

SUMMARY
While therapeutic modulation of miRNAs provides a promising approach for numerous diseases, the promiscuous nature of miRNAs raises concern over detrimental off-target effects. miR-33 has emerged as a likely target for treatment of cardiovascular diseases. However, the deleterious effects of long-term anti-miR-33 therapies and predisposition of miR-33−/− mice to obesity and metabolic dysfunction exemplify the possible pitfalls of miRNA-based therapies. Our work provides an in-depth characterization of miR-33−/− mice and explores the mechanisms by which loss of miR-33 promotes insulin resistance in key metabolic tissues. Contrary to previous reports, our data do not support a direct role for SREBP-1-mediated lipid synthesis in promoting these effects. Alternatively, in adipose tissue of miR-33−/− mice, we observe increased pre-adipocyte proliferation, enhanced lipid uptake, and impaired lipolysis. Moreover, we demonstrate that the driving force behind these abnormalities is increased food intake, which can be prevented by pair feeding with wild-type animals.
In Brief
While anti-miR-33 therapies offer promise for treating cardiovascular disease, deletion of miR-33 causes obesity and metabolic dysfunction. Price et al. elucidate how miR-33 deficiency affects metabolic functions in different tissues. Rather than finding that dysregulation of SREBP-1-mediated lipid synthesis primarily drives these effects, they demonstrate that these changes depend on increased food consumption.
INTRODUCTION
The spread of obesity across first-world nations and into developing countries has reached epidemic levels, and estimates indicatethat by 2025 nearly 3 billion people worldwide willbe overweight (Lam and LeRoith, 2012). This will make up nearly half of the total population, and of these over one-third will be either obese or severely obese. Obesity is the single biggest risk factor in a multitude of metabolic diseases, including certain cancers, diabetes, and cardiovascular disease (Grundy, 2016; Lam and LeRoith, 2012). As such, the dramatic rise in obesity has put a serious strain on public health networks across the globe. Although lifestyle changes are sufficient to combat many of the negative effects associated with obesity, genetic differences also play an important role in the development of these conditions. As such, our incomplete understanding of the mechanisms that regulate the development of obesity and metabolic dysfunction have impeded our ability to combat this growing public health issue.
Over the years, researchers have made substantial progress in trying to understand the many layers of regulation involved in the development of this condition, and recent work has demonstrated microRNAs (miRNAs) and other non-coding RNAs compose yet another level of regulation that is important for the development of obesity and metabolic dysfunction (Rottiers and Näär, 2012). miRNAs are small non-coding RNAs that can bind to specific regions of target genes leading to mRNA degradation or translational repression and have been shown to play an important role in the regulation of nearly all biological processes (Ambros, 2004; Chen, 2009; Fernández-Hernando et al., 2011, 2013). Among these, miRNAs have been demonstrated to regulate key functions in a variety of important metabolic organs including the liver, brain, pancreas, muscle, and adipose tissue, which can have a substantial impact on the development of obesity and metabolic dysfunction (Fernández-Hernando et al., 2013). Indeed, impairment of miRNA processing in adipose tissue was found to have dramatic effects on fat accumulation and distribution (Kim et al., 2014; Mori et al., 2014), and a number of individual miRNAs have been demonstrated to regulate adipocyte differentiation and function directly (Price and Fernández-Hernando, 2016). Moreover, work done in vivo has demonstrated that either genetic ablation or pharmacologic inhibition of a number of miRNAs, including Let-7, miR-33, miR-93, miR-130b, miR-143/145, miR146b, and miR-375 can have a major impact on the development of obesity and metabolic dysfunction (Ahn et al., 2013; Cioffi et al., 2015; Frost and Olson, 2011; Horie et al., 2013; Jordan et al., 2011; Pan et al., 2015; Poy et al., 2009; Price et al., 2016). Among these, miR-33 is of particular interest because of its well-established role in the regulation of lipid metabolism and the development of atherosclerosis (Marquart et al., 2010; Najafi-Shoushtari et al., 2010; Rayner et al., 2010).
The miR-33 family of miRNAs have been demonstrated to be important regulators of lipid metabolism by targeting a number of genes involved in reverse cholesterol transport (RCT) (Marquart et al., 2010; Najafi-Shoushtari et al., 2010; Rayner et al., 2010). The RCT pathway is the only mechanism by which peripheral tissues, including the macrophages that make up atherosclerotic plaques, can remove excess cholesterol for transportation to and degradation by the liver (Rosenson et al., 2012). Through its ability to target genes involved in cholesterol efflux (ABCA1, ABCG1), high-density lipoprotein (HDL) biogenesis (ABCA1), and bile acid metabolism, miR-33 has been established as an important regulator of this process (Allen et al., 2012; Li et al., 2013; Marquart et al., 2010; Najafi-Shoushtari et al., 2010; Rayner et al., 2010). Consistent with this, antagonism or loss of miR-33 has been shown to increase circulating levels of HDL-C in both rodents and non-human primates (Horie et al., 2010; Marquart et al., 2010; Najafi-Shoushtari et al., 2010; Rayner et al., 2010, 2011a; Rottiers et al., 2013). Moreover, a number of studies have demonstrated that treatment with anti-miR-33 therapeutic agents can significantly reduce plaque burden in mouse models of atherosclerosis (Rayner et al., 2011b; Rotllan et al., 2013).
In addition to its role in the control of lipid metabolism, miR-33 has been found to target genes involved in a number of other important metabolic functions including fatty acid metabolism (CPT1, CROT, HADHβ), insulin signaling (IRS2), and mitochondrial function (AMPK, PGC1α) (Dávalos et al., 2011; Gerin et al., 2010; Karunakaran et al., 2015; Ouimet et al., 2015, 2017). The ability of miR-33 to regulate such a wide array of different metabolic functions suggests that miR-33 may be involved in regulation of different metabolic functions in different tissues. This is especially interesting because miR-33a and miR-33b are intronic miRNAs encoded within intronic regions of the SREBP-2 and SREBP-1 genes. The SREBP transcription factors themselves are some of the most important regulators of cellular cholesterol and fatty acid metabolism, and have been demonstrated to be differentially regulated in a number of different metabolic tissues under conditions of obesity and insulin resistance (Horton et al., 2002).
Although early reports suggest that anti-miR-33 therapy may be an effective approach for the treatment of cardiovascular disease, some long-term studies with anti-miR-33 therapy have demonstrated the development of adverse outcomes including hypertriglyceridemia and hepatic steatosis (Allen et al., 2014; Goedeke et al., 2014). Our recent work has demonstrated that hematopoietic specific loss of miR-33 in the Ldlr−/− mouse model of atherosclerosis promotes RCT in vivo and reduces plaque burden. However, in whole body knockout mice these beneficial effects in plaque macrophages are offset by pro-atherogenic changes in circulating lipids as well as increased body weight and insulin resistance (Price et al., 2017). These findings are consistent with an earlier metabolic characterization of mice genetically deficient in miR-33, which showed a dramatic predisposition to the development of diet induced obesity and insulin resistance (Horie et al., 2013). These surprising effects were attributed to dysregulation of SREBP-1 in the liver, as loss of miR-33 was not found to impact body weight or hepatic steatosis in heterozygous SREBP-1 knockout mice (Horie et al., 2013). While these findings are interesting, and highlight the importance of lipid metabolism in the liver for maintaining metabolic function, prior work suggests that additional mechanisms may be primarily responsible for the obesity phenotype. Although expression of SREBP-1 and its targets are often altered in cells and tissues lacking miR-33, our previous findings do not indicate direct targeting of the SREBP-1 gene by miR-33 (Goedeke et al., 2014). Additionally, SREBP-1 is primarily regulated at a post-translational level, and even overexpression of a mature form of SREBP-1, that does not require cleavage for entry into the nucleus, does not result in the form of obesity observed in miR-33-deficient mice (Shimano et al., 1997). These findings, coupled with the near complete loss of miR-33 in the liver of animals treated with miR-33 inhibitors, indicate that other mechanisms and possibly other organs may be involved in the development of these phenotypes.
In this work, we provide a more complete characterization of the metabolic dysfunction observed in miR-33-deficient mice and explore additional mechanisms by which loss of miR-33 leads to the development of obesity and insulin resistance. In addition to the metabolic dysfunction previously reported in miR-33-deficient mice fed a high-fat diet (HFD) (Horie et al., 2013), we demonstrate that even in mice fed a chow diet (CD), loss of miR-33 results in impaired responsiveness to insulin in multiple metabolic organs including the liver, white adipose tissue (WAT), and skeletal muscle. By performing hyperinsulinemic-euglycemic clamp studies in our miR-33 knockout (miR-33−/−) mice, we establish that loss of miR-33 results in diminished glucose uptake into WAT and skeletal muscle and an impaired ability to suppress glucose production in the liver and hydrolysis of free fatty acids (FFA) from WAT in response to insulin. We further observe that following HFD feeding, miR-33−/− mice have increased circulating levels of triglycerides (TAGs), and increased incorporation of lipids into the liver. Importantly, our data do not support a direct role for SREBP-1 in promoting these changes, as neither the induction nor suppression of FASN, ACC, and other SREBP-1 target genes involved in lipid synthesis were elevated in miR-33−/− mice under CD or HFD fed conditions during fasting/refeeding experiments. Additionally, SREBP-1 processing in response to fasting and refeeding was not altered in miR-33−/− mice. Consistent with earlier work (Horie et al., 2013), we did not observe differences in fuel preference or energy expenditure that would explain the predisposition of miR-33−/− mice to diet induced obesity. However, we find that in young mice, loss of miR-33 promotes more efficient uptake of lipids into adipose tissue, while the ability to hydrolyze free fatty acids is impaired. These findings suggest that an increased propensity to store lipids may contribute to the initial increase in body weight (BW) observed in miR-33−/− mice. Metabolic cage studies show that 7-month-old miR-33−/− mice on a CD have increased food intake, and further assessment of feeding behavior confirms that miR-33−/− mice maintain a higher level of food intake than wild-type (WT) animals when fed a HFD. Moreover, pair-feeding experiments demonstrate that this is likely the primary factor contributing to the obesity and metabolic dysfunction, as miR-33−/− mice no longer gain more weight when forced to eat the same amount as control animals. Similarly, pair fed miR-33−/− mice no longer exhibited the severe metabolic dysfunction observed in miR-33−/− mice fed ad libitum. Together these findings provide novel insight into the specific role of miR-33 in regulation of metabolic function in different tissues. Our findings also dispute earlier claims that changes in SREBP-1 mediated lipid synthesis in the liver are responsible for driving the obesity phenotype of miR-33−/− mice (Horie et al., 2013), and demonstrate that changes in feeding behavior are primarily responsible for these effects.
RESULTS
Loss of miR-33 Predisposes Mice to the Development of Obesity and Insulin Resistance
Consistent with previous work (Horie et al., 2013), we find that feeding of miR-33−/− mice with a HFD results in a dramatic increase in BW compared to control animals, especially during the first few weeks of HFD feeding. CD animals lacking miR-33 also developed increased BW although at a much more gradual rate (Figures 1A and 1B). This increase in BW was primarily due to increased fat mass, which was significantly elevated in 7-month-old mice on either a CD or HFD (Figure 1C). We further show that HFD fed miR-33−/− mice have impaired regulation of glucose homeostasis as demonstrated by an impaired ability to restore blood glucose levels during a glucose tolerance test (GTT) (Figure S1A). While CD-fed animals did not have any differences in the ability to regulate blood glucose levels during a GTT (Figure S1B), these mice have nearly a 2-fold increase in fasted insulin levels suggesting that increased insulin production may be compensating for impaired insulin sensitivity during this test (Figure S1C). To explore this in more detail, we performed hyperinsulinemic-euglycemic clamp studies to directly assess responsiveness to insulin in different tissues. Consistent with the elevated fasted plasma insulin levels, we find that the glucose infusion rate needed to maintain normal blood glucose concentrations after insulin administration was dramatically reduced in miR-33−/− mice (Figures 1D and S1D), indicating an overall impairment in insulin stimulated glucose uptake (Figure 1E). We further demonstrate that the capacity of both skeletal muscle and WAT to take up glucose in response to insulin is impaired, as incorporation of 2-deoxyglucose, a non-metabolizable form of glucose, was significantly reduced in miR-33−/− mice (Figures 1F and 1G). Similarly, the ability to suppress insulin-stimulated glucose production in the liver and hydrolysis of FFA from WAT was also dramatically impaired in miR-33−/− mice (Figures 1H and 1I). Together, these findings indicate that even on a CD, mice lacking miR-33−/− develop insulin resistance in numerous key metabolic tissues.
Figure 1. Genetic Ablation of MiR-33 Results in Obesity and Whole-Body Insulin Resistance.

(A) Body weight of WT and miR-33−/− mice fed a chow diet (CD) or high-fat diet (HFD) for 20 weeks. (n = 20 for CD and ≥ 30 for HFD.)
(B) Representative images of WT and miR-33−/− mice fed a CD or HFD for 20 weeks.
(C) Fat mass of WT and miR-33−/− mice fed a CD or HFD for 20 weeks (n = 6).
(D) Glucose infusion rate (GIR) during hyperinsulinemic-euglycemic clamp in WT and miR-33−/− mice fed a CD for 20 weeks (n = 9).
(E) Whole-body glucose uptake during the hyperinsulinemic-euglycemic clamp in WT and miR-33−/− mice fed a CD for 20 weeks (n = 9).
(F and G) Muscle (F) and adipose tissue (G) 2-deoxyglucose uptake during hyperinsulinemic-euglycemic clamp in WT and miR-33−/− mice fed a CD for 20 weeks (n = 9).
(H) Endogenous glucose production (EGP) measured in the basal period and during the hyperinsulinemic-euglycemic clamp in WT and miR-33−/− mice fed a CD for 20 weeks (n = 9).
(I) Circulating non-esterified fatty acids in WT and miR-33−/− mice fed a CD for 20 weeks (n = 9). All data represent the mean ± SEM, and an asterisk indicates p < 0.05 comparing miR-33−/− with WT mice on the same diet.
Mice Lacking miR-33 Have Elevated Levels of Both HDL-C and Total Cholesterol
As anticipated from previous work (Horie et al., 2013), we find that loss of miR-33 raised plasma HDL-C levels on both a CD and HFD. Fast protein liquid chromatography (FPLC) fractionation analysis revealed a higher HDL-C peak for miR-33−/− mice under both conditions (Figures 2A and 2B), which was confirmed by quantification of circulating HDL-C in individual samples (Figures 2C and 2D). However, unlike previous descriptions of total cholesterol (TC) levels in miR-33-deficient mice (Horie et al., 2013), we find that these levels were also elevated in miR-33−/− animals under both CD and HFD conditions (Figures 2C and 2D), as were the cholesterol levels in FPLC fractions containing low-density lipoproteins (LDL) particles (Figures 2A and 2B). We further demonstrate that the protein levels of the apolipoproteins associated with HDL (ApoA1) and LDL/VLDL (ApoB) were also elevated in miR-33−/− mice, indicating that the total numbers of these particles are likely elevated (Figures 2E and 2F). Although the host gene of miR-33 is an important regulator of cholesterol metabolism, the excision of miR-33 did not have any impact on Srebp-2 expression levels, indicating that an unintentional dysregulation of Srebp-2 is not involved in mediating these effects (Figure S1E).
Figure 2. Absence of miR-33 Increases Circulating HDL-C and LDL-C.

(A and B) Lipoprotein profile analysis from pooled plasma samples obtained from WT and miR-33−/− mice fed a chow diet (CD) (A) or HFD (B) for 20 weeks.
(C and D) Plasma total cholesterol, HDL-C, and triglyceride (TAG) levels from WT and miR-33−/− mice fed a CD (C) or HFD (D) for 20 weeks. Data represent the mean ± SEM (n = 7–10), and an asterisk indicates p < 0.05 comparing miR-33−/− with WT mice on the same diet.
(E and F) Western blot analysis (representative of two blots) of plasma APOB and APOA1 in the FPLC-fractionated lipoproteins shown in (A) and (B) from WT and miR-33−/− mice fed a chow diet (CD) (E) or HFD (F) for 20 weeks. Lanes 1–13 correspond to the following pooled fractions: 1 (28–30), 2 (31–33), 3 (34–36), 4 (37–39), 5 (40–42), 6 (43–45), 7 (46–48), 8 (49–51), 9 (52–54), 10 (55–57), 11 (58–60), 12 (61–63), and 13 (64–66).
Loss of miR-33 Causes Increased Lipid Accumulation in Liver but Does Not Affect Expression of Genes Involved in Hepatic Lipid Synthesis
Accumulation of lipids in peripheral tissues can promote insulin resistance (Samuel and Shulman, 2016; Shulman, 2014) and may contribute to the impaired insulin sensitivity of miR-33−/− mice. Consistent with this, we observe increased levels of TAGs in the liver of miR-33−/− mice after 2 months of HFD feeding (Figure S2A). Levels of DAGs and ceramides were also elevated (Figures S2B and S2C) and membrane associated PKCε was increased (Figure S2D). Similarly, 7-month-old miR-33−/− mice on a CD also had increased levels of hepatic TAGs, DAGs, and membrane PKCε, although ceramide levels were not altered (Figures 3A–3D). While these changes likely contribute to the impaired insulin responsiveness observed in miR-33−/− mice, they do not necessarily indicate that alterations in liver function are responsible for the metabolic dysfunction observed.
Figure 3. Loss of MiR-33 Increases Liver Triglyceride and Diacylglycerol Content and PKCε Activation.

(A–C) Liver triglyceride (TAGs) (A), diacylglycerol (DAGs) (B), and ceramide content (C) in WT and miR-33−/− mice fed a chow diet (CD) for 20 weeks after overnight feeding (n = 6–7).
(D) Western blot analysis of PKCε translocation in WT and miR-33−/− mice fed a CD for 20 weeks after overnight feeding. GAPDH and ATPase were used as loading controls for cytosolic and membrane fractions respectively. Quantification is shown in right panels (n = 6–7). All data represent the mean ± SEM, and an asterisk indicates p < 0.05 comparing miR-33−/− with WT mice on the same diet.
While previous work has suggested that the metabolic dysfunction of miR-33-deficient mice may be due to derepression of SREBP-1, resulting in increased hepatic lipid synthesis (Horie et al., 2013), our data indicate that the increased liver weight of miR-33−/− mice is proportional to their increase in BW (Figures 4A and 4B). Conversely, SREBP-1 transgenic mice have been shown to have increased hepatic lipid production resulting in fatty liver, but not the dramatic obesity phenotype found in miR-33−/− mice (Shimano et al., 1997). Moreover, gene expression patterns of mice in response to changes in nutrient conditions do not reveal any difference in the ability of miR-33−/− mice to regulate SREBP-1-responsive genes involved in hepatic lipid synthesis on either CD or HFD (Figure 4C). To perform these analyses, mice fed either CD or HFD for 20 weeks were sacrificed in the morning after either overnight feeding, 24-hr fasting, or 24-hr fasting followed by 12 hr refeeding. Microarray analysis was then performed on tissue from the livers of these animals. To determine whether regulation of SREBP-1 target genes was significantly altered, we used ingenuity pathway analysis (IPA) software to generate a network of SREBP-1-interacting genes and overlaid this with an analysis of all genes significantly altered in response fasting. Among these nutrient-responsive SREBP-1-interacting genes, none were found to be significantly altered between WT and miR-33−/− mice, indicating that there are not any differences in the regulation of SREBP-1 target genes, including those involved in fatty acid synthesis (Figure 4C). While qPCR analysis did show an increase in mRNA levels of Srepb-1 in the livers of miR-33−/− mice on HFD under fed conditions, it was unaltered under all other conditions (Figure 4D). Moreover, western blot analysis of SREBP-1 processing in livers of WT and miR-33−/− mice demonstrated a clear reduction in the mature form of SREBP-1 upon fasting and a reinduction upon refeeding, but did not reveal any significant differences between WT and miR-33−/− mice in either the precursor or mature forms of SREBP-1 under different nutrient states (Figure 4E). As expected, expression of SREBP-1-regulated genes, such as fatty acid synthase (Fasn) and acetyl-CoA carboxylase (Acc), and their regulation in response to fasting/refeeding were unchanged, demonstrating that the activity of SREBP-1 was not altered in miR-33−/− mice (Figures 4F and 4G). Similarly, western blot analysis of the protein levels of FASN and ACC under different nutrient conditions did not show increased expression in miR-33−/− mice (Figure 4H).
Figure 4. Lack of MiR-33 Does Not Influence Hepatic SREBP-1 Activation or Promote Inflammatory Gene Expression.

(A and B) Relative liver weight/body weight (A) and liver weight/femur length (B) quantification of WT and miR-33−/− mice fed a chow diet (CD) or HFD for 20 weeks. Data represent the mean ± SEM (n = 6).
(C) Heatmap analysis of nutrient-responsive genes (fast versus fed p > 0.05) that directly interact with SREBP-1 in WT and miR-33−/− mice fed a CD for 20 weeks fasted or fed overnight. Data show no significant differences between both groups of mice.
(D) qRT-PCR analysis of Srebp-1 expression in liver samples isolated from WT and miR-33−/− mice fed a CD or HFD for 20 weeks and fed; fasted for 24 hr; or fasted for 24 hr and refed for 12 hr. Data represent the mean ± SEM of relative expression levels normalized to fed animals on CD (n = 4).
(E) Western blot analysis of the precursor (P) and nuclear (N) form of SREBP-1 in liver samples isolated from WT and miR-33−/− mice fed a CD or HFD for 20 weeks and fed; fasted for 24 hr; or fasted for 24 hr and refed for 12 hr. Vinculin is used as a loading control.
(F and G) qRT-PCR analysis of Acc (F) and Fasn (G) expression in liver samples isolated from WT and miR-33−/− mice fed a CD or HFD for 20 weeks and fed; fasted for 24 hr; or fasted for 24 hr and refed for 12 hr. Data represent the mean ± SEM of relative expression levels normalized to fed animals on CD (n = 4).
(H) Western blot analysis of FASN and ACC in liver samples isolated from WT and miR-33−/− mice fed a CD or HFD for 20 weeks and fed; fasted for 24 hr; or fasted for 24 hr and refed for 12 hr. Vinculin is used as a loading control.
To further assess what other effects may be associated with these gene expression changes, we used IPA to predict specific functions and gene networks likely to be affected by these alterations. Consistent with the severe insulin resistance and metabolic dysfunction observed in HFD fed mice, our microarray data show a dramatic impairment in the ability to repress genes involved in fatty acid oxidation (Figure S3A) and glucose production (Figure S3B) under fed conditions. Somewhat surprisingly, assessment of the top regulatory networks identified by IPA as being altered in the livers of these animals under fasted conditions shows that miR-33−/− mice actually have reduced expression of gene networks associated with liver disease, cholestasis (Figure S4A), and inflammation (Figure S4B). Taken together, these results suggest that the effect of miR-33 on regulation of lipid and glucose metabolism is independent of its role in regulating hepatic SREBP-1 expression and activity.
Direct Changes in Adipose Tissue May Contribute to Rapid Expansion of Fat Mass in miR-33−/− Mice
Next, we determined whether the absence of miR-33 influences adipocyte size and infiltration of monocytes/macrophages in WAT of HFD fed animals, which has been associated with insulin resistance and obesity (McNelis and Olefsky, 2014). Characterization of WAT demonstrated that miR-33−/− mice had significantly higher mRNA expression of Cd68, a macrophage marker, in WAT of HFD-fed animals (Figure 5A). Similarly, immunofluorescence analysis of CD68-positive cells revealed a substantial accumulation of macrophages in WAT of miR-33−/− mice fed a HFD (Figure 5B). The increased accumulation of macrophages in the WAT of miR-33-deficient mice results in an increase in tumor necrosis factor alpha (Tnf-α) expression, a pro-inflammatory cytokine associated with insulin resistance and type 2 diabetes (Figure 5A). The increased abundance of macrophages in the WAT of miR-33-deficient mice was independent of numbers of circulating leukocytes, which were similar between both groups of mice (Figures S5A–S5C). Average adipocyte size was significantly larger in miR-33−/− mice fed a HFD compared to WT adipocytes (Figure 5C). While CD-fed mice showed a similar trend toward increased adipocyte size, these changes did not reach statistical significance, despite the increased BW and total fat mass of these animals (Figure 5C). To determine whether increased adipocyte number may also contribute to this effect, we directly assessed populations of adipocyte precursor (AP) cells in young animals by flow cytometry (Figure S5D). Following 1 week on HFD, along with bromodeoxyuridine (BrdU) administration via the drinking water, miR-33−/− mice showed a trend toward enhanced BrdU incorporation into adipocyte-precursor cells (AP) (Figure 5D), resulting in a significant increase in the total number of APs (Figure 5E). Consistent with this, we observed a significant increase in the expression of HMGA2, a chromatin remodeling factor involved in regulation of pre-adipocyte clonal expansion (Figure 5F) (Anand and Chada, 2000). These findings suggest that both adipocyte hypertrophy and hyperplasia may contribute to the increase in adipose mass in miR-33−/− mice. Even in young animals with minimal differences in BW, we find that loss of miR-33 resulted in a significant increase in percent body fat (Figure 5G). These effects are exacerbated by feeding a HFD, resulting in significant increases in both fat mass and total BW after only 2 weeks HFD feeding (Figure 5G). These rapid changes in fat mass may be in part due to an increased capacity for WAT to store lipids. Consistent with this, we find that the clearance of circulating lipids during a fat tolerance test (FTT) is increased in miR-33−/− mice (Figure 5H). Furthermore, when we perform a similar experiment using radioactively labeled lipid [H3]-Triolein, we find that the incorporation of radioactively labeled FFA into both WAT and BAT was significantly increased, including nearly a 3-fold increase in uptake into WAT (Figure 5I). These findings suggest that the enhanced clearance of circulating lipids during FTT are due to increased uptake into adipose tissue. In agreement with this finding, we observed that the expression of lipoprotein lipase (LPL), the enzyme that hydrolyzes circulating TAG-rich lipoproteins, was significantly upregulated in WAT of mice lacking miR-33 (Figure S6A). The augmented lipid clearance noted in miR-33−/− mice was independent of TAG absorption, since we did not observe any differences in the accumulation of radioactively labeled lipids in circulation when uptake into tissues is blocked by treatment with an inhibitor of LPL (Figure S6B). In addition to increased lipid uptake capacity, we find that the ability of adipose tissue to mobilize lipids in times of nutrient deprivation may also be impaired. Ex vivo analysis of lipolysis in WAT from WT and miR-33−/− mice demonstrates that the ability of WAT to release FFA in response to the beta-adrenergic receptor agonist isoproterenol was reduced in animals lacking miR-33 (Figure 5J). Western blot and qPCR analysis further demonstrates that the lipases hormone-sensitive lipase (HSL) and adipose triglyceride lipase (ATGL), which are responsible for mobilization of FFA, were downregulated in WAT of miR-33−/− mice at both the protein and the mRNA levels (Figures 5K and 5L). These findings are consistent with our clamp studies which demonstrate that while the ability of miR-33−/− mice to suppress FFA release in response to insulin was severely impaired, the basal levels of circulating FFA under fasting conditions were actually significantly reduced, indicating reduced lipolysis from WAT (Figure 1I).
Figure 5. Loss of MiR-33 Enhances Adipose Tissue Inflammation, Expansion, and Lipid Uptake and Retention.

(A) qRT-PCR of Cd68 and Tnf-α expression in WAT samples isolated from WT and miR-33−/− mice fed a CD or HFD for 20 weeks. Data represent the relative expression levels normalized to WT mice (n = 5–6).
(B) Representative pictures of WAT of WT and miR-33−/− mice fed a HFD for 20 weeks. Pictures show an accumulation of macrophages (CD68-positive cells) (green) in regions of high cellularity (white arrows) in WAT isolated from miR-33−/− mice. DAPI (blue) stains DNA and caveolin-1 (red) labels adipocyte membranes.
(C) Representative H&E-stained sections of WAT isolated from WT and miR-33−/− mice fed a chow diet (CD) (top panels) or HFD (bottom panels) for 20 weeks. Quantification of adipocyte size (right) (n = 4–6).
(D) Flow cytometry analysis of BrdU incorporation into adipocyte precursors (AP) from WAT of WT and miR-33−/− mice following 1 week of HFD and BrdU treatment. (n = 6–9).
(E) Flow cytometry analysis of AP from WAT of WT and miR-33−/− mice following 1 week of HFD (n = 6–9).
(F) Western blot analysis of HMGA2 expression in WAT isolated from WT and miR-33−/− mice fed a CD or HFD for 1 week. In the quantification (right graphs), data are expressed as relative total protein levels normalized by the loading control HSP90 (n = 7).
(G) Body weight (right panel) and percentage fat mass (left panel) in 2-old-month WT and miR-33−/− mice before and after feeding mice a HFD for 2 weeks (n = 8).
(H) Triglyceride (TAG) clearance in WT and miR-33−/− mice. Mice were gavaged with olive oil, and circulating TAGs were assessed at the indicated times (n = 6).
(I) [3H]-triolein tissue uptake analysis in 2-old-month WT and miR-33−/− mice (n = 3).
(J) Ex vivo lipolysis of WAT isolated from WT and miR-33−/− mice and treated or not with isoproterenol (Iso) to activate lipolysis (n = 3).
(K) Western blot analysis of HSL and ATGL expression in WAT isolated from WT and miR-33−/− mice fed a CD and HFD for 1 week. HSP90 was used as a loading control.
(L) qRT-PCR of Hsl and Atgl expression in liver samples isolated from WT and miR-33−/− mice fed a CD for 2 months. Data represent the relative expression levels normalized to WT mice (n = 5–6). All data represent the mean ± SEM, and an asterisk indicates p < 0.05 comparing miR-33−/− with WT mice on the same diet.
Metabolic Characterization of miR-33−/− Mice Reveals Increase in Food Intake
To attempt to determine the underlying mechanisms behind the increased BW and insulin resistance observed in miR-33−/− mice, we performed metabolic cage analysis on CD fed animals. Surprisingly, miR-33−/− mice actually had increased oxygen (O2) consumption and carbon dioxide (CO2) production (Figures S6C and S6D) at some time points during the metabolic profiling resulting in increased in energy expenditure at these time points (Figure S6E), although the overall changes in these parameters were not found to be statistically significant. Moreover, these changes would be expected to make animals more resistant to the development of diet induced obesity, in direct opposition to the phenotype we observe. We further did not find any differences in the respiratory exchange ratio of miR-33−/− mice, indicating that these mice do not have any alterations in fuel utilization (Figure S6F). Additionally, we did not find any statistical differences in the overall activity of miR-33−/− mice, although there was decreased activity in these animals during some of the peak periods (Figure S6G). However, we found that miR-33−/− mice had enhanced feeding, especially during some of the peak feeding times just before the end of the dark cycle, which result in a significant increase in overall food intake (Figure 6A). Moreover, careful assessment of feeding behavior in individually housed mice during the early stages of HFD feeding demonstrates that miR-33−/− mice consume significantly more food than control animals beginning on the second day of HFD feeding, leading to a significant increase in BW by day 10 of HFD feeding (Figures 6B and 6C). To determine what factors may contribute to this phenotype, we assessed levels of circulating factors responsible for communicating signals of hunger/satiety in young animals. Although we did not observe any differences in levels of ghrelin under fasted conditions, but did find increased levels of ghrelin in fed miR-33−/− mice compared to WT mice, suggesting that miR-33−/− mice may have impaired suppression of ghrelin production in the stomach after feeding (Figure 6D) Alternatively, levels of corticosterone were not altered in miR-33−/− mice (Figure 6E), while fed levels of leptin were significantly increased in miR-33−/− mice (Figure 6F). Although leptin is a satiety hormone generally promoting a reduction in food intake, consistently high levels can also promote leptin resistance which could have the opposite effect (Könner and Bruning, 2012). As leptin is produced and secreted from adipose tissue, these changes are likely due to increased adipose mass in these animals. We further assessed circulating levels of fibroblast growth factor 21 (FGF21), but did not observe any significant changes in either circulating levels of FGF21 (Figure 6G) or its mRNA expression in the liver (Figure S7A) To determine whether changes in feeding behavior are directly responsible for the obesity and metabolic dysfunction observed in miR-33−/− mice, we performed pair-feeding experiments in which WT and miR-33−/− mice are fed the same amount based on the daily food consumption of a separate group of WT animals. Under these conditions miR-33−/− mice no longer gained weight more rapidly than controls, clearly demonstrating that the increased food intake was primarily responsible for the obesity phenotype (Figure 6H). Similarly, the increased fat mass of miR-33−/− mice following 12 weeks of HFD feeding (Figure S7B) was also negated by pair feeding (Figure 6I). We further demonstrate that the increased food intake and associated obesity of miR-33−/− mice are directly responsible for promoting the metabolic dysfunction observed in these animals, as we no longer detect any differences in circulating levels of insulin or regulation of glucose homeostasis (Figures 6J and 6K). Additionally, circulating levels of cholesterol and TAGs were not different in pair fed miR-33−/− mice compared to controls (Figures 6L and 6M). This is in stark contrast to ad libitum fed miR-33−/− mice after a similar amount of time on HFD, which had a dramatic impairment in regulation of glucose homeostasis (Figure S1A), and elevated levels of fasting insulin and cholesterol, while TAGs were not altered (Figures S7C–S7E).
Figure 6. Germline Deletion of miR-33 Enhances Food Intake.

(A) Metabolic cage analysis of food intake in WT and miR-33−/− mice fed a CD for 20 weeks (n = 5–6).
(B and C) Food intake (B) and change in body weight (BW) (C) in 2-month-old WT and miR-33−/− mice individually housed and fed a HFD for 2 weeks (n = 7–10).
(D–F) Circulating ghrelin (D), corticosterone (E), and leptin (F) in serum of 2-month-old WT and miR-33−/− mice fed a CD in fed and fasting conditions (n = 5–7).
(G) Circulating levels of FGF21 in serum of fasted WT and miR-33−/− mice fed a CD or HFD for 20 weeks.
(H) Every other day measurements of body weight in pair fed WT and miR-33−/− mice fed a HFD for 12 weeks (n = 10).
(I and J) Measurements of fat mass (I) and fasting insulin (J) in pair-fed WT and miR-33−/− mice fed a HFD for 12 weeks (N = 10).
(H and K) Measurements of blood glucose levels (H) and calculation of area under the curve (K) during a glucose tolerance test in pair fed WT and miR-33−/− mice fed a HFD for 12 weeks (N = 10).
(L and M) Measurements of circulating total cholesterol (L) and triglyceride (TAG) levels (M) in fasted pair fed WT and miR-33−/− mice fed a HFD for 12 weeks (N = 10). All data represent the mean ± SEM, and an asterisk indicates p < 0.05 comparing miR-33−/− with WT mice on the same diet.
DISCUSSION
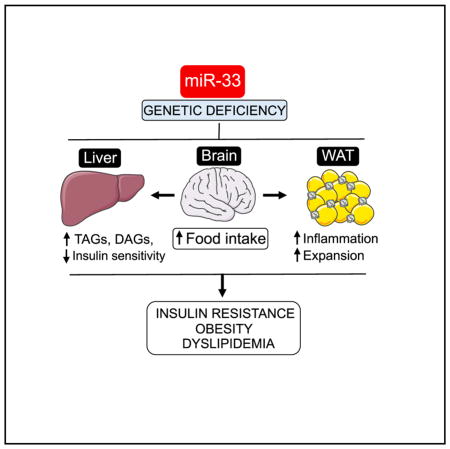
In this work, we perform an in-depth characterization of the metabolic changes that occur in miR-33−/− mice and provide novel insights into the specific mechanisms by which miR-33 regulates metabolic function, which will be necessary to better understand the possible clinical implications of treating patients with anti-miR-33 therapies (Figure 7). The most salient finding is the demonstration that alterations in feeding behavior are primarily responsible for the obesity and metabolic dysfunction observed in miR-33 mice, which might be enhanced by changes in WAT lipid uptake and mobilization. Importantly, our work does not indicate that alterations in lipid synthesis in the liver play a major role in driving this phenotype. As regulation of HDL biogenesis and bile acid metabolism in the liver are believed to contribute to the athero-protective effects of miR-33 inhibitors, these findings may have a direct impact on the development and utilization of anti-miR-33 therapeutics for the treatment of cardiovascular disease.
Figure 7.

Schematic Representation of the Role of MiR-33 in Lipid and Glucose Metabolism
Similar to previous work, we find that miR-33-deficient mice have increased BW and impaired metabolic function after feeding of a HFD. We further demonstrate that even CD-fed animals have impaired metabolic function as demonstrated by the diminished responsiveness to insulin in hyperinsulinemic-euglycemic clamp studies. These findings demonstrate that miR-33−/− mice have insulin resistance in a number of key metabolic tissues including the liver, WAT, and skeletal muscle. Consistent with this, we find that accumulation of TAGs, DAGs and ceramides is increased in the liver of miR-33−/− mice after HFD feeding and hepatic TAGs and DAGs were also elevated in CD fed mice. Increased incorporation of TAGs has been shown to promote insulin resistance in a number of different tissues, as has the accumulation of other lipid subspecies such as DAGs and ceramides (Shulman, 2014). Both DAGs and ceramides (Chavez and Summers, 2012; Holland et al., 2011) have been proposed to mediate insulin resistance, the former through activation of novel PKCs, which in turn inhibits insulin signaling at a proximal step (Samuel and Shulman, 2012) and the latter through the impairment of Akt translocation and activation. Specifically, the accumulation of DAGs promotes activation and translocation of PKCε (in the liver), thereby inhibiting insulin receptor kinase activity through phosphorylation of the insulin receptor at Thr1160 (Petersen et al., 2016) or phosphorylation of IRS1 (e.g., Ser1101) (Li et al., 2004), respectively. All of these effects may contribute to the impaired metabolic function observed in HFD fed miR-33−/− mice, as levels of DAGs, membrane PKCε, and ceramides were all found to be elevated in the liver miR-33−/− animals fed a HFD.
While these findings provide insight into some of the mechanisms by which loss of miR-33 may promote insulin resistance in the liver, they do not necessarily demonstrate that the changes in this organ are responsible for driving the development of metabolic dysfunction in these animals. Indeed, in depth microarray profiling of the livers of WT and miR-33−/− mice under different nutrient conditions revealed only minimal differences in the regulation of nutrient-responsive genes between WT and miR-33−/− mice. More importantly, our data do not show any increase in the levels of SREBP-1-regulated genes involved in lipid synthesis at either the mRNA or protein level. This is in direct contradiction to earlier work that concluded that derepression of SREBP-1 in the liver of miR-33-deficient mice promoted obesity and insulin resistance through increased hepatic lipid synthesis (Horie et al., 2013). While the mRNA expression of SREBP-1 may be altered under some conditions in miR-33−/− mice, its activity does not appear to be altered, as we do not observe any differences in the expression of SREBP-1-responsive genes, consistent with our earlier work which failed to identify SREBP-1 as a miR-33 target gene. These findings are not surprising, as SREBP-1 is known be regulated primarily at the post-transcriptional level, and even transgenic mice overexpressing a constitutively active form of SREBP-1 develop primarily a fatty liver phenotype, as opposed to the dramatic increase in fat mass and body weight found in miR-33−/− animals (Shimano et al., 1997). Western blot analysis of SREBP-1 processing also did not reveal any differences between WT and miR-33−/− mice. While some of the differences between our work and that of Horie et al. (2013) may be due to differences in the HFD that was used (45% versus 60%), the type of fat was the same, other aspects of the dietary composition were very similar, and even on a CD miR-33−/− mice have increased body weight and impaired metabolic function. Additionally, numerous in vivo studies have demonstrated that anti-miR-33 therapeutic approaches are very effective at inhibiting miR-33 in the liver (Marquart et al., 2010; Najafi-Shoushtari et al., 2010; Rayner et al., 2010). However, these animals also do not develop the dramatic obesity phenotype found in miR-33−/− mice, suggesting additional organs may be primarily responsible for driving this phenotype. If alterations in liver function are primarily responsible for the obesity and metabolic dysfunction of miR-33−/− mice, it may suggest that the passenger strand (miR-33*), which regulates glucose and lipid metabolism in concert with miR-33 (Goedeke et al., 2013), may be primarily responsible for these effects as this would not be targeted by anti-miR-33 therapeutics.
Analysis of visceral WAT revealed a significant increase in both adipocyte size and the infiltration of macrophages into adipose tissue, which are both indicators of insulin resistance (Yao et al., 2017). These findings are consistent with the impaired glucose uptake and suppression of FFA hydrolysis observed in clamp studies. Additionally, when we isolate tissue from young animals we observe an impairment in lipolytic function, which may contribute to the rapid increase in fat mass in miR-33−/− animals. Moreover, we find that the capacity of miR-33−/− mice to take up circulating lipids is enhanced even in young animals. This may be in part due to an increase in the number of adipocyte precursor cells, which were found to be elevated in the WAT of miR-33−/− mice after short-term HFD feeding. These findings support our previous finding that miR-33 can regulate pre-adipocyte proliferation and differentiation in vitro through regulation of HMGA2 (Price et al., 2016). Consistent with this, we demonstrate that protein levels of HMGA2 are elevated in WAT of miR-33−/− mice compared to WT mice. Together the increased capacity for lipid uptake and decreased lipolytic activity of miR-33-deficient mice may contribute to the rapid increase in BW and fat mass following HFD feeding.
While our data demonstrate that changes in a number of key metabolic tissues are involved in the metabolic dysfunction observed in miR-33−/− mice, we find that the primary factor responsible for the obesity phenotype appears to be an increase in food intake. While this difference in food intake is not dramatic, it does appear to be sufficient to drive the rapid weight gain and metabolic dysfunction, as miR-33−/− mice that were pair fed based on the food consumption of WT mice did not show any differences in BW or metabolic regulation compared to controls even after 3 months of HFD feeding. This is in stark contrast to ad libitum fed miR-33−/− mice which consistently gain more BW during the first few weeks on HFD and had dramatic impairments in regulation of glucose homeostasis and elevated insulin levels after 3 months on a HFD. Assessment of circulating factors involved in regulation of hunger and satiety reveals levels of ghrelin and corticosterone were not altered in miR-33−/− mice, while leptin levels are increased. Although this would generally be expected to reduce food intake, it could also promote leptin resistance (Könner and Bruning, 2012). While a number of different organs are involved in the regulation of food intake, these data do raise the interesting possibility that miR-33 may have direct effects upon the hypothalamic neurons that regulate feeding behavior (Waterson and Horvath, 2015). Previous work has shown that miR-33 is highly expressed in the brain and can affect state dependent memory by regulation of GABAergic receptors (Jovasevic et al., 2015). As the hypothalamic POMC and AgRP neurons that regulate food intake are also GABAergic neurons, they may be directly affected by alterations in miR-33. This could also explain some of the discrepancies between genetic and pharmacologic inhibition of miR-33, as anti-miR-33 therapeutics would not be expected to cross the blood-brain barrier.
Overall this work provides a more complete description of the metabolic dysfunction that occurs in animals lacking miR-33, including in depth characterization of the impact on specific metabolic functions in different key tissues. While we find that loss of miR-33 results in alteration of metabolic function in a number of different tissues, including the liver, our data do not support the conclusion that derepression of SREBP-1 and increased lipid synthesis is responsible for the predisposition of miR-33−/− mice to obesity and metabolic dysfunction. Alternatively, we find that differences in the feeding behavior of miR-33−/− mice are primarily responsible for this phenotype. This is in direct contradiction to earlier work, which still demonstrated an increase in body weight in pair-feeding experiments (Horie et al., 2013). While differences in the methodologies used in performing these experiments may explain the discrepancies between these findings, the limited description of conditions used in previous work has limited our ability to assess what specific alterations may be responsible. While this work has considerably improved our understanding of how loss of miR-33 affects the function of different metabolic tissues, the use of whole body miR-33−/− mice limits our ability to determine if these changes are directly due to effects of miR-33 in an individual tissue or secondary to overall changes in metabolic regulation. Further work using tissue-specific knockout models will be necessary to provide greater insight into the specific mechanisms behind these changes, and will be necessary to properly assess the potential of anti-miR-33 therapeutic approaches.
EXPERIMENTAL PROCEDURES
Further details and an outline of resources used in this work can be found in the Supplemental Information. Briefly, microarray analysis was performed as previously described (Fang et al., 2017), using Agilent SurePrint G3 Mouse 8×60K Microarray. Gene network and pathway analysis were carried out using Qlucore Omics Explorer (v.3.2) (Qlucore) and ingenuity pathway analysis (Ingenuity Systems QIAGEN). Animal studies were approved by the Institutional Animal Care and Use committee of Yale University School of Medicine. WT C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Generation of miR-33−/− mice was accomplished with the assistance of Cyagen Biosciences. Induction of diet-induced obesity was accomplished by feeding a HFD (60% fat) (D12492; Research Diets, New Brunswick, NJ, USA). Measurements of metabolic function, circulating lipids, and adipocyte proliferation/function were carried out using established techniques. Statistical differences were measured using an unpaired two-sided Student’s t test or one-way ANOVA with Bonferroni correction for multiple comparisons. Normality was checked using the Kolmogorov-Smirnov test. A nonparametric test (Mann-Whitney) was used when data did not pass the normality test. p < 0.05 was considered statistically significant.
Supplementary Material
Highlights.
Genetic loss of miR-33 results in severe insulin resistance, even in chow-fed mice
Livers from miR-33−/− mice have ↑ lipids but no changes in SREBP-1 target genes
miR-33−/− mice have ↑ adipocyte precursors, ↑ lipid uptake in WAT, and ↓ lipolysis
Pair feeding prevents obesity and metabolic dysfunction in miR-33−/− mice
Acknowledgments
For microarray analysis, the Laboratory of Genetics and Genomics, NIA IRP, and NIH performed the sample QC, labeling, hybridization, feature extraction, and GEO submission (Dr. Elin Lehrmann) and also provided bioinformatic analysis (Dr. Yongqing Zhang) within the microarray facility directed by Dr. Kevin Becker. This work was supported by grants from the NIH (R35HL135820 to C.F.-H.; R01HL105945 and R01HL135012 to Y.S.; 5F32DK10348902 to N.L.P.; and R01DK40936 to G.I.S.), the American Heart Association (16EIA27550005 to C.F.-H.; 16GRNT26420047 to Y.S.; and 17SDG33110002 to N.R.), the American Diabetes Association (1-16-PMF-002 to A.C.-D.), and the Foundation Leducq Transatlantic Network of Excellence in Cardiovascular Research MIRVAD (to C.F.-H.).
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the microarray data reported in this study is GEO: GSE109055.
Supplemental Information includes seven figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.01.074.
AUTHOR CONTRIBUTIONS
N.L.P. and C.F.-H. conceived and designed the study and wrote the manuscript. N.L.P., A.K.S., N.R., A.C.-D., L.G., A.D.-R., E.A., A.W., J.P.-C., A.B., and Y.S. performed the experiments and analyzed the data. Y.S., M.S.R., R.d.C., and G.I.S. assisted with the experimental design and the data interpretation.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Ahn J, Lee H, Jung CH, Jeon TI, Ha TY. MicroRNA-146b promotes adipogenesis by suppressing the SIRT1-FOXO1 cascade. EMBO Mol Med. 2013;5:1602–1612. doi: 10.1002/emmm.201302647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RM, Marquart TJ, Albert CJ, Suchy FJ, Wang DQ, Ananthanarayanan M, Ford DA, Baldán A. miR-33 controls the expression of biliary transporters, and mediates statin- and diet-induced hepatotoxicity. EMBO Mol Med. 2012;4:882–895. doi: 10.1002/emmm.201201228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RM, Marquart TJ, Jesse JJ, Baldán A. Control of very low-density lipoprotein secretion by N-ethylmaleimide-sensitive factor and miR-33. Circ Res. 2014;115:10–22. doi: 10.1161/CIRCRESAHA.115.303100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- Anand A, Chada K. In vivo modulation of Hmgic reduces obesity. Nat Genet. 2000;24:377–380. doi: 10.1038/74207. [DOI] [PubMed] [Google Scholar]
- Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metab. 2012;15:585–594. doi: 10.1016/j.cmet.2012.04.002. [DOI] [PubMed] [Google Scholar]
- Chen XM. MicroRNA signatures in liver diseases. World J Gastroenterol. 2009;15:1665–1672. doi: 10.3748/wjg.15.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioffi M, Vallespinos-Serrano M, Trabulo SM, Fernandez-Marcos PJ, Firment AN, Vazquez BN, Vieira CR, Mulero F, Camara JA, Cronin UP, et al. MiR-93 controls adiposity via inhibition of Sirt7 and Tbx3. Cell Rep. 2015;12:1594–1605. doi: 10.1016/j.celrep.2015.08.006. [DOI] [PubMed] [Google Scholar]
- Dávalos A, Goedeke L, Smibert P, Ramírez CM, Warrier NP, Andreo U, Cirera-Salinas D, Rayner K, Suresh U, Pastor-Pareja JC, et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci USA. 2011;108:9232–9237. doi: 10.1073/pnas.1102281108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang EF, Waltz TB, Kassahun H, Lu Q, Kerr JS, Morevati M, Fivenson EM, Wollman BN, Marosi K, Wilson MA, et al. Tomatidine enhances lifespan and healthspan in C. elegans through mitophagy induction via the SKN-1/Nrf2 pathway. Sci Rep. 2017;7:46208. doi: 10.1038/srep46208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Hernando C, Suárez Y, Rayner KJ, Moore KJ. MicroRNAs in lipid metabolism. Curr Opin Lipidol. 2011;22:86–92. doi: 10.1097/MOL.0b013e3283428d9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Hernando C, Ramírez CM, Goedeke L, Suárez Y. MicroRNAs in metabolic disease. Arterioscler Thromb Vasc Biol. 2013;33:178–185. doi: 10.1161/ATVBAHA.112.300144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost RJ, Olson EN. Control of glucose homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proc Natl Acad Sci USA. 2011;108:21075–21080. doi: 10.1073/pnas.1118922109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerin I, Clerbaux LA, Haumont O, Lanthier N, Das AK, Burant CF, Leclercq IA, MacDougald OA, Bommer GT. Expression of miR-33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. J Biol Chem. 2010;285:33652–33661. doi: 10.1074/jbc.M110.152090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedeke L, Vales-Lara FM, Fenstermaker M, Cirera-Salinas D, Chamorro-Jorganes A, Ramírez CM, Mattison JA, de Cabo R, Suárez Y, Fernández-Hernando C. A regulatory role for microRNA 33* in controlling lipid metabolism gene expression. Mol Cell Biol. 2013;33:2339–2352. doi: 10.1128/MCB.01714-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedeke L, Salerno A, Ramírez CM, Guo L, Allen RM, Yin X, Langley SR, Esau C, Wanschel A, Fisher EA, et al. Long-term therapeutic silencing of miR-33 increases circulating triglyceride levels and hepatic lipid accumulation in mice. EMBO Mol Med. 2014;6:1133–1141. doi: 10.15252/emmm.201404046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy SM. Metabolic syndrome update. Trends Cardiovasc Med. 2016;26:364–373. doi: 10.1016/j.tcm.2015.10.004. [DOI] [PubMed] [Google Scholar]
- Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest. 2011;121:1858–1870. doi: 10.1172/JCI43378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie T, Ono K, Horiguchi M, Nishi H, Nakamura T, Nagao K, Kinoshita M, Kuwabara Y, Marusawa H, Iwanaga Y, et al. MicroRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (Srebp2) regulates HDL in vivo. Proc Natl Acad Sci USA. 2010;107:17321–17326. doi: 10.1073/pnas.1008499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie T, Nishino T, Baba O, Kuwabara Y, Nakao T, Nishiga M, Usami S, Izuhara M, Sowa N, Yahagi N, et al. MicroRNA-33 regulates sterol regulatory element-binding protein 1 expression in mice. Nat Commun. 2013;4:2883. doi: 10.1038/ncomms3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan SD, Kruger M, Willmes DM, Redemann N, Wunderlich FT, Brönneke HS, Merkwirth C, Kashkar H, Olkkonen VM, Böttger T, et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat Cell Biol. 2011;13:434–446. doi: 10.1038/ncb2211. [DOI] [PubMed] [Google Scholar]
- Jovasevic V, Corcoran KA, Leaderbrand K, Yamawaki N, Guedea AL, Chen HJ, Shepherd GM, Radulovic J. GABAergic mechanisms regulated by miR-33 encode state-dependent fear. Nat Neurosci. 2015;18:1265–1271. doi: 10.1038/nn.4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunakaran D, Thrush AB, Nguyen MA, Richards L, Geoffrion M, Singaravelu R, Ramphos E, Shangari P, Ouimet M, Pezacki JP, et al. Macrophage mitochondrial energy status regulates cholesterol efflux and is enhanced by anti-mir33 in atherosclerosis. Circ Res. 2015;117:266–278. doi: 10.1161/CIRCRESAHA.117.305624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Cho H, Alexander R, Patterson HC, Gu M, Lo KA, Xu D, Goh VJ, Nguyen LN, Chai X, et al. MicroRNAs are required for the feature maintenance and differentiation of brown adipocytes. Diabetes. 2014;63:4045–4056. doi: 10.2337/db14-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Könner AC, Bruning JC. Selective insulin and leptin resistance in metabolic disorders. Cell Metab. 2012;16:144–152. doi: 10.1016/j.cmet.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Lam DW, LeRoith D. The worldwide diabetes epidemic. Curr Opin Endocrinol Diabetes Obes. 2012;19:93–96. doi: 10.1097/MED.0b013e328350583a. [DOI] [PubMed] [Google Scholar]
- Li Y, Soos TJ, Li X, Wu J, Degennaro M, Sun X, Littman DR, Birnbaum MJ, Polakiewicz RD. Protein kinase C Theta inhibits insulin signaling by phosphorylating IRS1 at Ser(1101) J Biol Chem. 2004;279:45304–45307. doi: 10.1074/jbc.C400186200. [DOI] [PubMed] [Google Scholar]
- Li T, Francl JM, Boehme S, Chiang JY. Regulation of cholesterol and bile acid homeostasis by the cholesterol 7α-hydroxylase/steroid response element-binding protein 2/microRNA-33a axis in mice. Hepatology. 2013;58:1111–1121. doi: 10.1002/hep.26427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquart TJ, Allen RM, Ory DS, Baldán A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc Natl Acad Sci USA. 2010;107:12228–12232. doi: 10.1073/pnas.1005191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity. 2014;41:36–48. doi: 10.1016/j.immuni.2014.05.010. [DOI] [PubMed] [Google Scholar]
- Mori MA, Thomou T, Boucher J, Lee KY, Lallukka S, Kim JK, Torriani M, Yki-Järvinen H, Grinspoon SK, Cypess AM, Kahn CR. Altered miRNA processing disrupts brown/white adipocyte determination and associates with lipodystrophy. J Clin Invest. 2014;124:3339–3351. doi: 10.1172/JCI73468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Näär AM. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet M, Ediriweera HN, Gundra UM, Sheedy FJ, Ramkhelawon B, Hutchison SB, Rinehold K, van Solingen C, Fullerton MD, Cecchini K, et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J Clin Invest. 2015;125:4334–4348. doi: 10.1172/JCI81676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet M, Ediriweera H, Afonso MS, Ramkhelawon B, Singaravelu R, Liao X, Bandler RC, Rahman K, Fisher EA, Rayner KJ, et al. microRNA-33 regulates macrophage autophagy in atherosclerosis Arterioscler. Thromb Vasc Biol. 2017;37:1058–1067. doi: 10.1161/ATVBAHA.116.308916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S, Yang X, Jia Y, Li Y, Chen R, Wang M, Cai D, Zhao R. Intravenous injection of microvesicle-delivery miR-130b alleviates high-fat diet-induced obesity in C57BL/6 mice through translational repression of PPAR-γ. J Biomed Sci. 2015;22:86. doi: 10.1186/s12929-015-0193-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, Madiraju AK, Gassaway BM, Marcel M, Nasiri AR, Butrico G, Marcucci MJ, Zhang D, Abulizi A, Zhang XM, et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J Clin Invest. 2016;126:4361–4371. doi: 10.1172/JCI86013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poy MN, Hausser J, Trajkovski M, Braun M, Collins S, Rorsman P, Zavolan M, Stoffel M. miR-375 maintains normal pancreatic alpha-and beta-cell mass. Proc Natl Acad Sci USA. 2009;106:5813–5818. doi: 10.1073/pnas.0810550106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NL, Fernández-Hernando C. miRNA regulation of white and brown adipose tissue differentiation and function. Biochim Biophys Acta. 2016;1861(12 Pt B):2104–2110. doi: 10.1016/j.bbalip.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NL, Holtrup B, Kwei SL, Wabitsch M, Rodeheffer M, Bianchini L, Suárez Y, Fernández-Hernando C. SREBP-1c/microRNA 33b genomic loci control adipocyte differentiation. Mol Cell Biol. 2016;36:1180–1193. doi: 10.1128/MCB.00745-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NL, Rotllan N, Canfrán-Duque A, Zhang X, Pati P, Arias N, Moen J, Mayr M, Ford DA, Baldán Á, et al. Genetic dissection of the impact of miR-33a and miR-33b during the progression of atherosclerosis. Cell Rep. 2017;21:1317–1330. doi: 10.1016/j.celrep.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner KJ, Suárez Y, Dávalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernández-Hernando C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner KJ, Esau CC, Hussain FN, McDaniel AL, Marshall SM, van Gils JM, Ray TD, Sheedy FJ, Goedeke L, Liu X, et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature. 2011a;478:404–407. doi: 10.1038/nature10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011b;121:2921–2931. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenson RS, Brewer HB, Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–1919. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotllan N, Ramírez CM, Aryal B, Esau CC, Fernández-Hernando C. Therapeutic silencing of microRNA-33 inhibits the progression of atherosclerosis in Ldlr−/− mice–brief report. Arterioscler Thromb Vasc Biol. 2013;33:1973–1977. doi: 10.1161/ATVBAHA.113.301732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottiers V, Näär AM. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol. 2012;13:239–250. doi: 10.1038/nrm3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottiers V, Obad S, Petri A, McGarrah R, Lindholm MW, Black JC, Sinha S, Goody RJ, Lawrence MS, deLemos AS, et al. Pharmacological inhibition of a microRNA family in nonhuman primates by a seed-targeting 8-mer antimiR. Sci Transl Med. 2013;5:212ra162. doi: 10.1126/scitranslmed.3006840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest. 2016;126:12–22. doi: 10.1172/JCI77812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest. 1997;99:846–854. doi: 10.1172/JCI119248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med. 2014;371:1131–1141. doi: 10.1056/NEJMra1011035. [DOI] [PubMed] [Google Scholar]
- Waterson MJ, Horvath TL. Neuronal regulation of energy homeostasis: beyond the hypothalamus and feeding. Cell Metab. 2015;22:962–970. doi: 10.1016/j.cmet.2015.09.026. [DOI] [PubMed] [Google Scholar]
- Yao F, Zhang M, Chen L. Adipose tissue-specialized immunologic features might be the potential therapeutic target of prospective medicines for obesity. J Diabetes Res. 2017;2017:4504612. doi: 10.1155/2017/4504612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.