, TDT −. | 10 | 1 | Yes |
| 3a | 54 | CD2−, sCD3−, CytoCD3−, CD5−, CD7−, CD10−, CD13+, CD14−, CD15partial +, CD19−, CD33+, CD34+, CD38+/decreased, CD41−, CD45dim+, CD56−, CD64 −, CD117+, HLA-DR+/decreased, MPO+, TDT −. | 4 | 2 | No |
| 4 | 87 | CD7−, CD10−, CD13+, CD14−, CD19−, CD33+, CD34+, CD38+, CD45dim+, CD56−, CD64 partial+, CD117+, HLA-DR+, MPO+, TDT −. | 2 | 0 | No |
| 5 (3) | 20 | CD7−, CD10−, CD13+, CD14−, CD19−, CD33+, CD34 partial+, CD38+, CD45dim+, CD56−, CD64 −, CD117+, HLA-DR+, MPO partial+, TDT −. | 5 | 7 | Yes |
| 6 (1) | 30 | CD7−, CD10−, CD13+, CD14−, CD19−, CD33+, CD34 partial+, CD38+/decreased, CD45dim+, CD56−, CD64 partial+, CD117+, HLA-DR+/decreased, MPO partial+, TDT −. | 22 | 19 | Yes |
| 7 (2) | 20 | N/A | 30 | 9 | Yes |
| 8 (4) | 40 | CD2−, CD7−, CD10−, CD13+, CD19−, CD33+/increased, CD34 small subset+, CD64+, HLA-DR+ | 11 | 2 | Yes |
| 9a | 20 | CD2−, sCD3−, CytoCD3−, CD4−, CD5−, CD7−, CD10−, CD13+, CD14−, CD15partial +, CD19−, CD22−, CD33+/decreased, CD34+, CD36−, CD38+/decreased, CD41−, CD45dim+, CD56−, CD64−, CD117+, CD123+, HLA-DR+/decreased, MPO+, TDT dim+. | 5 | 16 | No |
| 10 | 21 | CD2−, CD5−, CD7−, CD10−, CD13+, CD14−, CD15 partial+, CD19−, CD33+/decreased, CD34 partial+, CD38+, CD45dim+, CD56−, CD64 −, CD117+, HLA-DR+/decreased, MPO partial+, TDT −. | 2 | 0 | No |
These patients were previously treated with chemotherapy.
Intensities are reported in comparison with normal myeloid blasts.
Patients 5–8 are those previously reported by Merzianu, M et al. (Am J Clin Pathol. 2005 Nov;124(5):807–14.). Designations in that article are provided parenthetically.
3.4 | Cytogenetics results
Detailed cytogenetic results at the time of initial presentation and at the time of co-detection of BCR-ABL1 and CBFB rearrangements are provided in Table 3. Case #1 had two clones at initial diagnosis: a clone with inv(16) only as stemline and a clone with both inv(16) and t(9;22) as a sideline (Figure 3A); Case #2 had inv(16) at initial diagnosis, 14 months later the patient developed clonal evolution with acquisition of t(9;22); Case #3 and #4 had only one clone with t(9;22) and inv(16) detected simultaneously; The remaining six cases (cases #5–10) had t(9;22) at initial diagnosis of CML and acquired inv(16) during blast crisis.
TABLE 3.
Summary of cytogenetic and molecular features
| Case | Karyotype at initial diagnosis | Karyotype at BCR-ABL1 and CBFB rearrangementco-occurrence | % nuclei positive for BCR-ABL1:and CBFB rearrangement by FISH | BCR-ABL1 fusion transcript | ABL1 kinase mutation | Other mutations |
|---|---|---|---|---|---|---|
| 1 | 46,XY,inv(16)(p13.1q22)[3] 46,idem,t(9;22)(q34;q11.2)[17] |
Same | NA:NA | e1a2 | ND | None (next generation sequencing, multiple genes.) |
| 2 | 46,XY,inv(16)(p13.1q22) | 46,XY,inv(16)(p13q22)[2] 48,idem,t(9;22)(q34;q11.2),+13,+22[16] 47~48,idem,+13[cp2] |
4.5%:92% | e1a2 | p.G254E and p.E329G | None (tested for FLT3, KRAS, NRAS, NPM1, KIT) |
| 3 | NA | 46,XX,t(9;22)(q34;q11.2),inv(16)(p13.1q22)[19] 46,XX[1] |
91.5%:81% | e1a2 | ND | None (tested for FLT3, KRAS, NRAS, KIT) |
| 4 | NA | 46,XX,t(9;22)(q34;q11.2),inv(16)(p13.1q22)[20] | 99.5%:83% | b3a2 and b2a2 | None | None (tested for FLT3, KRAS, NRAS) |
| 5 (3) | 46,XX,t(9;22)(q34;q11.2)[20] | 46,XX,t(9;22)(q34;q11.2)[3] 46,XX,idem,inv(16)(p13q22)[7]/47,idem,+8[2] 47,idem,+8,inv(16)(p13q22)[4] 46~47,XX,t(9;22)(q34;q11.2),inv(16)(p13q22)[cp2] /46,XX[2] |
59%:57.5% | b3a2 and b2a2 | None | None (tested for FLT3) |
| 6 (1) | 46,XX,t(9;22)(q34;q11.2) | 46,XX,t(9;22)(q34;q11.2),inv(16)(p13q22)[20] | 91%:NA | b2a2 | ND | ND |
| 7 (2) | 46,XY,t(9;22)(q34;q11.2)[20] | 46,XY,t(9;22)(q34;q11.2),inv(16)(p13q22)[6]/46,XY[14] | NA:NA | b2a2 | None | ND |
| 8 (4) | 46,XY,t(9;22)(q34;q11.2)[20] | 46,XY,t(9;22)(q34;q11),inv(16)(p13q22)[25] | NA:NA | b2a2 | ND | ND |
| 9 | 46,XY,t(9;22)(q34;q11.2)[10] | 46,XY,t(9;22)(q34;q11.2),inv(16)(p13.1q22)[8] 47,idem,+der(22)t(9;22)[4] 46,XY[8] |
52%:45% | b3a2 and b2a2 | None | ND |
| 10 | 46,XX,t(9;22)(q34;q11.2) | 46,X,-Y,+8,t(9;22)(q34;q11.2)[5] 46,idem,inv(16)(p13q22)[7] 47,XY,+Y[2] 46,idem,t(3;18)(p21;q23)[1] 46,XY[5] |
41%: 59.5% | b3a2 | None | ND |
Abbreviations: NA: not available; ND: not done.
+++ Patients 5–8 are those previously reported by Merzianu, M et al. (Am J Clin Pathol. 2005 Nov;124(5):807–14.). Designations in that article are provided parenthetically.
FIGURE 3.

Karyotype and FISH analysis of case #1. A. Karyotype of 46,XY,t(9;22)(q34;q11.2),inv(16)(p13q22). B. FISH analysis using LSI CBFB breakapart probe on a metaphase. Chromosome with break signal (red & green, marked by arrow) indicates CBFB rearrangement; C. FISH analysis using LSI BCR/ABL1 ES probe on a metaphase. Two fusion signals (yellow, marked by arrow head) indicate BCR/ABL1 rearrangement. [Color figure can be viewed at wileyonlinelibrary.com]
FISH analysis using LSI BCR/ABL1 ES probe and CBFB breakapart probe showed equal or similar percentages of nuclei positive for BCR-ABL1 and CBFB rearrangement (Figure 3B, C) in all cases except case #2 who showed CBFB rearrangement in 92% of nuclei and BCR-ABL1 rearrangement in 4.5% of the nuclei. This patient presented with AML with CBFB rearrangement and later acquired BCR-ABL1 as a secondary genetic alteration.
3.5 | Molecular results
A p210 kD BCR-ABL1 product was identified in all cases in which BCR-ABL1 fusion preceded CBFB rearrangement, whereas a p190 kD product was identified in the other three cases. Among six cases analyzed for ABL1 kinase domain mutations, one case showed two distinct mutations (p.G254E and p.E329G). All evaluated cases were negative for FLT3 (n =5), NRAS and KRAS (n =4), and KIT (n =3) mutations.
3.6 | Treatment and outcome
Treatment details are provided in Table 1. Briefly, two patients were treated with the FLAG-IDA regimen (fludarabine, cytarabine, idarubicin, and G-CSF) and TKIs; one of which ultimately received an allogeneic stem cell transplant. Seven patients were treated with cytarabine-based regimens and TKIs; in two of these patients treatment was followed by allogeneic stem cell transplant. One patient was treated with ponatinib alone.
Seven of 10 patients were dead at time of last follow-up [median, 16 months; range 2–85 from the time of t(9;22) and inv(16)]. Of the three patients alive, two received FLAG-IDA and TKI; one had CML-BP and another had AML with both alterations discovered simultaneously; the sequence of events is unknown in the third patient. The latter two patients harbored the e1a2 fusion transcript and the former had a b3a2/b2a2 fusion.
4 | DISCUSSION
We describe 10 patients with simultaneous occurrence of BCR-ABL1 and CBFB rearrangement. The co-occurrence of BCR-ABL1 and CBFB rearrangement is an extremely rare event with less than a total of 20 cases reported in the literature.18,20,29–38 There is a male predominance, and the disease can present in any age, although patients tend to be older than those with AML with inv(16).13
These patients can generally be classified into two major groups: those who present with chronic phase CML and progress to CML-BP by means of acquiring inv(16)(p13q22) and those who present with de novo AML in which both genetic alterations are discovered simultaneously or, in very rare cases, inv(16) precedes BCR-ABL1. Among the patients included in this study, those with an antecedent history of CML carried the p210 kD fusion protein whereas all patients with de novo AML with inv(16) and BCR-ABL1 carried the p190 fusion protein. These results are similar to what has those reported by previous authors19,21 and suggests that the biology of the two processes is distinct. Although rare patients with CML-BP with inv(16) have been reported to carry the p190 fusion protein, there were no cases in this study group.21
Although flow cytometry is not routinely used for follow-up of patients with CML, aberrant myeloid blasts were identified in all cases included in this study suggesting that flow cytometry panels designed for the detection of minimal residual acute myeloid leukemia may be useful in monitoring disease in patients with concurrent inv(16) and BCR-ABL1. This approach may be useful in patient where RNA may not be readily available for minimal residual disease detection by PCR.
Among six patients evaluated for the ABL1 kinase domain mutation, one showed two distinct mutations whereas the other five had wild-type ABL1. Of note, the patient with mutated ABL1 presented initially with AML with inv(16) and had not been treated with tyrosine kinase inhibitors prior to the discovery of the ABL1 kinase domain mutations. Mutations in NRAS, KIT and FLT3 have been reported to occur in 35% of cases of AML with inv(16).39,40 All of the cases evaluated in this series were wild type for these three genes, suggesting that the potent leukemogenic effect executed by the simultaneous presence of both chromosomal alterations precludes the need for additional somatic mutations for survival of the leukemic clone.
Based on their review of the literature, Ninomiya and colleagues suggested that de novo AML with BCR-ABL1 has a favorable prognosis when compared with other acute myeloid leukemias, similar to that of AML with inv(16) alone.21 Two such patients were included in this cohort; one patient was treated with the FLAG-Ida regimen plus dasatinib and had a favorable clinical course and was in remission 21 months following presentation. The second patient was treated with the 3 +7 regimen and tyrosine kinase inhibitors (details included in Table 1), relapsed 8 months after presentation and succumbed to disease two years following initial diagnosis. The prognosis of patients with CML and inv(16) as a secondary genetic alteration on the other hand seems to be very poor.21 In fact, six of seven patients included in our series died despite intensive chemotherapy and targeted therapy with tyrosine kinase inhibitors (median OS from the time of inv(16): 14 months, range 4–85 months). One patient was alive and free of disease at the time of last follow-up; this patient was treated with the FLAG-IA regimen plus ponatinib and later received an allogeneic stem cell transplant. Although the number of patients in this series, and in the literature in general is too small to draw a definite conclusion, it appears that patients with concurrent inv(16) and BCR-ABL1 may benefit from intensive chemotherapy regimens such as the FLAG-Ida plus tyrosine kinase inhibitors.
Footnotes
CONFLICT OF INTEREST DISCLOSURE
The authors have no relevant conflicts of interest to disclose.
AUTHOR CONTRIBUTIONS
Design and conception: JDK; Data gathering and analysis: AS, LJM, GT, SL, JDK; Manuscript preparation: AS, LJM, GT, SL, WW, JDK. All authors have read and approved the final version of this manuscript.
References
- 1.Vardiman JW, Melo JV, Baccarani M, et al. Chronic myelogenous leukaemia, BCR-ABL1 positive. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 32–37. [Google Scholar]
- 2.Konoplev S, Yin CC, Kornblau SM, et al. Molecular characterization of de novo Philadelphia chromosome-positive acute myeloid leukemia. Leuk Lymphoma. 2013;54:138–144. doi: 10.3109/10428194.2012.701739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jabbour E, Kantarjian H, Ravandi F, et al. Combination of hyper-CVAD with ponatinib as first-line therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukaemia: a single-centre, phase 2 study. Lancet Oncol. 2015;16:1547–1555. doi: 10.1016/S1470-2045(15)00207-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2016 update on diagnosis, therapy, and monitoring. Am J Hematol. 2016;91:252–265. doi: 10.1002/ajh.24275. [DOI] [PubMed] [Google Scholar]
- 5.Huang X, Cortes J, Kantarjian H. Estimations of the increasing prevalence and plateau prevalence of chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Cancer. 2012;118:3123–3127. doi: 10.1002/cncr.26679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang W, Cortes JE, Tang G, et al. Risk stratification of chromosomal abnormalities in chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Blood. 2016;127:2742–2750. doi: 10.1182/blood-2016-01-690230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Z, Shao C, Wang W, et al. Cytogenetic landscape and impact in blast phase of chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Leukemia. 2017;31:585–592. doi: 10.1038/leu.2016.231. [DOI] [PubMed] [Google Scholar]
- 8.Wang W, Chen Z, Hu Z, et al. Clinical significance of trisomy 8 that emerges during therapy in chronic myeloid leukemia. Blood Cancer J. 2016;6:e490. doi: 10.1038/bcj.2016.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang W, Ali S, Tang Z, et al. Constitutional pericentric inversion of chromosome 9 has no impact on survival in chronic myelogenous leukemia. Ann Hematol. 2016;95:657–659. doi: 10.1007/s00277-016-2592-3. [DOI] [PubMed] [Google Scholar]
- 10.Chen Z, Cortes JE, Jorgensen JL, et al. Differential impact of additional chromosomal abnormalities in myeloid vs lymphoid blast phase of chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Leukemia. 2016;30:1606–1609. doi: 10.1038/leu.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang W, Tang G, Cortes JE, et al. Chromosomal rearrangement involving 11q23 locus in chronic myelogenous leukemia: a rare phenomenon frequently associated with disease progression and poor prognosis. J Hematol Oncol. 2015;8:32. doi: 10.1186/s13045-015-0128-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang W, Cortes JE, Lin P, et al. Impact of trisomy 8 on treatment response and survival of patients with chronic myelogenous leukemia in the era of tyrosine kinase inhibitors. Leukemia. 2015;29:2263–2266. doi: 10.1038/leu.2015.96. [DOI] [PubMed] [Google Scholar]
- 13.Arber DA, Brunning RD, Le Beau MM, et al. Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: International Agency for Research on Cancer; 2008. pp. 110–123. [Google Scholar]
- 14.Sun X, Zhang W, Ramdas L, et al. Comparative analysis of genes regulated in acute myelomonocytic leukemia with and without inv (16)(p13q22) using microarray techniques, real-time PCR, immunohistochemistry, and flow cytometry immunophenotyping. Mod Pathol. 2007;20:811–820. doi: 10.1038/modpathol.3800829. [DOI] [PubMed] [Google Scholar]
- 15.Sun X, Medeiros LJ, Lu D, et al. Dysplasia and high proliferation rate are common in acute myeloid leukemia with inv(16)(p13q22) Am J Clin Pathol. 2003;120:236–245. doi: 10.1309/PGNT-8LGN-9AR4-QVAJ. [DOI] [PubMed] [Google Scholar]
- 16.Richter L, Wang Y, Hyde RK. Targeting binding partners of the CBFbeta-SMMHC fusion protein for the treatment of inversion 16 acute myeloid leukemia. Oncotarget. 2016;7:66255–66266. doi: 10.18632/oncotarget.11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwind S, Edwards CG, Nicolet D, et al. inv(16)/t(16;16) acute myeloid leukemia with non-type A CBFB-MYH11 fusions associate with distinct clinical and genetic features and lack KIT mutations. Blood. 2013;121:385–391. doi: 10.1182/blood-2012-07-442772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merzianu M, Medeiros LJ, Cortes J, et al. inv(16)(p13q22) in chronic myelogenous leukemia in blast phase: a clinicopathologic, cytogenetic, and molecular study of five cases. Am J Clin Pathol. 2005;124:807–814. doi: 10.1309/3HFE-16DK-MB1D-BFMN. [DOI] [PubMed] [Google Scholar]
- 19.Cividin M, Brizard F, Sorel N, et al. p190(BCR-ABL) rearrangement as a secondary change in a case of acute myelomonocytic leukemia with inv(16)(p13q22) Leuk Res. 2004;28:97–99. doi: 10.1016/s0145-2126(03)00161-9. [DOI] [PubMed] [Google Scholar]
- 20.Heim S, Christensen BE, Fioretos T, et al. Acute myelomonocytic leukemia with inv(16)(p13q22) complicating Philadelphia chromosome positive chronic myeloid leukemia. Cancer Genet Cytogenet. 1992;59:35–38. doi: 10.1016/0165-4608(92)90154-z. [DOI] [PubMed] [Google Scholar]
- 21.Ninomiya S, Kanemura N, Tsurumi H, et al. Coexistence of inversion 16 and the Philadelphia chromosome comprising P190 BCR/ABL in chronic myeloid leukemia blast crisis. Int J Hematol. 2011;93:806–810. doi: 10.1007/s12185-011-0854-3. [DOI] [PubMed] [Google Scholar]
- 22.Thiele J, Kvasnicka HM, Facchetti F, et al. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90:1128–1132. [PubMed] [Google Scholar]
- 23.Khoury JD, Sen F, Abruzzo LV, et al. Cytogenetic findings in blastoid mantle cell lymphoma. Hum Pathol. 2003;34:1022–1029. doi: 10.1053/s0046-8177(03)00412-x. [DOI] [PubMed] [Google Scholar]
- 24.Shaffer LG, McGowan-Jordan J, Schmid M. ISCN (2013): An International System for Human Cytogenetic Nomenclature. Basel, Switzerland: S. Karger; 2013. [Google Scholar]
- 25.Luthra R, Medeiros LJ. TaqMan reverse transcriptase-polymerase chain reaction coupled with capillary electrophoresis for quantification and identification of bcrabl transcript type. Methods Mol Biol. 2006;335:135–145. doi: 10.1385/1-59745-069-3:135. [DOI] [PubMed] [Google Scholar]
- 26.Gabert J, Beillard E, van der Velden VH, et al. Standardization and quality control studies of ’real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—A Europe Against Cancer program. Leukemia. 2003;17:2318–2357. doi: 10.1038/sj.leu.2403135. [DOI] [PubMed] [Google Scholar]
- 27.Zhang L, Singh RR, Patel KP, et al. BRAF kinase domain mutations are present in a subset of chronic myelomonocytic leukemia with wild-type RAS. Am J Hematol. 2014;89:499–504. doi: 10.1002/ajh.23652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warren M, Luthra R, Yin CC, et al. Clinical impact of change of FLT3 mutation status in acute myeloid leukemia patients. Mod Pathol. 2012;25:1405–1412. doi: 10.1038/modpathol.2012.88. [DOI] [PubMed] [Google Scholar]
- 29.Mecucci C, Noens L, Aventin A, et al. Philadelphia-positive acute myelomonocytic leukemia with inversion of chromosome 16 and eosinobasophils. Am J Hematol. 1988;27:69–71. doi: 10.1002/ajh.2830270118. [DOI] [PubMed] [Google Scholar]
- 30.Secker-Walker LM, Morgan GJ, Min T, et al. Inversion of chromosome 16 with the Philadelphia chromosome in acute myelomonocytic leukemia with eosinophilia. Report of two cases. Cancer Genet Cytogenet. 1992;58:29–34. doi: 10.1016/0165-4608(92)90129-v. [DOI] [PubMed] [Google Scholar]
- 31.Enright H, Weisdorf D, Peterson L, et al. Inversion of chromosome 16 and dysplastic eosinophils in accelerated phase of chronic myeloid leukemia. Leukemia. 1992;6:381–384. [PubMed] [Google Scholar]
- 32.Asou N, Sanada I, Tanaka K, et al. Inversion of chromosome 16 and bone marrow eosinophilia in a myelomonocytic transformation of chronic myeloid leukemia. Cancer Genet Cytogenet. 1992;61:197–200. doi: 10.1016/0165-4608(92)90086-n. [DOI] [PubMed] [Google Scholar]
- 33.Anastasi J, Feng J, Le Beau MM, et al. The relationship between secondary chromosomal abnormalities and blast transformation in chronic myelogenous leukemia. Leukemia. 1995;9:628–633. [PubMed] [Google Scholar]
- 34.Evers JP, Bagg A, Himoe E, et al. Temporal association of marrow eosinophilia with inversion of chromosome 16 in recurrent blast crises of chronic myelogenous leukemia. Cancer Genet Cytogenet. 1992;62:134–139. doi: 10.1016/0165-4608(92)90251-3. [DOI] [PubMed] [Google Scholar]
- 35.Colovic M, Jankovic G, Bila J, et al. Inversion of chromosome 16 in accelerated phase of chronic myeloid leukaemia: report of a case and review of the literature. Med Oncol. 1998;15:199–201. doi: 10.1007/BF02821939. [DOI] [PubMed] [Google Scholar]
- 36.Myint H, Ross FM, Hall JL, et al. Early transformation to acute myeloblastic leukaemia with the acquisition of inv(16) in Ph positive chronic granulocytic leukaemia. Leuk Res. 1997;21:473–474. doi: 10.1016/s0145-2126(97)00070-2. [DOI] [PubMed] [Google Scholar]
- 37.Tsuboi K, Komatsu H, Miwa H, et al. Lymphoid blastic crisis of chronic myelogenous leukaemia with inv(16)(p13;q22) Leuk Res. 2002;26:771–774. doi: 10.1016/s0145-2126(01)00199-0. [DOI] [PubMed] [Google Scholar]
- 38.Mohamed AN, Pemberton P, Zonder J, et al. The effect of imatinib mesylate on patients with Philadelphia chromosome-positive chronic myeloid leukemia with secondary chromosomal aberrations. Clin Cancer Res. 2003;9:1333–1337. [PubMed] [Google Scholar]
- 39.Faber ZJ, Chen X, Gedman AL, et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet. 2016;48:1551–1556. doi: 10.1038/ng.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim HG, Kojima K, Swindle CS, et al. FLT3-ITD cooperates with inv (16) to promote progression to acute myeloid leukemia. Blood. 2008;111:1567–1574. doi: 10.1182/blood-2006-06-030312. [DOI] [PMC free article] [PubMed] [Google Scholar]