

Abstract
Transplant recipients developing donor specific HLA class II antibodiesare at higher risk for antibody mediated rejection (AMR) and transplant vasculopathy (TV). To understand how HLA class II antibodies cause AMR and TV, we determined the signaling events triggered in vascular endothelial cells (EC) following antibody ligation of HLA class II molecules. HLA class II expression in EC was induced by adenoviral-vector expression of the class II transactivator or by pretreatment with TNFα/IFNγ. Antibody ligation of class II stimulated EC proliferation and migration. Class II antibody also induced activation of key signaling nodes Src, FAK, PI3K and ERK that regulated downstream targets of the mTOR pathway Akt, S6K, and S6RP. Pharmacological inhibitors and siRNA showed the protein kinases Src, FAK, PI3K/Akt, and MEK/ERK regulate class II antibody-stimulated cell proliferation and migration. Treatment with rapalogs for 2h did not affect HLA II antibody-induced phosphorylation of ERK, instead mTORC1 targets were dependent on activation of ERK. Importantly, suppression of mTORC2 for 24h with rapamycin or everolimus or treatment with mTOR active-site inhibitors enhanced HLA II antibody-stimulated phosphorylation of ERK. Furthermore, knockdown of Rictor with siRNA caused over-activation of ERK while abolishing phosphorylation of Akt Ser473 induced by class II antibody. These data are different from HLA class I antibody-induced activation of ERK, which is mTORC2 dependent. Our results identify a complex signaling network triggered by HLA II antibody in EC and indicate that combined ERK and mTORC2 inhibitors may be required to achieve optimal efficacy in controlling HLA II antibody-mediated AMR.
Keywords: Endothelial cells, MHC, Anti-HLA II antibodies, Signal transduction, proliferation, migration
Introduction
Solid organ transplant recipients developing donor specific HLA antibodies (DSA) are at a higher risk for acute and chronic antibody mediated rejection (AMR) and graft loss (1-3). Acute AMR is estimated to affect 10-15% of allografts (1, 4-6), whereas chronic AMR occurs in as many as 50% of allografts by 10 years after transplant (7-9). Notably, chronic antibody-mediated allograft injury shares common histologic features across all transplanted organs and manifests as an insidious vascular disease known as transplant vasculopathy (TV). The affected vessels of the donor organ exhibit neointimal growth and perivascular fibrosis (10-15). Cell proliferation and angiogenic processes appear to be a central mechanism for the formation of these vascular lesions(16, 17).
Although strong clinical evidence supports the association between HLA DSA, chronic AMR and TV, the exact mechanism(s) where by DSAs cause neointimal hyperplasia and fibrosis are largely unknown. Vascular endothelial cell injury mediated by complement fixing DSA was thought to mediate chronic AMR and graft failure (18, 19). However, recent studies indicate that DSA can contribute to alterations in EC function through complement-independent mechanisms by transducing intracellular signals (20-24). Studies by our group and others have shown that crosslinking of HLA class I molecules with antibody on the surface of endothelial cells (EC) promotes diverse biological functions, including cellular proliferation and survival in clinically relevant in vivo and in vitro models of AMR(25, 26). Engagement of class I molecules byHLA antibodies stimulates phosphorylation of protein kinases Src, focal adhesionkinase (FAK), and paxillin and assembly of focal adhesions and activation of the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB/Akt) pathway (27-29). The activation of PI3K and Akt leads to up-regulation of anti-apoptotic Bcl-2 and Bcl-XL protein expression in EC (27). Ligation of class I molecules on EC results in cell proliferation(28, 30-32) via activation of the mammalian target of rapamycin (mTOR) complex 1 (mTORC1) and downstream signal targets including p70 ribosomal S6 kinase (S6K) and S6 ribosomal protein (S6RP) (31, 33, 34); and the mTORC2 signaling targets Akt and ERK (31, 33, 35).
HLA class II molecules, in addition to their classical role in antigen presentation, have been reported to regulate various cellular processes, including proliferation, maturation, cytokine production, and apoptosis, in macrophages, B cells, and dendritic cells (36, 37). These functions of HLA class II have been shown to engage various intracellular signaling events, in antigen presenting cells through agonistic actions after engagement by T cell receptors, including activation of protein kinases Src, Syk, PKC, the mitogen activated kinase (MAPK) p38, and ERK (36, 38). Allograft recipients may form antibodies against any mismatched HLA antigens carried by the donor, but DSA to HLA II molecules vastly predominate, particularly in the late post-transplant period (39-43). However, despite the strong correlation between DSA to HLA II and poor graft outcome across solid organs, very little is known about the intracellular signaling in graft vascular cells activated by HLA II antibody binding and how they contribute to allograft injury and the process of TV.
Under physiological conditions, most human vascular EC do not express HLA class II molecules and vascular endothelial cells in culture rapidly lose HLA II expression. Inflammatory insults, occurring during the process of transplantation including surgical trauma, and ischemia/reperfusion injury, as well as rejection, produce proinflammatory cytokines such as tumor necrosis factor (TNF)-α interleukin (IL)-1β and interferon (IFN)-γ. In turn, cytokines like IFNγ activate the HLA class II transactivator (CIITA), turn on transcription, and induce HLA class II molecule expression on EC (44, 45).
In this study, we aimed to elucidate the role of HLA class II DSA in intracellular signal transduction, cell proliferation, and migration in vascular EC—the angiogenic processes thought to drive TV. To overcome historical limitations of studying HLA II in human EC, we constructed and transfected an adenovirus-based vector encoding CIITA (Ad-CIITA)into primary human aortic EC or pretreated EC with cytokines TNFα and IFN-γ to induce HLA class II expression. Antibody ligation of HLA class II molecules on EC triggered a network of intracellular signals including activation of protein kinases Src, FAK, PI3K/Akt; the mTOR signaling cascade including mTOR, S6K, S6RP, and the mitogen activated protein kinase (MAPK) ERK. HLA II antibodies also stimulated angiogenic responses in EC including proliferation and migration. Studies using pharmacological inhibitors and siRNA demonstrated that FAK/Src, PI3K, PDK1/Akt and ERK function as upstream signaling elements regulating downstream targets of the mTOR pathway. Disruption of signaling events elicited through Src/FAK, ERK or mTOR prevented class II-mediated EC proliferation and migration. Importantly, pharmacological or siRNA suppression of mTORC2, blocked AKT at Ser-473 and lead to hyper-phosphorylation of ERK in response to antibody ligation of HLA class II molecules in EC. These results identify a novel feedback loop in EC stimulated with HLA class II antibodies and underscore a major functional difference in the signaling networks that modify endothelial cell function through HLA class I and class II molecules (33). Our results identify mechanisms of HLA class II antibody-mediated vascular injury and suggest that novel combinations of mTORC2 and MEK inhibitors may be clinically useful to treatAMR and TV.
Materials and Methods
Antibodies and Chemicals
Cell culture reagents were from Invitrogen Life Technologies. Purified mouse monoclonal antibodies against HLA II (clone ID: F002-6C6G1, IgG2a), DR (clone ID: FM5203, IgG1), DQ (clone ID: SPVL3, IgG2a), and DP (clone ID: BRA, IgG2b) were generously provided by Dr. Jar-how Lee, One Lambda Inc. (Canoga Park, CA). Plasmid pcDNA3 myc CIITA (P#808) was a gift from Matija Peterlin (Addgene plasmid # 14650, Cambridge, MA). Plasmid pENTR™ 4, plasmid pAd/PL-DEST Gateway®, DH5 competent cells, HEK293A cell line, Gateway LR Clonase II enzyme mix, Lipofectamine 2000, OPTI-MEM® I medium were from Invitrogen (Carlsbad, CA). Adeno-XTM Rapid Titer Kits were from Clontech (Mountain View, CA). Rapamycin, everolimus, and dasatinib were purchased from Sigma (St. Louis, MO). PP242 was from Chemdea (Ridgewood, NJ). PP2, PP3, LY294002, U0126, and U0124 were from Calbiochem (La Jolla, CA). A66 and anti-integrin αvβ3 antibody (mIgG 1)were obtained from R&D System (Minneapolis, MN). anti-CD105 antibody (Clone 43A3, mIgG 1) was from BioLegend (San Diego, CA). MK-2206, GDC-0068 and PD0325901 were obtained from Selleckchem (Houston, TX). Polyclonal Ab against phospho-Src (Tyr418), phospho-FAK (Tyr576), and phospho-FAK (Tyr577) were obtained from Invitrogen/BioSource International, Inc. (Camarillo, CA). Rabbit polyclonal Ab against phospho-mTOR (Ser2448), phospho-S6K (Thr389), phospho-S6K (Thr421/Ser424), phospho-S6RP (Ser235/236), phospho-S6RP (Ser240/300), phosphor-PI3 Kinase p85 (Tyr458)/p55 (Tyr199), phospho-Akt (Thr308), phospho-Akt (Ser473), phospho-c-Raf (Ser259), phospho-ERK (Thr202/Tyr204), mTOR, S6K, S6RP, Akt, MEK, ERK, and β-actin Ab were from Cell Signaling Technology (Beverly, MA). Anti-mTOR, Rictor, and Raptor mAb were from Millipore/Upstate (San Diego, CA). Anti-mTOR (A300-504A), Raptor (A300-506A), and Rictor (A300-459A) rabbit Ab were from Bethyl Laboratory (Montgomery, TX). The rabbit polyclonal Ab against c-Src (SRC2), β-Tubulin (H-235), the goat anti-rabbit horseradish peroxidase (HRP), goat anti-mouse HRP Ab, and protein A/G plus-agarose were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal anti-Vinculin (Clone Hvin-1) and Mitomycin C (M4287) were from SIGMA-ALDRKCH (St. Louis, MO). FITC-conjugated donkey anti-rabbit IgG was purchased from Jackson Immuno Research Laboratories, Inc. (West Grove, PA). Vybrant ™ CFDA SE Cell Tracer Kit (V-12883) was purchased from Molecular Probes (Eugene, OR). BD Pharmingen BrdU Flow Kit was from BD Biosciences (San Jose, CA).
Cell Culture
Primary human aortic endothelial cells (EC) were isolated from the aortic rings of explanted donor hearts, as described previously (46), or commercial EC (Lot number: EC5555) were obtained from Lonza/Clonetics (Walksville, MD), andcultured in M199 medium (Mediatech, Inc., Manassas, VA) supplemented with 20% (v/v) FBS (Hyclone), penicillin-streptomycin(100 units/ml and 100 μg/ml, respectively, both from Invitrogen),sodium pyruvate (1 mmol/liter), heparin (90 μg/ml, Sigma),and endothelial cell growth supplement (20 μg/ml, BD Bioscience). Cells from passage 3 to 8 were used at a confluence of 80%. Prior to use in experiments, cells were grown for 6 h in medium M199 containing 0.2% FBS.
Adeno-CIITA Construct
The DNA fragment encoding Class II Transactivator (CIITA), a transcription factor regulating HLA class II expression, was sub-cloned from plasmid pcDNA3 myc CIITA into the adenovirus-based vector pAd/PL-DEST. Recombinant adenovirus encoding CIITA were then produced, amplified and tittered. Primary cultures of human aortic endothelial cells were infected with adenovirus encoding CIITA. Expression of HLA class II in endothelial cells was determined by flow cytometric analysis. CIITA expressing endothelial cells were stimulated with HLA class II antibodies and protein phosphorylation of distinguished signaling pathways was examined by Western blot.
HLA class II Expression
Primary cultures of human aortic endothelial cells (EC) were infected with adenovirus encoding CIITA (Ad-CIITA) at a multiplicity of infection (MOI) of 2 or were pretreated with 200U of TNFα and 500U of IFNγ for two days. Expression of HLA class II antigens on EC was determined by flow cytometric analysis using anti-human HLA class II antibodies. MOI is the ratio of infectious agents, virus particles, to infection targets, EC. As virus particles increased, the ratio of infected cells with at least one viral particle also increased, MOI is 1.0, the percentage of infected is 63.2%; MOI=2.0, the percentage of infected is 86.5%; MOI=3.0, the percentage of infected is 95.0%; and MOI=5.0, the percentage of infected is 99.3% (47).
siRNA Transfection
The human mTOR small inferring RNA (siRNA) duplexes(5′- CCA AAG UGC UGC AGU ACU AUU-3′ and 5′-UAG UAC UGC AGC ACU UUG GUU-3′), human Raptor siRNA duplexes (5′- GGA CAA CGG CCA CAA GUA CdTdT-3′ and 5′-GUA CUU GUG GCC GUU GUC CdTdT-3′), human Rictor siRNA duplexes (5′-ACU UGU GAA GAA UCG UAU CdTdT-3′ and 5′-GUA ACG AUU CUU CAC AAG UdTdT-3′), human FAK siRNA duplexes (5′-GGU UCA AGC UGG AUU AUU U-3′and 5′-AAA UAA UCC AGC UUG AAC C-3′), human c-Src siRNA SMARTpool, human MEK siRNA SMARTpooland control non-targeting siRNA duplexes (5′-UAG CGA CUA AAC ACA UCA AUU-3′ and 5′-AAU UGA UGU GUU UAG UCG CUA-3′) were synthesized by Dharmacon, Inc. (Lafayette, CO). Transfection of siRNA for EC was previously described (28, 31). Briefly, EC were plated at a density of 70% confluency in 35mm dishes in FBS free medium M199 and transfected with siRNA using Mirus TransIT-TKO® transfection reagents. For each transfection, 6 μl of the Mirus transfection reagent was mixed with 200 μl of serum-free medium OPTI-MEM®I in a 5 ml tube and incubated for 5 min at room temperature. Following the incubation, 100 nM siRNA was added into the mixture and incubated for 5 min at room temperature. Fresh complete medium was added to the cells 5 h after transfection and experiments were conducted 48 h post-transfection. Immunoblotting with anti-mTOR, Raptor, Rictor, or FAK antibody was performed to monitor the efficiency of siRNA knockdown. Anti-Vinculin, β-tubulin, or β-actin Ab was used to confirm equal loading of cellular proteins in lysates.
Preparation of Cell Lysates and Western Blot
Cell lysates for Western blot were prepared as previously described (29). Briefly, EC were seeded in 35 mm dishes coated with 0.1% gelatin. Serum and growth factor-starved cells were treated with anti-HLA class II antibody or control IgG at 37°C, washed 2 times with ice-cold phosphate-buffered saline (PBS) containing 1mM sodium orthovanadate (Na3VO4) and lysed in buffer (containing 20 mM Tris, pH 7.9, 137 mM NaCl, 5 mM ethylenediamine tetraacetic acid (EDTA), 1 mM ethyleneglycol-bis-(b1aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), 10% glycerol, 1% Triton X-100, 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM Na3VO4, 10 μg/ml aprotinin, and 10 μg/ml leupeptin) for 10 min on ice. The cell lysates were centrifuged at 15,000 × g for 10 min at 4°C, and the supernatants were collected and pre-cleared for 2 hours with 15 μl of protein A/G plus-agarose at 4°C. The total protein content was measured using the Bradford Protein Assay method (Pierce) using bovine serum albumin (BSA) as standard. Proteins in whole cell lysates were heated for 5 min at 95°C in 6 x sodium dodecylsulfate (SDS) sample buffer, electrophoresed on SDS polyacrylamide gels (SDS-PAGE), and transferred to polyvinylidene difluoride (PVDF) membrane. The membranes were blocked using 5% non-fat dry milk in Tris-buffered saline, pH 7.4, containing 0.05% Tween 20 (TBST) for 1 h at room temperature, and incubated with appropriate primary antibody overnight at 4°C. The blots were washed with TBST followed by incubation in HRP-conjugated secondary antibody for 1 h at room temperature. The blots were subsequently washed with TBST and developed with an enhanced chemiluminescense (ECL, Amersham). The phosphorylated protein bands were scanned using the EPSON Perfection V700 Photo Scanner (EPSON) and were quantified using the software ImageJ (NIH, Bethesda, MD).
Cell Proliferation Assays by CFSE Labeling
EC were labeled with carboxy fluoresce in diacetate, succinimidyl ester (CFSE) (Molecular Probes, Eugene, OR) according to the manufacturer's protocol. Briefly, EC were grown in 35 mm culture dishes coated with 0.1% gelatin up to 70% confluence. The cells were starved in medium M199 without FBS for 6 h, labeled with 2 μM CFSE at 37°C for 15 min, and washed twice in warm M199. CFSE labeled cells were stimulated with anti-HLA class II antibody for 48 h in M199 with 2%FBS. Cells were detached using 0.125% Trypsin/0.05% EDTA, washed with PFA (2.5%FBS, 0.1% Sodium Azide, in PBS), and analyzed by flow cytometry. Analysis was performed on 10,000 cells/sample using a FACScalibur (Becton Dickinson, Mountain View, CA). Freshly CFSE labeled EC and CFSE labeled EC grown in complete medium were used as negative and positive controls for staining, respectively. Data was analyzed using CellQuestPro (Becton Dickinson, Mountain View, CA) and Modfit LT software (Verity Software House, Topsham, ME). Cell proliferation was calculated using the Proliferation Wizard Model (Verity Software House, Topsham, ME). The proliferation index (PI) is the sum of the cells in all generations divided by the computed number of original parent cells present at the start of the experiment (33). Our previous studies verified that endothelial cells do not exhibit dye extrusion up to 72 hours after CFSE labeling(33, 48).
Cell Proliferation Assays by BrdU Incorporation
DNA synthesis was measured by bromide oxyuridine (BrdU) incorporation using the BD Pharmingen BrdU Flow Kits (BD Biosciences, San Jose, CA), according to the manufacturer's protocol. Briefly, EC grown to 70% confluence in 35 mm culture dishes coated with 0.1% gelatin in medium M199 containing 2.0% FBS were treated with anti-HLA class II antibody or isotype control IgG for 48 h. During the final 2 h of incubation, 10 μM BrdU was added to the cell culture. The cells were detached with accutase (Innovative Cell Technologies, Inc., San Diego, CA), fixed, and permeabilized at room temperature. DNA was denatured by incubation for 60 min at 37°C with 50 μl DNase. The cells were incubated with 20 μl FITC-anti-BrdU antibody for 20 min at room temperature, and then total DNA was stained with 7-AAD. Thereafter, the cells were analyzed for simultaneous green (FL1) and red (FL3) fluorescence emission on a FACScaliber flow cytometer (Becton Dickinson).
Cell migration assays by wound healing
In Vitro wound healing assay was performed as previously described (33, 48). Briefly, EC were grown in 35 mm culture dishes coated with 0.1% gelatin up to confluence. Starved cells were pretreated with 10 μg/ml of mitomycin C for 2 h to inhibit cell proliferation. A scratch wound was created with a sterile 200-μl pipette tip. The dishes were rinsed with M199 to remove detached cells. Wounded cells were treated with anti-HLA class II mAb for 16 h. The cells were fixed with 10% neutral buffered formalin, stained with Wright-Giemsa (Sigma-aldrich), and wound closure was monitored by microscopy. The cell number between two initiated front edges was counted. Migration rate was analyzed by calculating the cell number between two initiated front edges of class II-stimulated EC divided by the cell number between two initiated front edges of control EC.
Cell migration assays by transwell insert system
The migration of EC was measured in a transwell insert system (8.0 μm pore size, Corning). EC were grown in 24-well plate coated with 0.1% gelatin and were infected with Ad-CIITA or pretreated with TNFα/IFNγ for 2 days. Cells were trypsinized, resuspended, and 30,000 cells were seeded on the upper chamber of the insert and stimulated with anti-HLA II antibody in M199 + 2% FBS. After 16 h, cells on the upper surface of the membrane were removed, and the migrated cells were fixed with methanol and stained with crystal violet. Three fields per insert were photographed with 10 x objective lens, and were counted (33).
Statistical analysis
Each experiment was repeated three times independently. Unless otherwise noted, data are presented as mean ±SEM. Differences in protein phosphorylation, cell proliferation, or cell migration were calculated using the Student's t test or the one way analysis of variance (ANOVA) with Fisher's least significant difference (LSD). All p values were two-sided, and less than 0.05 was considered significant.
Results
Induction of HLA class II expression in human endothelial cells by Ad-CIITA infection and TNFα/IFNγ treatment
HLA class II molecules are not constitutively expressed on primary EC in culture, but can be up-regulated by inflammatory cytokines such as TNFα and IFNγ (44). However, treatment of EC with cytokines induces intracellular signaling cascades and phenotypic changes that may interfere and/or confound interpretation of the signaling pathways and crosstalk mechanisms induced by engagement of HLA II. To overcome this limitation, we engineered an adenoviral vector to express the class II transactivator (CIITA), a master regulator of HLA II gene expression in primary EC (Fig S1A). Prior to initiation of experiments, we optimized the pretreatment time and concentration of cytokines TNFα/IFNγ and Ad-CIITA to achieve HLA II expression. We determined a multiplicity of infection (MOI) of 2.0 Ad-CIITA achieved high levels of HLA II expression with low background. Pretreatment of EC with a combination of 200 U of TNFα and 500 U of IFNγ for 48 h induced expression of CIITA and HLA II (Fig S1B). Infection of EC with Ad-CIITA (Fig S1C) or pretreatment of EC with TNFα and IFNγ (Fig S1D) induced CIITA protein production, but not on LacZ-transfected or un-stimulated EC (Fig S1E, F). The bar graph shows that median fluorescence intensity (MFI) of HLA II molecules increased on Ad-CIITA transfected EC and cytokine pretreated EC as antibody binding to EC increased, with saturation at about 1.0 μg/ml (Fig S1G, H). Expression of HLA-DR was the highest followed by nearly equivalent levels of DQ and DP (Fig S1I, J).
Ligation of HLA II with antibody stimulates EC proliferation, migration and an array of early signaling events
Having established conditions to generate EC expressing HLA II, we determined whether antibody-mediated ligation of HLA II stimulates proliferation and migration. Prior to initiation of experiments, we performed dose response experiments and established 1.0 μg/ml of HLA II antibody to elicit the highest degree of EC proliferation and migration (data not shown). This concentration was then used in subsequent experiments. EC from three different donors were infected with Ad-CIITA or pretreated with TNFα/IFNγ for 48 h to up-regulate HLA II expression, and then treated with HLA II antibody for 48 h. Significantly, more DNA synthesis was seen in Ad-CIITA infected EC exposed to HLA II antibodies compared with isotope control mouse IgG (mIgG)(Fig 1A, B). In contrast, ligation of LacZ-infected EC with anti-HLA II antibody failed to stimulate cell proliferation (Fig 1A, B), consistent with lack of HLA II expression (Fig S1E, G). Likewise, ligation of HLA II on TNFα/IFNγ pretreated EC with a saturating dose of 1μg/mL of antibody stimulated a 2.7-fold increase in cell proliferation compared with isotype control mIgG (Fig 1A, C). Similar results were observed when cell proliferation was assayed by CFSE labeling in Ad-CIITA transfected or TNFα/IFNγ pretreated EC in response to ligation of HLA II (Fig 1D, E, F). Specifically, more endothelial cells underwent several rounds of division, as measured by dilution of CFSE, a 1.5-fold increase in cell proliferation in Ad-CIITA infected EC or a 2.7-fold increase in TNFα/IFNγ pretreated EC, in the presence of HLA II antibodies compared with mIgG (Fig 1D, E, F).
Fig. 1. HLA class II antibody stimulates EC proliferation and migration.

EC wereinfected with Ad-CIITA or pretreated with TNFα/IFNγ in 35 mm dishes coated with 0.1% gelatin for 48 h. A, EC were stimulated with 1.0 μg/ml HLA class II antibody for 48 h, incorporated with BrdU for 2 h, and harvested. EC proliferation was measured by flow cytometry. DNA synthesis S phase was gated, and proliferation index (PI) is presented as fold increase in the percent of cells positive for BrdU normalized to untreated control. D. EC were labeled with CFSE, stimulated with 1.0 μg/ml HLA class II antibody for 48 h, and harvested. EC proliferation was measured by flow cytometry and analyzed by Modfit LT software. EC proliferation was calculated using the Proliferation Wizard Model. The proliferation index is the sum of the cells in all generations divided by the computed number of original parent cells present at the start of the experiment. G. EC were pretreated with 10 μg/ml of mitomycin C for 2 h to inhibit cell proliferation before being assayed for their ability to migrate. A scratch wound was created with a sterile 200-μl pipette tip. Wounded cells were stimulated with 1.0 μg/ml of anti-HLA class II antibody for 16 h. Representative microscopy fields are shown. The cell number between two initiated front edges was counted; migration rate was analyzed by calculating the cell number between two front edges of class II-stimulated EC divided by the cell number between two front edges of control EC. J. Cell migration was measured in a transwell insert system. EC infected with Ad-CIITA or pretreated with TNFα/IFNγwere seeded to the upper chamber of insert and stimulated with 1.0 μg/ml class II antibody. After incubation for 16 h at 37°C, the cells on the upper surface of the insert membrane were removed with a cotton swab, the migrated cells on the reverse side of the insert membrane were fixed with methanol and stained with crystal violet, three middle fields per insert were photographed with 10x objective lens, and migrated cells were counted. Fluorescence 10 x microscopy images are presented. B,C, E, F. The bar graphs show the mean ± SEM fold change of proliferation index. H, I, K, L. The bar graph shows the mean ± SEM number of migrated cells. *p<0.05, **p<0.01, and ***p<0.001 were analyzed by one way ANOVA with Fisher's LSD. Data represent a minimum of three independent experiments.
To determine whether HLA II signaling stimulates EC migration, primary aortic endothelial cells from three different donors were infected with Ad-CIITA or pretreated with TNFα/IFNγ for 48 h, then treated with anti-HLA II antibody for 16 h. Antibody ligation of HLA II on Ad-CIITA infected EC stimulated a 1.9-fold increase in cell migration (wound healing assay) compared with mIgG-treated EC (Fig 1G, H). Ligation of HLA II on LacZ infected EC with anti-HLA II antibody failed to stimulate detectable EC migration (Fig 1G, H). Ligation of HLA II on TNFα/IFNγ pretreated EC with antibody stimulated a 4.7-fold increase in cell migration compared with isotype control mIgG (Fig 1 G, I). These results were corroborated by using the transwell migration assay. Ligation of HLA-II on Ad-CIITA infected EC stimulated a 1.9-fold increase in cell migration (Fig 1 J, K). Again, no effect of HLA II antibody was seen in LacZ infected EC. Furthermore, ligation of HLA-II on TNFα/IFNγ pretreated EC with antibody stimulated a 2.9-fold increase in cell migration compared with isotope control mIgG(Fig 1 J, L). Endothelial cell binding antibodies not directed against HLA, anti-CD105 antibody or anti-integrin αvβ3 antibody, did not significantly stimulate cell migration in wound healing and transwell migration assays (Fig S4 C-F). These results demonstrate that ligation of HLA class II with antibodies stimulates proliferation and migration in primary human endothelial cells.
Ligation of HLA II with antibody stimulates an array of early signaling events
We next sought to identify the intracellular signaling pathways that lead to HLA II-induced EC proliferation and migration. To characterize HLA II antibody-triggered activation of intracellular signal transduction networks, we initially established the time and concentration of HLA II antibody necessary to elicit optimal antibody-stimulated activation of signal transduction pathways in Ad-CIITA infected and TNFα/IFNγpretreated EC (Fig S2, S3). Stimulation of Ad-CIITA infected EC or TNFα/IFNγpretreated EC with HLA II antibody at various concentrations (Fig. S2) and times (Fig. S3) induced a marked increase in the phosphorylation of Src Tyr-418, FAK Tyr-577, mTOR Ser-2448, S6K Thr-389, a site directly targeted by mTORC1, S6RP Ser-240/244, a site directly downstream to S6K, Akt Thr-308, a site targeted by PDK1 in response to PI3K, Akt Ser-473, targetedby mTORC2, and ERK Thr-202/Tyr-204 compared with mIgG. No increased phosphorylation of any of these targets was detected in LacZ infected EC (Fig S2, S3). Endothelial cell binding non-HLA antibodies, against CD105 and integrin αvβ3 also did not significantly increase phosphorylation of these signal molecules (Fig S4 A, B). Given that treatment for 15 min using HLA II antibody at a concentration of 0.1 μg/ml produced optimal responses with increased phosphorylation of all signaling molecules tested, these conditions were adopted in subsequent experiments. The results shown in Fig S2 and Fig S3 prompted us to use multiple inhibitors and siRNAs to unravel the cause-effect relationships and regulatory feedback loops in this set of signaling events.
HLA II antibody-stimulated signaling through mTORC1 and mTORC2 in EC
The Akt/mTOR network is a key signaling pathway in the regulation of cell metabolism, migration, survival, and proliferation (49) that plays a critical role in HLA class I antibody-induced EC activation, survival, proliferation and migration(27, 31, 33, 34). However, the function of this pathway in HLA II antibody-induced EC signaling, proliferation and migration is unknown. Consequently, we explored the in depth relationship between mTOR complexes (mTORC1 and mTORC2) and their downstream targets, including S6K, S6RP, and Akt, following ligation of HLA class II molecules with antibodies.
Pretreatment of EC with rapamycin for 2 h, which inhibits mTORC1 but does not interfere with mTORC2 signaling, prevented HLA II-induced phosphorylation of mTOR Ser-2448, S6K Thr-389, and S6RP Ser-240/244, but did not have any detectable effect on the phosphorylation of Akt Ser-473 or ERK Thr-202/Tyr-204 in either Ad-CIITA infected or TNFα/IFNγpretreated EC (Fig 2A, B). In contrast, long-term treatment (24 h) with rapamycin, which blocks both mTORC1 and mTORC2 in EC(33), and additionally abolished phosphorylation of Akt Ser-473, in line with the notion that this residue is targeted by mTORC2 (Fig 2A, B). Interestingly, long-term exposure to rapamycin caused hyper-phosphorylation of ERK in Ad-CIITA infected (Fig 2 A, C) or TNFα/IFNγpretreated EC (Fig 2B, D), and did not affect phosphorylation of c-Raf-1 Ser-259. Importantly, either short-term or long-term treatment with rapamycin of Ad-CIITA infected or TNFα/IFNγpretreated EC did not affect HLA II antibody-stimulated phosphorylation of Src Tyr-418, FAK Tyr-576, p85 PI3K Tyr-458 or Akt Thr-308 (Fig 2), implying that Src, FAK, and PI3K/Aktfunction upstream of the mTOR complexes in the signaling network triggered by engagement of HLA II molecules in EC.
Fig. 2. mTOR regulates HLA class II antibody-stimulated activation of intracellular signal networks, cell proliferation and migration.
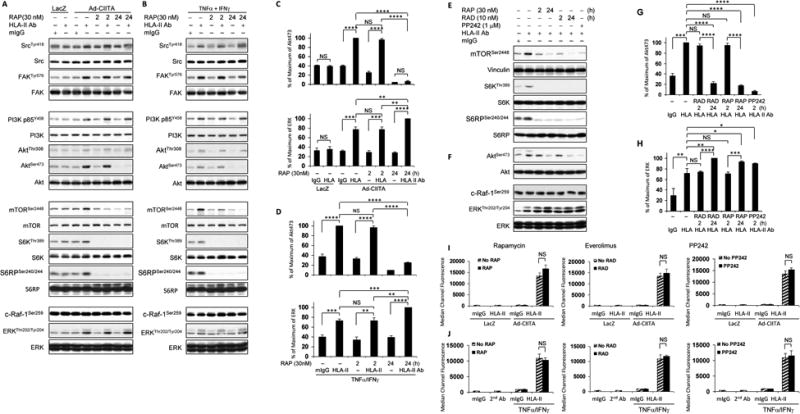
EC wereinfected with Ad-LacZ A, C, I or Ad-CIITA or B, D, E, F, J pretreated with TNFα/IFNγ for 48 h. A, B, E, F StarvedEC were pretreated with 30 nM of rapamycin for 2 h or 24 h, or E, F with 10 nM of everolimus for 2 h or 24 h;or I, J with 30 nM of rapamycin or 10 nM of everolimus for 24 h; or E, F, I, J with 1.0 μM of PP242 for 2 h. A, B, E, F Cells were stimulated with 0.1 μg/ml HLA II antibody or control mIgG for 15 min. Proteins in the precleared cell lysates were separated by 6∼15% SDS-PAGE followed by immunoblotting with A, B anti-phospho-Src Tyr418, FAK Tyr576, p85 PI3K Tyr458, Akt Thr308, A, B, E, F Akt Ser473, mTOR Ser2448, S6K Thr389, S6RP Ser240/244, c-Raf-1 Ser259, or ERK1/2 Thr202/Tyr204 antibodies. The membrane was re-probed with anti-vinculin, S6K, S6RP, Akt, ERK or A, B Src, FAK, PI3K, mTOR, or β-actin total antibodies to confirm equal loading of proteins. C, D, G, H The bar graphs show phosphorylated protein bands shown in A, B, E, F were quantified by densitometry scan analysis, and results are expressed as the mean ± SEM percentage of maximal increase in phosphorylation above control values. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 were analyzed by one way ANOVA with Fisher's LSD. I, J EC were detached with 0.125% trypsin/0.05% EDTA, washed with PBS containing 2.5% FBS and 0.1% sodium azide (PFA), and incubated with 1.0 μg/ml of anti-HLA-II monoclonal antibodies for 30 min at 4°C. EC were washed 3 times with PFA and incubated with FITC-conjugated goat anti-mouse F(ab’)2 fragment for 30 min at 4°C. The fluorescence intensity was measured by LSRFortessa flow cytometry using FACSDiva program (Becton Dickinson, Mountain View, CA). The bar graphs show the mean ± SEM of median channel fluorescence values of HLA-II expression on EC and were analyzed by one way ANOVA with Fisher's LSD. Data represent at least three independent experiments.
Rapamycin and everolimus are allosteric inhibitors of mTOR that act via FKBP-12 (33), whereas the compound PP242 acts at the catalytic site of mTOR thereby inhibiting both mTORC1 and mTORC2 (50). Exposure to everolimus or PP242 prevented the phosphorylation of mTOR Ser-2448, S6K Thr-389, and S6RP Ser-240/244 (Fig 2E), indicating that allosteric or active-site inhibitors of mTOR blocked the mTORC1/S6K signal cascade. Long-term treatment with allosteric inhibitors or exposure to PP242 abolished phosphorylation of Akt Ser-473 (Fig 2F, G), in line with the notion that these interventions blocked mTORC2. In agreement with data shown in Fig 2A-D, long-term treatment with rapamycin or everolimus or exposure to PP242 caused over-activation of ERK (Fig 2F, H) with no effects on HLA-II expression (Fig 2I, J), and did not affect phosphorylation of c-Raf-1 Ser-259 (Fig 2F). These results imply that mTORC2 mediates a novel negative feedback loop, which reduces the activation of ERK in HLA II-stimulated EC cells.
We next determined whether inhibition of mTOR in these cells prevents HLA II antibody-stimulated cell proliferation and migration. Treatment with everolimus, rapamycin or PP242 blocked HLA II antibody-stimulated cell proliferation in either Ad-CIITA infected EC (Fig 3A) or TNFα/IFNγ pretreated EC (Fig. 3B).
Fig. 3. mTOR regulates HLA class II antibody-stimulated EC proliferation and migration.

EC were infected with Ad-LacZ or Ad-CIITA (A, C, E) or pretreated with TNFα/IFNγ for 48 h (B, D, F). A, B Cells were stimulated with 1.0 μg/ml HLA class II antibody or control mIgG for 48 h. Cells were incorporated with BrdU for 2 h and harvested. EC proliferation was measured by flow cytometry, DNA synthesis S phase was gated, and proliferation index (PI) is presented as fold increase in the percent of cells positive for BrdU normalized to negative control. C, D Cells were pretreated with 10 μg/ml of mitomycin C for 2 h to inhibit cell proliferation before being assayed for their ability to migrate. A scratch wound was created with a sterile 200-μl pipette tip. Wounded cells were stimulated with 1.0 μg/ml of anti-HLA class II antibody for 16 h. The cell number between two initiated front edges was counted; migration rate was analyzed by calculating the cell number between two initiated front edges of class II-stimulated EC divided by the cell number between two initiated front edges of control EC. E, F Cell migration was measured in a transwell insert system. EC infected with Ad-CIITA or pretreated with TNFα/IFNγwere added to the upper chamber of insert and stimulated with 1.0 μg/ml class II antibody. After incubation for 16 h at 37°C, the cells on the upper surface of the insert membrane were removed with a cotton swab, the migrated cells on the bottom of the insert membrane were fixed with methanol and stained with crystal violet, three middle fields per insert were photographed with 10 x objective lens, and migrated cells were counted. The bar graphs show the mean ± SEM fold change of A, B proliferation index; or C, D, E, F migrated cells. *p<0.05, ***p<0.001, and ****p<0.0001 were analyzed by one way ANOVA with Fisher's LSD. Data represent at least three independent experiments.
Similarly, pretreatment with these pharmacological agents inhibited HLA-II Ab-stimulated cell migration, in Ad-CIITA infected EC by (Fig 3 C) or TNFα/IFNγ pretreated EC (Fig. 3D). These results were corroborated using a transwell migration assay (Fig 3 E, F). Collectively the results indicate that mTORC1 and mTORC2 play a critical role in HLA II antibody-stimulated EC proliferation and migration.
Knockdown of mTOR, Raptor, or Rictor with siRNA inhibits HLA II antibody-stimulated EC proliferation and signaling
To corroborate our findings with pharmacological inhibitors of mTOR in HLA II antibody-stimulated EC proliferation and signaling, we next determined the effect of siRNA-mediated knockdown of mTOR, Raptor (mTORC1), or Rictor (mTORC2) protein expression on cell proliferation. Initially, we characterized the efficiency of siRNA knockdown. As shown in Fig 4A, transfection of EC with siRNA targeting mTOR siRNA markedly reduced mTOR protein expression but did not alter the expression of Raptor, Rictor, S6K, Akt, ERK, Vinculin, β-tubulin, and β-actin in the same cell lysates. Furthermore, transfection of EC with non-targeting control siRNA had no effect on mTOR expression (Fig 4A). Transfection of EC with siRNAs targeting Raptor and Rictor also caused a marked decrease in the expression of these proteins (Fig 4A). Importantly, siRNA-mediated knockdown of mTOR, Raptor, or Rictor did not reduce HLA-II expression level on Ad-CIITA infected EC (Fig 4B) and TNFα/IFNγ pretreated EC (Fig 4C) measured by flow cytometry, completely blocked HLA II antibody-stimulated cell proliferation (Fig 4D, E) supporting the notion that mTORC1 and mTORC2 play key roles in EC proliferation triggered by engagement of HLA II.
Fig. 4. Effects of mTOR, Raptor, and Rictor siRNA knockdown on HLA II antibody-stimulated EC proliferation and activation of signal transduction networks.
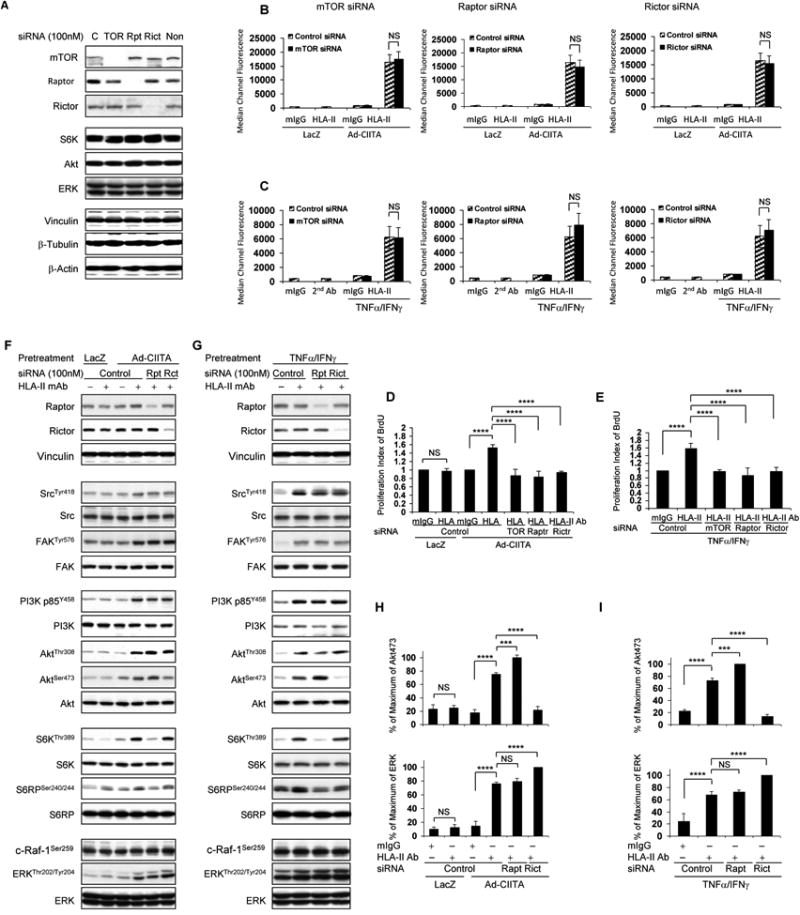
A, B, C, D, E EC were transfected with 100 nM mTOR, or A, B, C, D, E, F, G Raptor, Rictor, or control siRNA for 48 h. A, F, G Protein in pre-cleared cell lysates were separated by 6∼15% SDS-PAGE followed by immunoblotting with anti-mTOR, Raptor, Rictor, S6K, Akt, ERK, Vinculin, β-tubulin, and β-actin. B, D, F EC wereinfected with Ad-LacZ or Ad-CIITA or C, E, G pretreated with TNFα/IFNγ for 48 hours. B, C EC were detached with 0.125% trypsin/0.05% EDTA, washed with PFA, and incubated with 1.0 μg/ml HLA class II antibody for 30 min at 4°C. EC were washed 3 times with PFA and incubated with FITC-conjugated goat anti-mouse F(ab’)2 fragment for 30 min at 4°C. The fluorescence intensity was measured on anLSRFortessa flow cytometer and calculated using the FACSDiva program (Becton Dickinson, Mountain View, CA). The bar graphs show the mean ± SEM of median channel fluorescence values of HLA-II expression on EC and were analyzed by one way ANOVA with Fisher's LSD. D, E Transfected EC were stimulated with 1.0 μg/ml HLA class II antibody or mIgG for 48 h. EC were incorporated with BrdU for 2 h and harvested. EC proliferation was measured by flow cytometry, DNA synthesis S phase was gated, and proliferation index (PI) is presented as fold increase in the percent of cells positive for BrdU normalized to negative control. The bar graphs show the mean ± SEM fold change of proliferation index. F, G Transfected ECwere stimulated with0.1 μg/ml HLA class II antibody for 15 min. Treatment of EC with mIgG serves as negative control. Proteins in the pre-cleared cell lysates were separated by 6∼15% SDS-PAGE followed by immunoblotting withantibodies to Raptor, Rictor for knockdown efficiency, anti-phospho-Src Tyr418, FAK Tyr576, p85 PI3K Tyr458, Akt Thr308, Akt Ser473, S6K Thr389, S6RP Ser240/244, c-Raf-1 Ser259, or ERK Thr202/Tyr204. The membranes were re-probed with anti-Src, FAK, PI3K, Akt, S6K, S6RP, ERK, or Vinculin antibodies to confirm equal loading of proteins. H, I Phosphorylated protein bands shown in F and G were quantified by densitometry scan analysis and results are expressed as the mean ± SEM percentage of maximal increase in phosphorylation above control values. ***p<0.001, and ****p<0.0001 was analyzed by one way ANOVA with Fisher's LSD. Data represent at least three independent experiments.
To further characterize the mechanism(s) by which mTOR complexes regulate HLA II antibody-stimulated activation of signal transduction pathways, we determined the effect of siRNA knockdown of Raptor and Rictor on HLA II antibody-induced signaling. Similar to short term exposure to Rapamycin (Fig 2A, B, E), knockdown of Raptor with siRNA blocked HLA II antibody-induced phosphorylation of S6K Thr-389 and S6RP Ser-240/244 (Fig 4 F, G). Interestingly, transfection of Ad-CIITA infected EC and TNFα/IFNγ pretreated EC with Raptor siRNA to dissect mTORC1 caused hyper-activation of Akt Ser-473 (Fig 4 F-I), suggesting that mTORC1 mediates a negative feedback loop, reducing the activity of mTORC2. Conversely, siRNA-mediated knockdown of Rictor to block mTORC2 abolished HLA II antibody-mediated phosphorylation of Akt Ser-473 and caused over-activation of ERK (Fig 4 F, G, H, I), and did not affect phosphorylation of c-Raf-1 Ser-259 (Fig 4 F, G),thus confirming our results with long-term treatment with allosteric inhibitors or catalytic inhibitor of mTOR (Fig 2A-H). As expected, transfection of EC with Rictor siRNA did not inhibit mTORC1-mediated phosphorylation of S6K Thr-389 and S6RP Ser-240/244 (Fig 4 F, G). It is noteworthy that knockdown of Raptor or Rictor did not affect HLA II antibody-stimulated phosphorylation of Src Tyr-418, FAK Tyr-576, p85 PI3K Tyr-458 or Akt Thr-308, indicating that Src, FAK, PI3K, and PDK1/Akt function upstream of the mTOR signaling pathway.
PI3K activation leads to Akt and mTOR activation but is not necessary for FAK/Src or ERK activation in HLA class II antibody-stimulated EC
As PI3K/Akt is a major pathway leading to mTOR activation, we determined whether HLA class II antibody-stimulated mTOR activation also proceeds through PI3K/Akt in human endothelial cells. Pretreatment of starved Ad-CIITA infected and TNFα/IFNγ pretreated EC with either the dual PI3K/mTOR inhibitor LY294002 or the compound A66, a specific and potent inhibitor of the p110α catalytic subunit of PI3K (51, 52) suppressed HLA class II antibody-stimulated phosphorylation of p85 PI3K Tyr-458, Akt Thr-308, and Ser-473, and also abolished phosphorylation of mTOR Ser-2448, S6K Thr-389, and S6RP Ser-240/244 (Fig. 5 A, B). Exposure of EC to either LY294002 or A66 did not affect HLA class II antibody-induced phosphorylation of Src Tyr-418 or FAK Tyr-576 (Fig. 5 A, B), implying that the FAK/Src kinases function upstream or in parallel to PI3K/Akt/mTOR in EC.
Fig. 5. PI3K regulates HLA class II antibody-mediated signal transduction pathways, cell proliferation, and migration.
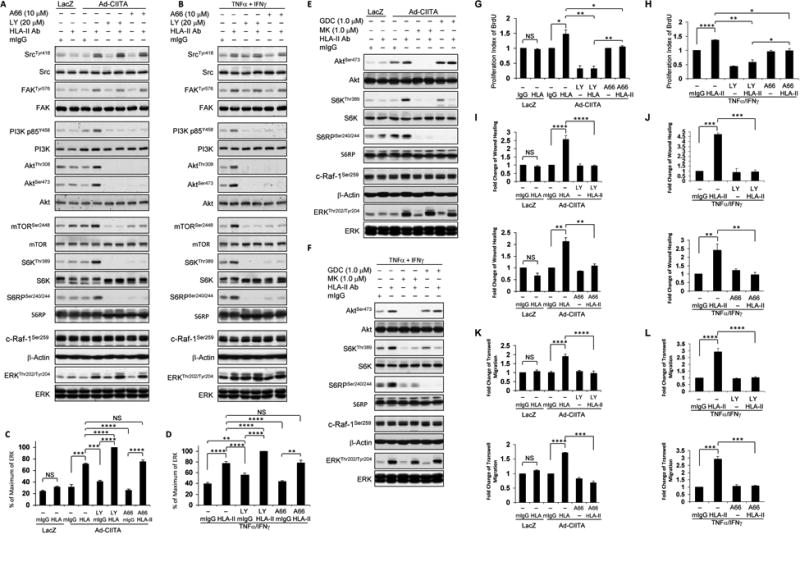
EC were A, C, E, G, I, K infected with Ad-LacZ or Ad-CIITA or B, D, F, H, J, L pretreated with TNFα/IFNγ for 48 h. A, B, C, D, G, H, I, J, K, L Starved cells were pretreated with 20 μM LY294002, or with 10 μM A66, or E, F with 1.0 μM MK-2206 or with 1.0 μM GDC-0068 for 60 min. A, B, E, F Pretreated ECwere stimulated with0.1 μg/ml of HLA class II antibody or mIgG control for 15 min. Proteins in the pre-cleared cell lysates were separated by 6∼15% SDS-PAGE followed by immunoblotting with anti-phospho-Src Tyr418, FAK Tyr576, p85 PI3K Tyr458, Akt Thr308, Akt Ser473, mTOR Ser2448, S6K Thr389, S6RP Ser240/244, MEK Ser217/221, c-Raf-1 Ser259, or ERK Thr202/Tyr204. The membranes were re-probed with anti-Src, FAK, PI3K, Akt, mTOR, S6K, S6RP, ERK, β-actin antibodies to confirm equal loading of proteins. C, D Phosphorylated protein bands shown in A and B were quantified by densitometry scan analysis and results are expressed as the mean ± SEM percentage of maximal increase in phosphorylation above control values. **p<0.01, ***p<0.001, and ****p<0.0001 were analyzed by one way ANOVA with Fisher's LSD. G, H Starved cells were stimulated with 1.0 μg/ml HLA class II antibody for 48 h, incorporated with BrdU for 2 h, and harvested. EC proliferation was measured by flow cytometry, DNA synthesis S phase was gated, and proliferation index is presented as fold increase in the percent of cells positive for BrdU normalized to negative control. The bar graphs show the mean ± SEM fold change of proliferation index. I, J Cells were pretreated with 10 μg/ml of mitomycin C for 2 h to inhibit cell proliferation before being assayed for their ability to migrate. A scratch wound was created with a sterile 200-μl pipette tip. Wounded cells were stimulated with 1.0 μg/ml of anti-HLA class II antibody, or mIg for 16 h. The cell number between two initiated front edges was counted; migration rate was analyzed by calculating the cell number between two initiated front edges of class II-stimulated EC divided by the cell number between two initiated front edges of negative control EC. K, L. Cell migration was measured in a transwell insert system. EC K infected with Ad-CIITA or L pretreated with TNFα/IFNγwere seeded to the upper chamber of insert and stimulated with 1.0 μg/ml HLA class II antibody or mIgG for 16 h at 37°C. After incubation, the cells on the upper surface of the insert membrane were removed with a cotton swab, the migrated cells on the bottom of the insert membrane were fixed with methanol and stained with crystal violet, three middle fields per insert were photographed with 10 x objective lens, and migrated cells were counted. G, H The bar graphs show the mean ± SEM fold change of proliferation index. I, J, K, L The bar graph shows the mean ± SEM number of migrated cells. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 were analyzed by one way ANOVA with Fisher's LSD. Data represent at least three independent experiments.
Interestingly, treatment with LY294002 enhanced ERK phosphorylation induced by antibody-mediated ligation of HLA II in either Ad-CIITA infected EC (Fig 5 A, C) or TNFα/IFNγpretreated EC (Fig 5 B, D) and did not affect phosphorylation of c-Raf-1 Ser-259 (Fig 5 A, B). These results reinforce the notion (posited above) that mTORC2 mediates negative feedback on ERK activation.
Akt may inhibit RAF-1 by directly phosphorylating c-Raf-1 at Ser259 (53). However, this negative cross talk is controversial(54, 55). Therefore, we determined whether ERK over-activation induced by mTORC2 and PI3K inhibition is mediated by reduction of c-Raf-1 phosphorylation at Ser-259. Starved Ad-CIITA infected EC and TNFα/IFNγ-EC were pretreated with allosteric (MK-2206) or active site (GDC-0068) inhibitors of Akt. Inhibition of Akt blocked PI3K/Akt targeting molecules S6K Thr-389 and S6RP Ser-240/244, but did not affectconstitutive phosphorylation of c-Raf-1 at Ser259. Moreover, hyperphosphorylation of ERK at Thr-202/Tyr-204 was not observed under Akt inhibition (Fig. 5E, F). Our results indicate that PI3K inhibition with LY294002 suppresses anegative feedback loop via mTORC2, thereby leading to enhanced MEK/ERK pathway activity in endothelial cells.
Next, we determined if PI3K was also required for class II antibody-induced cell proliferation and migration, as it would be expected in the light of our previous results with mTOR inhibitors and knockdown of Raptor and Rictor. To test this prediction, starved Ad-CIITA infected EC and TNFα/IFNγ pretreated EC were exposed to LY294002 or A66 and then stimulated with anti-HLA II antibody. Cell proliferation was measured by BrdU incorporation, and cell migration was measured by wound healing and transwell migration assays. Pretreatment with LY294002 or A66 completely prevented class II antibody-stimulated cell proliferation in Ad-CIITA infected EC (Fig 5 G) and TNFα/IFNγ pretreated EC (Fig 5 H). Pretreatment with LY294002 also blocked class II antibody-mediated EC migration in Ad-CIITA infected EC (Fig 5 I, K) and TNFα/IFNγ pretreated EC (Fig 5 J, L) by wound healing or transwell migration assay. These data indicate that PI3K regulates class II antibody-stimulated mTOR, and thereby leads to cell proliferation and migration.
Protein Tyrosine kinase Src regulates HLA II antibody-activated signal transduction networks
Thus far, our analysis of HLA II signaling shows that multiple pharmacological inhibitors or transfection of siRNAs targeting Raptor and Rictor do not affect the activation of Src (scored by phosphorylation at Tyr-418) or FAK (at Tyr-576/577). These findings imply that these kinases are likely to function as upstream elements in the signaling cascade triggered by engagement of HLA II in EC. In order to explore this hypothesis, we next evaluated the impact of Src inhibition on phosphorylation of FAK, PI3K, mTOR, S6K, S6RP, Akt, and ERK. Exposure of Ad-CIITA infected or TNFα/IFNγ pretreated EC to the Src inhibitor PP2 prevented class II antibody-stimulated phosphorylation of Src Tyr-418, FAK Tyr-576/577, p85 PI3KTyr-458, Akt Thr-308, Akt Ser-473, and also abolished phosphorylation of mTOR Ser-2448, S6K Thr-389, and S6RP Ser-240/244 with no effect on HLA-II expression level (Fig. 6 C, D). Importantly, the Src inhibitor also prevented phosphorylation of ERK at Thr-202/Tyr-204 in both Ad-CIITA infected and TNFα/IFNγ pretreated EC (Fig. 6 A, B).
Fig. 6. Src regulates HLA class II antibody-stimulated activation of intracellular signal networks, cell proliferation, and migration.
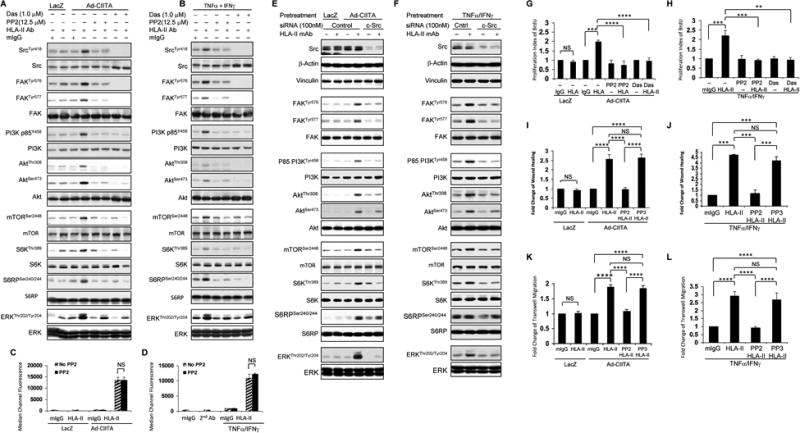
EC were A, C, E, G, I, K infected with Ad-LacZ or Ad-CIITA or B, D, F, H, J, L pretreated with TNFα/IFNγ for 48 h. A, B, C, D, G, H, I, J, K, L Starved cells were pretreated with 12.5 μM PP2, or A, B, G, H with 1.0 μM dasatinib, or I, J, K, L with PP2 inactive analog PP3 for 30 min. E, F, Starved EC were transfected with 100 nM of c-Src, or control siRNA for 48 hours. A, B, E, F EC were stimulated with0.1 μg/ml HLA class II antibody or mIgG control for 15 min. Treatment of EC with mIgG serves as negative control. Proteins in the pre-cleared cell lysates were separated by 6∼15% SDS-PAGE followed by immunoblotting withanti-phospho-Src Tyr418, FAK Tyr576, FAK Tyr577, p85 PI3K Tyr458, Akt Thr308, Akt Ser473, mTOR Ser2448, S6K Thr389, S6RP Ser240/244, or ERK Thr202/Tyr204. The membranes were re-probed with anti-Src, FAK, PI3K, Akt, mTOR, S6K, S6RP, ERK, or β-actin antibodies to confirm equal loading of proteins. C, D EC were detached, incubated with 1.0 μg/ml HLA class II antibody for 30 min at 4°C, after wash 3 times with PFA the cells were incubated with FITC-conjugated goat anti-mouse F(ab’)2 fragment for 30 min at 4°C. The fluorescence intensity was measured by LSRFortessa flow cytometry and calculated using FACSDiva program. The bar graphs show the mean ± SEM of median channel fluorescence values of HLA-II expression on EC and were analyzed by one way ANOVA with Fisher's LSD. G, H Starved cells were stimulated with 1.0 μg/ml HLA class II antibody for 48 h, incorporated with BrdU for 2 h, and harvested. EC proliferation was measured by flow cytometry, DNA synthesis S phase was gated, and proliferation index (PI) is presented as fold increase in the percent of cells positive for BrdU normalized to negative control. I, J Cells were pretreated with 10 μg/ml of mitomycin C for 2 h to inhibit cell proliferation before being assayed for their ability to migrate. A scratch wound was created with a sterile 200-μl pipette tip. Wounded cells were stimulated with 1.0 μg/ml of anti-HLA class II antibody for 16 h. The cell number between two initiated front edges was counted; migration rate was analyzed by calculating the cell number between two initiated front edges of class II-stimulated EC divided by the cell number between two initiated front edges of negative control EC. K, L Cell migration was measured in a transwell insert system. EC infected with Ad-CIITA or pretreated with TNFα/IFNγwere seeded to the upper chamber of insert and stimulated with 1.0 μg/ml HLA class II antibody for 16 h at 37°C. After incubation, the cells on the upper surface of the insert membrane were removed with a cotton swab, the migrated cells on the bottom of the insert membrane were fixed with methanol and stained with crystal violet, three middle fields per insert were photographed with 10 × objective lens, and migrated cells were counted. G, H The bar graphs show the mean ± SEM fold change of proliferation index. I, J, K, L The bar graph shows the mean ± SEM number of migrated cells. **p<0.01, ***p<0.001, and ****p<0.0001 were analyzed by one way ANOVA with Fisher's LSD. Data represent at least three independent experiments.
To substantiate that Src family kinases are required for triggering the signaling network characterized in this study in response to HLA II ligation, we next tested dasatinib, a second-generation tyrosine kinase inhibitor, including the Src family, recently used in clinical trials in solid tumors and leukemia (56, 57). Dasatinib, like PP2, abolished HLA II antibody-stimulated phosphorylation of Src Tyr-418, FAK Tyr576/577, p85 PI3K Tyr-458, Akt Thr-308, Akt Ser-473, mTOR Ser-2448, S6K Thr-389, and S6RP Ser-240/244 and ERK Thr-202/Tyr204 in both Ad-CIITA infected and TNFα/IFNγ pretreated EC (Fig. 6 A, B).
There are nine members of Src family, including c-Src, Fyn, and Yes expressed in EC (29). Different members of Src family may play a different role in HLA-II antibody-triggered signal networks. To confirm our observation using pharmacological inhibitors of Src, we next determined the effect of c-Src siRNA on HLA-II antibody-induced Src activation and its downstream targets. First, we determined the efficiency of c-Src siRNA on c-Src expression. As shown in Fig 6 E, F, c-Src expression was markedly decreased by siRNA knockdown whereas transfection of EC with non-targeted siRNA had no effect on c-Src protein expression (Fig 6E, lane 1-4 and Fig 6 F, lane 1 and 2). To verify c-Src siRNA specificity, we determined the expression of several cytoskeletal proteins in the same cell lysates of EC transfected with c-Src siRNA. Transfection of EC with c-Src siRNA or control siRNA did not affect expression of vinculin and β-actin (Fig 6 E, F). Furthermore, transfection of Ad-CIITA infected and TNFα/IFNγ pretreated EC with c-Src siRNA prevented HLA-II antibody-stimulated phosphorylation of FAK Tyr-576/577, p85 PI3KTyr-458, Akt Thr-308, Akt Ser-473, and abolished phosphorylation of mTOR Ser-2448, S6K Thr-389, and S6RP Ser-240/244 (Fig. 6 E, F). Importantly, the c-Src siRNA also prevented phosphorylation of ERK at Thr-202/Tyr-204 in both Ad-CIITA infected and TNFα/IFNγ pretreated EC (Fig. 6 E, F).
In view of these results, we anticipated that Src should be essential for EC proliferation and migration induced by antibody-mediated ligation of HLA II. Pretreatment with PP2 or dasatinib completely inhibited class II antibody-stimulated cell proliferation in both Ad-CIITA infected and TNFα/IFNγpretreated EC (Fig 6 G, H). Furthermore, pretreatment of EC with PP2 abolished HLA II antibody-stimulated cell migration as determined by wound healing and transwell migration assays (Fig 6 I-L). In contrast, the PP2 inactive analog PP3 did not inhibit class II-stimulated cell migration in either Ad-CIITA infected EC or TNFα/IFNγ pretreated EC (Fig 6 I-L). Our results are consistent with the notion that Src activation is one of the earliest events in the HLA II antibody-triggered signal transduction network elucidated in this study and required for EC proliferation and migration.
Requirement of FAK for HLA II antibody-activated signal transduction in EC
The interaction between focal adhesion kinase (FAK) and Src plays a critical role in the activation of these kinases in response to a variety of stimuli, but the role of FAK in Src activation in EC remains unknown. Consequently, we next determined the role of FAK in HLA-II antibody-induced Src activation and its downstream targets. Initially, we determined the efficiency of FAK siRNA on FAK expression. As shown in Fig 7 A-C, FAK expression was markedly decreased by siRNA knockdown whereas transfection of EC with non-targeted siRNA had no effect on FAK protein expression (Fig 7A, lane 1-4, Fig 7 B, lane 1 and 2; Fig 7 C, lane 1). To verify FAK siRNA specificity, we determined the expression of several cytoskeletal proteins in the same cell lysates of EC transfected with FAK siRNA. Transfection of EC with FAK siRNA or control siRNA did not affect expression of vinculin, β-actin (Fig 7 A-C), and β-tubulin (Fig 7 C) and also did not affect HLA-II expression level on Ad-CIITA infected and TNFα/IFNγ pretreated EC (Fig 7 D, E). To determine whether FAK is associated with Src in HLA class II antibody-stimulated activation of Src, we examined the effect of FAK siRNA on HLA-II-induced phosphorylation of Src at Tyr-418, in the catalytic domain of Src. Transfection of Ad-CIITA infected and TNFα/IFNγ pretreated EC with FAK siRNA inhibited class II antibody-induced phosphorylation of Src at Tyr-418 (Fig. 7 A, B). In contrast, transfection of EC with non-target control siRNA had no effect on class II-mediated activation of Src.
Fig. 7. Effect of FAK siRNA transfection on HLA II antibody-stimulated activation of signal transduction networks and cell proliferation.
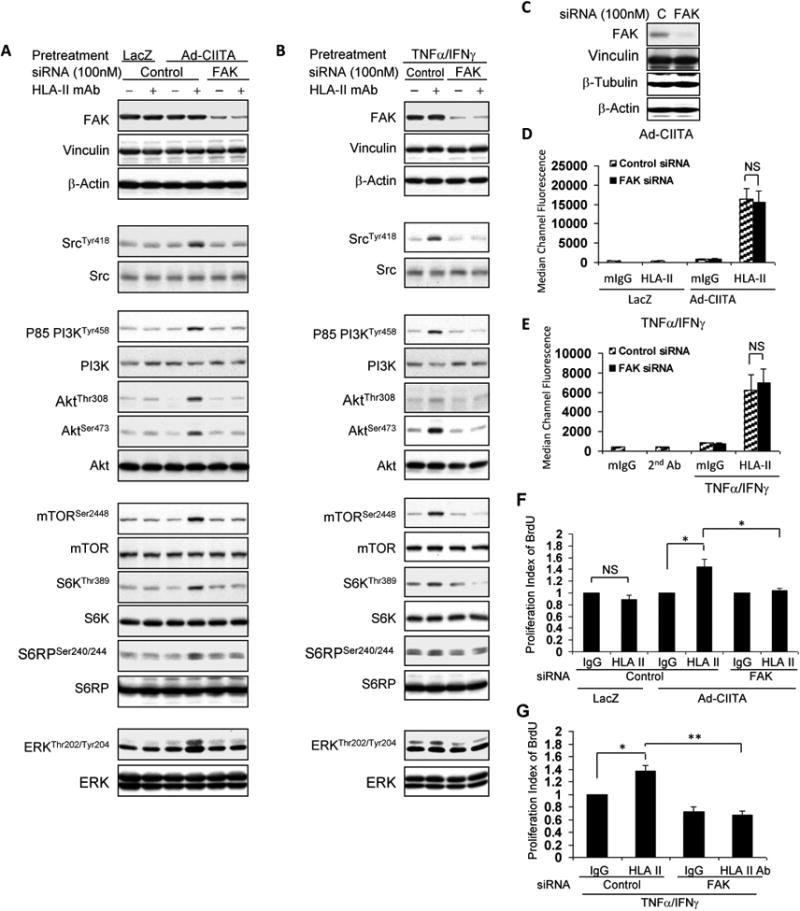
A, D, F EC were infected with Ad-LacZ or Ad-CIITA or B, E, G pretreated with TNFα/IFNγ for 48 hours. A, B, C, D, E, F, GStarved EC were transfected with 100 nM of FAK, or control siRNA for 48 hours. A, B Transfected EC were stimulated with 0.1 μg/ml HLA class II antibody or control mIgG for 15 min. A, B, C Proteins in the pre-cleared cell lysates were separated by 6∼15% SDS-PAGE followed by immunoblotting withanti-FAK antibody for siRNA knockdown efficiency, or A, B with anti-phospho-Src Tyr418, p85 PI3K Tyr458, Akt Thr308, Akt Ser473, mTOR Ser2448, S6K Thr389, S6RP Ser240/244, or ERK Thr202/Tyr204. The membranes were re-probed with anti-Vinculin, Src, PI3K, Akt, mTOR, S6K, S6RP, ERK, β-actin antibodies to confirm equal loading of proteins. Cβ The membrane was immunoblotted with anti-Vinculin, β-tubulin, or β-actin antibody for FAK siRNA knockdown specificity. D, E EC were detached, incubated with 1.0 μg/ml HLA class II antibody for 30 min at 4°C, washed 3 timesand incubated with FITC-conjugated goat anti-mouse F(ab’)2 fragment for 30 min at 4°C. The fluorescence intensity was measured on a LSRFortessa flow cytometer and calculated using FACSDiva program. The bar graphs show the mean ± SEM of median channel fluorescence values of HLA-II expression on EC and were analyzed by one way ANOVA with Fisher's LSD. F, G Starved cells were stimulated with 1.0 μg/ml HLA class II antibody for 48 h, incorporated with BrdU for 2 h, and harvested. EC proliferation was measured by flow cytometry, DNA synthesis S phase was gated, and proliferation index is presented as fold increase in the percent of cells positive for BrdU normalized to negative control. The bar graphs show the mean ± SEM fold change of proliferation index. *p<0.05 and **p<0.01 were analyzed by one way ANOVA with Fisher's LSD. Data represent at least three independent experiments.
Our results demonstrating that FAK plays a critical role in class II antibody-stimulated Src activation raised the possibility that FAK is required for the activation of the whole signaling network triggered by engagement of HLA II. In line with this possibility, transfection of Ad-CIITA infected and TNFα/IFNγ pretreated EC with FAK siRNA abolished HLA II antibody-induced phosphorylation of PI3K, Akt, mTOR, S6K, S6RP and ERK (Fig. 7 A, B). In addition, siRNA-mediated knockdown of FAK blocked HLA-II antibody-stimulated proliferation in Ad-CIITA infected EC (Fig 7 F) and in TNFα/IFNγ pretreated EC (Fig 7 G). In contrast, transfection of EC with non-target control siRNA had no effect on HLA-II antibody-mediated cell proliferation (Fig. 7 F, G). Collectively, our results imply that the activation of the FAK/Src complex is one of the earliest executes in the HLA class II antibody-activated intracellular signal network.
ERK inhibition blocked HLA II antibody-stimulated phosphorylation of p70S6K, S6RP, and ERK, but not Src, FAK, PI3K, Akt, nor mTOR in EC
Our results implicated FAK/Src in the activation of ERK but the role of ERK in HLA class II antibody-triggered activation of intracellular signal transduction network was not known. To clarify the role of ERK in HLA II signaling, starved Ad-CIITA infected EC or TNFα/IFNγ pretreated EC were pretreated with MEK inhibitors UO126 or PD0325901 followed by stimulation with class II antibody. Pretreatment of EC with either UO126 or PD0325901 had no effect on HLA class II antibody-induced phosphorylation of Src Tyr-418, FAK Tyr-576/577, p85 PI3K Tyr-458, Akt Thr-308, Akt Ser-473 and mTOR Ser-2448 but prevented phosphorylation of S6K Thr-389, S6K Thr-421/Ser424, S6RP Ser-240 and ERK Thr202/Tyr204 triggered by HLA II crosslinking with class II antibody in Ad-CIITA infected EC and TNFα/IFNγ pretreated EC (Fig. 8 A, B). These results indicate that ERK is required for full mTORC1 activation, as phosphorylation of S6K and S6RP was reduced when ERK was inactivated.
Fig. 8. ERK1/2 regulates HLA class II antibody-induced activation of intracellular signal transduction networks, cell proliferation, and migration.
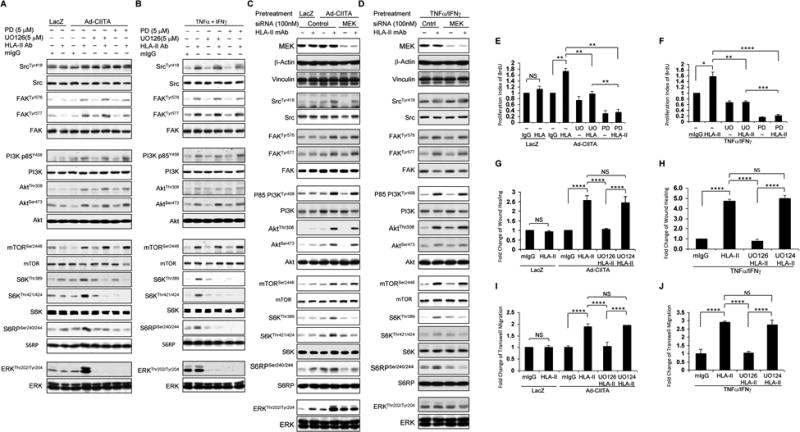
EC were A, C, E, G, I infected with Ad-LacZ or Ad-CIITA or B, D, F, H, J pretreated with TNFα/IFNγ for 48 h. A, B Starved cells were pretreated with 5 μM UO126, or E, F, G, H, I, Jwith 1.0 μM UO126, or A, B, with 5.0 μM PD0325901, or E, F with 1.0 μM PD0325901, or G, H, I, J with 1.0 μM UO124 for 60 min, or C, D, Starved EC were transfected with 100 nM of MEK, or control siRNA for 48 hours. A, B, C, D Pretreated ECwere stimulated with0.1 μg/ml of HLA class II antibody for 15 min. Treatment of EC with mIgG serves as a negative control. Proteins in the pre-cleared cell lysates were separated by 6∼15% SDS-PAGE followed by immunoblotting with anti-phospho-Src Tyr418, FAK Tyr576, FAK Tyr577, p85 PI3K Tyr458, Akt Thr308, Akt Ser473, mTOR Ser2448, S6K Thr389, S6K Thr421/424, S6RP Ser240/244, or ERK Thr202/Tyr204. The membranes were re-probed with anti-Src, FAK, PI3K, Akt, mTOR, S6K, S6RP, ERK, or β-actin antibodies to confirm equal loading of proteins. E, F Cells were stimulated with 1.0 μg/ml HLA class II antibody for 48 h, incorporated with BrdU for 2 h, and harvested. EC proliferation was measured by flow cytometry. DNA synthesis S phase was gated, and proliferation index (PI) is presented as fold increase in the percentage of cells positive for BrdU normalized to negative control. G, HCells were pretreated with 10 μg/ml of mitomycin C for 2 h to inhibit cell proliferation before being assayed for their ability to migrate. A scratch wound was created with a sterile 200-μl pipette tip. Wounded cells were stimulated with 1.0 μg/ml of anti-HLA class II antibody for 16 h. The cell number between two initiated front edges was counted; migration rate was analyzed by calculating the cell number between two initiated front edges of HLA class II antibody-stimulated EC divided by the cell number between two initiated front edges of negative control EC. I, J Cell migration was measured in a transwell insert system. EC infected with Ad-LacZ or Ad-CIITA or pretreated with TNFα/IFNγwere seeded to the upper chamber of insert, pretreated with UO126 or UO124 for 60 min, and stimulated with 1.0 μg/ml HLA class II antibody for 16 h at 37°C. After incubation, the cells on the upper surface of the insert membrane were removed with a cotton swab, the migrated cells on the bottom of the insert membrane were fixed with methanol and stained with crystal violet, three middle fields per insert were photographed with 10 × objective lens, and migrated cells were counted. The bar graphs show the mean ± SEM fold change of C, D. proliferation index, E, F, G, H migrated cells. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 were analyzed by one way ANOVA with Fisher's LSD. Data represent at least three independent experiments.
In order to confirm our observation using pharmacological inhibitors of MEK, we next determined the effect of MEK siRNA on HLA-II antibody-induced ERK activation and its targets. For this, we determined the efficiency of MEK siRNA on MEK expression first. As shown in Fig 8 C, D, MEK expression was markedly reduced by siRNA knockdown whereas transfection of EC with non-targeted siRNA had no effect on MEK protein expression (Fig 8 C, lane 1-4 and Fig 8 D, lane 1 and 2). To verify MEK siRNA specificity, we determined the expression of several cytoskeletal proteins in the same cell lysates of EC transfected with MEK siRNA. Transfection of EC with MEK siRNA or control siRNA did not affect expression of vinculin and β-actin (Fig 8 C, D). Transfection of starved Ad-CIITA infected and TNFα/IFNγ pretreated EC with MEK siRNA had no effect on HLA class II antibody-induced phosphorylation of Src Tyr-418, FAK Tyr-576/577, p85 PI3K Tyr-458, Akt Thr-308, Akt Ser-473 and mTOR Ser-2448 but abrogated phosphorylation of S6K Thr-389, S6K Thr-421/Ser424, S6RP Ser-240/244 and ERK at Thr-202/Tyr-204 in both Ad-CIITA infected and TNFα/IFNγ pretreated EC (Fig. 8 C, D).
Next we determined the impact of ERK inhibition on EC proliferation and migration. Pretreatment of starved Ad-CIITA infected and TNFα/IFNγ pretreated EC with UO126 or PD0325901 inhibited HLA II antibody-stimulated EC proliferation (Fig 8 E, F) and EC migration, as determined by wound healing (Fig 8 G, H) or transwell migration assays (Fig 8 I, J). In contrast, UO124, an inactive analog of UO126, had no effect on HLA class II antibody-induced cell migration (Fig 8 G-J). These data indicated that the ERK signaling contributes to HLA II antibody-stimulated mTORC1 activation, proliferation and migration of EC stimulated by antibody-mediated ligation of HAL II.
Treatment with MEK inhibitor abrogates over-activation of ERK mediated by suppression of mTORC2 in response to HLA II antibody in EC
Since long-term treatment with rapamycin or everolimus, exposure to PP242, or siRNA knockdown of Rictor inhibited Akt phosphorylation on Ser-473 but induced concomitant over-activation of ERK, we proposed that mTORC2 mediates negative feedback on ERK activation. To elucidate this mechanism, we next determined whether EC exposure to a MEK inhibitor prevents over-activation of ERK in response to mTORC2 inhibition. Treatment with UO126 abrogated hyper-phosphorylation of ERK induced by rapamycin 24h treatment (Fig 9 A, B) but did not prevent phosphorylation of Akt Ser473.
Fig. 9. Treatment of EC with MEK inhibitor abrogates mTORC2-mediated over-activation of ERK mediated in response to HLA II antibody.
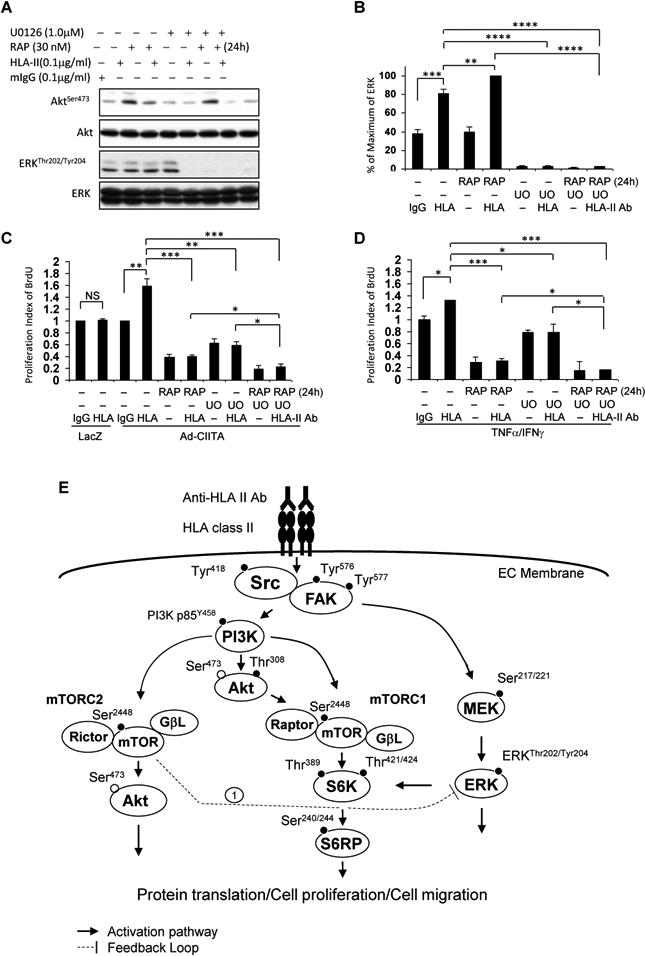
EC were C infected with Ad-LacZ or Ad-CIITA or A, B, D pretreated with TNFα/IFNγ for 48 h. Starved cells were pretreated with 30 nM rapamycin for 24 h and/or 1.0 μM UO126 for 60 min. A, B. Cells were stimulated with 0.1 μg/ml HLA class II antibody for 15 min. Treatment of EC with mIgG serves as negative control. Proteins in the precleared cell lysates were separated by 6∼15% SDS-PAGE followed by immunoblotting withanti-phospho-Akt Ser473, or ERK1/2 Thr202/Tyr204 antibodies. The membranes were re-probed with anti-Akt or ERK total antibodies to confirm equal loading of proteins. B Phosphorylated protein bands shown in A were quantified by densitometry scan analysis and results are expressed as the mean ± SEM percentage of maximal increase in phosphorylation above control values. **p<0.01, ***p<0.001, and ****p<0.0001 were analyzed by one way ANOVA with Fisher's LSD. C, D Cells were stimulated with 1.0 μg/ml HLA class II antibody for 48 h, incorporated with BrdU for 2 h, and harvested. EC proliferation was measured by flow cytometry. DNA synthesis S phase was gated, and proliferation index is presented as fold increase in the percentage of cells positive for BrdU normalized to negative control. The bar graphs show the mean ± SEM fold change of proliferation index. *p<0.05, **p<0.01, and ***p<0.001 were analyzed by one way ANOVA with Fisher's LSD. Data represent at least three independent experiments. E. Scheme representing HLA class II antibody-triggered activation of intracellular signal networks in EC leading to cell proliferation and migration. EC were infected with Ad-CIITA or pretreated with TNFα/IFNγ for 48 h to up-regulate HLA class II molecule expression. Ligation of HLA II molecules with antibody stimulates phosphorylation of protein tyrosine kinases, including Src Tyr418, FAK Tyr576 and Tyr577. Dissecting the upstream/downstream interactions using siRNA and pharmacological inhibitors, weshow that Src is the earliest protein kinase in the HLA II antibody-induced signaling cascade. Activated FAK/Src signaling complex mediates phosphorylation of p85 PI3K Tyr458, Akt Thr308, and Akt Ser473. The PI3K/Akt cascade stimulates phosphorylation of mTOR and assembly of mTORC1 and mTORC2. mTORC1 induces phosphorylation of S6K Thr389 and Thr421/Ser424. mTORC1/S6K signal axis phosphorylates S6RP at Ser240/244. mTORC2 mediates phosphorylation of Akt at Ser473. In parallel to the PI3K/Akt/mTOR signal pathways, activated FAK/Src complex also stimulates activation of the MEK/ERK pathway. These signal networks, solid lines, regulate HLA antibody-stimulated cell proliferation and migration. Long-term treatment with rapamycin, everolimus, anactive-site inhibitor or knockdown Rictor with siRNA mediates mTORC2 negatively feedbacks to enhance phosphorylation of ERK (negative feedback loop 1). Solid line with arrow presents stimulatory signal transduction pathways. The dotted line presents negative feedback loop.
To further determine whether the over-activation of the MEK/ERK pathway counterbalances the EC growth-suppressing effect of the mTORC2 inhibitor, we examined the proliferation (BrdU incorporation assay) of Ad-CIITA infected EC and TNFα/IFNγ pretreated EC treated with rapamycin and without or with UO126. Pretreatment with either rapamycin or UO126 inhibited HLA class II antibody-stimulated cell proliferation, but the combination of rapamycin and UO126 produced a further inhibitory effect on EC proliferation (Fig 9 C, D). These results indicate that co-targeting mTORC2 and MEK/ERK pathways mediates a profound inhibition of EC proliferation. Similar results were obtained when PD0325901 was used to inhibit MEK instead of UO126. Pretreatment of EC with PD0325901 also abolished over-activation of ERK in response to rapamycin 24h treatment, by which inhibits mTORC2. Similar results were seen in proliferation in Ad-CIITA infected EC and TNFα/IFNγ pretreated EC (data not shown). Our results show that over-activation of ERK induced by suppression of mTORC2 can be abrogated by administration of a MEK inhibitor in EC, a conclusion with potential translational implications.
Discussion
Endothelial cells line the inner surface of blood vessels and serve as the inter face between circulating blood and surrounding tissue. After solid organ transplantation, the endothelium in the transplanted organ forms a vast network that dynamically regulates vascular barrier functions. Physiologically, human artery endothelial cells do not express HLA class II antigens. During the process of transplantation, surgical trauma and ischemia/reperfusion injury induce an inflammatory process leading to TNFα, IL1β, and IFNγ production by EC (44). MHC-II promoters share a common set of cis-acting elements including the W/S, X1, X2, and Y boxes (S-X-Y module), which provides the appropriate interaction surface for the recruitment of CIITA. Upon IFNγ induction, CIITA is produced and induces or enhances the association of transcription factors with the promoter (45). Subsequently these inflammatory cytokines stimulate HLA II antigen expression on EC.
Due to the technical limitations of studying HLA II-mediated functions in cultured EC, studies of the effects of HLA II antibodies on EC have been limited and very little is known about the signaling cascades triggered by these antibodies in these cells. Recently, Taflin et al. described a model in which HLA-DR expression was induced in cultured EC through lentiviral delivery, and was sufficient to drive activation of memory CD4+ T cells (58). However, the forced expression of classical HLA II molecules without accessory HLA-DM, HLA-DO, and invariant chain expression, which are necessary for proper antigen presentation, may yield artificial CLIP-loaded HLA-DR at the cell surface. Moreover, this model did not permit investigation of other classical HLA II molecules, HLA-DQ and HLA-DP, which are clinically relevant in transplantation. Herein, we describe a new model of HLA II expression in human primary EC utilizing adenoviral vector-mediated expression of class II transactivating factor (CIITA), a transcription factor which promotes expression of both classical and non-classical HLA II molecules, as well as the invariant chain and cathepsin S (59). Adenovirus was chosen for higher transfection efficiency in primary endothelial cells. Importantly, this approach drives expression of endogenous alleles of HLA-DR, DQ, and DP. In parallel, we utilized endothelial cells treated with IFNγ and TNFα, to mimic inflammatory conditions under which EC might physiologically and pathologically upregulate HLA II antigens. Similar to the Ad-CIITA model, this parallel treatment of EC with cytokines also induced CIITA production and expression of HLA-DR, DQ, and DP on EC. Using these two model systems together with multiple chemical inhibitors and siRNA-mediated knockdown of signal transducers, we explored in depth the organization of the signaling network elicited by antibody-mediated ligation of class II molecules in primary EC that leads to proliferation and migration.
Transplant vasculopathy is caused by proliferation of both endothelial cells and smooth muscle cells, as evident from proliferating cell nuclear antigen staining in chronic lesions (60, 61). Angiogenic changes resulting from endothelial cell activation, proliferation, and migration, characterized by new microvessel formation under the hyperplastic neointima, occur in cardiac transplant recipients with TV (10). In this study, we found that ligation of HLA class II molecules with antibody in Ad-CIITA infected or TNFα/IFNγ pretreated EC stimulates the proliferation and migration of these cells, as demonstrated using different assays. Activated endothelial cells may also stimulate smooth muscle cells through paracrine effects promoting fibroproliferation, such as through production of VEGF(62).
Studies using pharmacological inhibitors and siRNAs to knockdown the expression of signal transducers demonstrate that the protein kinases Src/FAK, PI3K/Akt, mTOR, and ERK play critical roles in mediating class II antibody-stimulated EC proliferation and migration. Pretreatment of EC with either structurally unrelated Src inhibitors or siRNA-mediated knockdown of c-Src and FAK blocked HLA II antibody-stimulated activation of all signaling molecules detected in this study, including PI3K, Akt, mTORC1, mTORC2 and ERK. FAK and Src are non-receptor protein tyrosine kinases that function cooperatively. Indeed, many stimuli induce FAK auto-phosphorylation on Tyr-397, creating a high affinity binding site for the Src homology (SH)2 domain of Src. Upon binding to FAK, Src is autophosphorylated at Tyr-418 in its catalytic domain, thereby stabilizing its active conformation. Then, Src phosphorylates Tyr-576 and Tyr-577 within the activation loop of the kinase domain of FAK promoting maximal activation of FAK catalytic activity. In line with this model, knockdown of FAK inhibited HLA class II antibody-induced phosphorylation of Src at Tyr-418 and chemical inhibitors of Src family kinases abrogated the increase in the phosphorylation of FAK on Tyr-576 and Tyr-577. We therefore propose that FAK/Src activation is one of the earliest events leading to downstream signaling, migration and proliferation triggered by antibody-mediated ligation of HLA class II molecules in primary human aortic endothelial cells.
PI3K catalyzes the formation of PIP3 a second messenger that coordinates the localization and activation of several downstream signaling molecules, including Akt (28, 52). Pretreatment of starved Ad-CIITA infected or TNFα/IFNγ pretreated EC with either the dual PI3K/mTOR inhibitor LY294002 or A66, a selective antagonist of the p110α subunit of the PI3K, suppressed HLA class II antibody-stimulated phosphorylation of p85 PI3K Tyr-458, Akt Thr-308 and Akt Ser-473, and abolished phosphorylation of mTOR Ser-2448, S6K Thr-389, and S6RP Ser-240/244. Because in some cell types PI3K activation leads to Raf-mediated MEK/ERK(63), it is of interest that A66 di not prevented HLA II antibody-induced phosphorylation of ERK Thr202/Tyr204, indicating that PI3K does not contribute to ERK activation in EC in context of HLA II signaling. Interestingly, dual PI3K/mTOR inhibitor LY294002 caused ERK hyperphosphorylation, which suggests that there is negative feedback via mTORC2. Importantly, pretreatment with A66 or LY294002 did not affect HLA class II antibody-induced FAK/Src activation, reinforcing our conclusion that Src/FAK is upstream of PI3K/Akt in EC activated by ligation of HLA class II (Fig. 9 E). It has been reported that overactivation of ERK mediated by inhibition of mTORC2 was shown to involve Akt-mediated inhibitory phosphorylation of c-Raf (64). To test this hypothesis, we determined the effect of Akt inhibitors on HLA-II antibody-stimulated phosphorylation ofc-Raf-1 at Ser259. Inhibition of mTORC2 (by 24h exposure to rapamycin) inhibited AKT phosphorylation at Ser-473 completely but did not prevent HLA II-induced AKT phosphorylation at Thr-308, which is the key site in the T loop for AKT activation. Furthermore, treatment of EC with allosteric (MK-2206) or active-site (GDC-0068) inhibitors of Akt did not affect HLA-II antibody-stimulated phosphorylation of c-Raf-1 Ser259 or ERK Thr202/Tyr204 (Fig. 5E, F). Our data suggest the mechanism by which mTORC2 inhibition results in ERK overactivation is predominantly Akt-independent.
The PI3K/Akt/mTOR network is a key signaling pathway in the regulation of cell metabolism, migration, survival, and proliferation (49) that plays a critical role in HLA class I antibody-induced EC activation in chronic rejection (27, 31, 33, 34) but its function and regulation in the context of HLA II signaling was virtually unknown. mTOR functions as a catalytic subunit in two structurally distinct multiprotein complexes, mTORC1 and mTORC2(65, 66). mTORC1, a complex of mTOR characterized by the substrate binding subunit, phosphorylates and controls at leasttwo regulators of protein synthesis, the 40S ribosomal protein subunit S6kinase (S6K) and the inhibitor of protein synthesis 4E-binding protein 1, referred as 4EBP1(67-70). mTORC1 is acutely inhibited by rapamycin and everolimus whereas mTORC2, characterized by Rictor is not inhibited by short exposure to this agent (71, 72). Here, we demonstrate that antibody-mediated ligation of HLA II molecules expressed in Ad-CIITA infected or TNFα/IFNγ pretreated EC induces a marked increase in mTORC1 and mTORC2, as revealed by the increase in the phosphorylation of S6K Thr-389 and Akt Ser-473, respectively. Using pharmacological and genetic approaches, we demonstrate that mTOR plays a key role in proliferation and migration in EC stimulated by antibody-mediated ligation of HLA II molecules. Subsequently, we explored in depth the relationship between mTOR complexes (mTORC1 and mTORC2) and their upstream elements (FAK/Src, PI3K and ERK) and downstream targets, including S6K, S6RP and Akt, following ligation of HLA class II molecules. The upstream and downstream elements in mTOR signaling in EC stimulated by antibody-mediated ligation of HLA II molecules identified in this study are summarized in Fig 9 E.
Mounting evidence indicates that the mTOR complexes not only stimulate growth-promoting signaling but also mediate negative feedback loops that restrain signaling through upstream or parallel pathways in a cell-context specific manner(63). Suppression of these feedback loops by inhibitors of mTOR causes compensatory over-activation of signaling nodes, including ERK, that potentially oppose the anti-proliferative effects of the mTOR inhibitors thus leading to drug resistance. The elucidation of feedback loops that regulate the outputs of signaling networks has emerged as an area of fundamental importance for the rationale design of effective inhibitors or combination of inhibitors to prevent transplant rejection. In the present study, we produced several lines of evidence demonstrating that suppression of mTORC2 leads to over-activation of ERK in response to antibody-mediated ligation of class II molecules in EC. Specifically, long-term term exposure to either rapamycin or everolimus and treatment with the mTOR active-site inhibitor PP242 or the dual PI3K/mTOR inhibitor LY294002, all of which suppress both mTORC1 and mTORC2 enhanced ERK activation. In agreement with this conclusion, the siRNA-mediated knockdown of Rictor, an essential component of the mTORC2 complex, also promoted ERK over-activation. We verified that all these interventions blocked the phosphorylation of Akt at Ser-473, a site targeted by mTORC2. In contrast, short-term treatment with either rapamycin or everolimus or knockdown of Raptor, which inhibits mTORC1 but not mTORC2, did not have any detectable effect on ERK activation in response to antibody-mediated HLA II engagement in EC. It is noteworthy that long-term treatment with rapamycin, everolimus or knockdown Rictor with siRNA inhibited HLA class I antibody-induced phosphorylation of ERK(33, 35), thus implying an important difference in the feedback loops that fine-tune endothelial cell signaling through HLA I and HLA II molecules. Collectively, these findings identify a novel feedback loop in EC cells challenged with antibody directed against HLA II by which ERK activation is attenuated through a mTORC2-mediated pathway but is unleashed by multiple treatments that interfere with mTORC2 activity (Fig 9 E, feedback loop 1).
In antigen presenting cells, engagement of MHC II molecules by the T cell receptor (or mimicked by antibodies) induces bidirectional intracellular signaling. For example, in addition to activation of ERK, antibodies to HLA II increase p38 MAPK phosphorylation in monocytes leading to cytokine production (73). ERK and JNK are also strongly phosphorylated in B cells after treatment with anti-HLA II antibodies (74), as are the NFAT and c-Fos pathways(75). Dendritic cell apoptosis after ligation of HLA-DR is regulated by PKC(76), and super-antigen stimulation of MHC II in this cell type also activates NFκB/MyD88(77). Whether these pathways are also activated in allograft vascular endothelial cells in the presence of anti-donor HLA II antibodies remains to be determined.
Our studies also demonstrate that exposure of Ad-CIITA infected or TNFα/IFNγ pretreated EC to MEK inhibitors opposed the over-activation of ERK induced by suppression of mTORC2 inhibition. These results show that the enhanced ERK activation induced by long-term treatment with rapamycin or everolimus can be prevented by co-treatment with MEK inhibitors. Further studies are required to determine the effect of class II signaling in an in vivo allograft model. However, given the increasing importance of everolimus in combating transplant rejection, the findings presented in this study suggest a novel therapeutic strategy, co-treatment with PI3K/mTORC2 and MEK inhibitors in the prevention or treatment of HLA II antibody-mediated TV.
Supplementary Material
Acknowledgments
The authors hereby express their thanks for the cooperation of One Legacy and all of the organ and tissue donors and their families for giving the gift of life and the gift of knowledge, by their generous donation.
Abbreviations used in this paper
- Ab
Antibody
- AMR
antibody-mediated rejection
- ANOVA
analysis of variance
- EC
endothelial cells
- ERK
extracellular signal-regulated kinase
- FAK
focal adhesion kinase
- EC
human aortic EC
- HLA
human leukocyte antigen
- LSD
least significant difference
- mTOR
mammalian target of rapamycin
- PI3K
phosphatidylinositol 3-kinase
- S6K
p70 ribosomal S6 kinase
- S6RP
S6 ribosomal protein
- TV
transplant vasculopathy
Footnotes
This work was supported by the National Institute of Allergy and Infectious Diseases Grant RO1 AI 042819 and NIH U01 AI-124319-01, and the Novartis Investigator Initiated Research Grant (to EFR). GrantsR01DK100405, P30DK41301 and P01CA163200 and Department of Veterans Affair Merit Award 1I01BX001473support E. Rozengurt.
Disclosures: The authors have no financial conflict of interest.
References
- 1.O'Leary JG, Samaniego M, Barrio MC, Potena L, Zeevi A, Djamali A, Cozzi E. The Influence of Immunosuppressive Agents on the Risk of De Novo Donor-Specific HLA Antibody Production in Solid Organ Transplant Recipients. Transplantation. 2016;100:39–53. doi: 10.1097/TP.0000000000000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Terasaki PI, Ozawa M. Predicting kidney graft failure by HLA antibodies: a prospective trial. Am J Transplant. 2004;4:438–443. doi: 10.1111/j.1600-6143.2004.00360.x. [DOI] [PubMed] [Google Scholar]
- 3.Valenzuela NM, Reed EF. Antibody-mediated rejection across solid organ transplants: manifestations, mechanisms, and therapies. J Clin Invest. 2017;127:2492–2504. doi: 10.1172/JCI90597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chih S, Chruscinski A, Ross HJ, Tinckam K, Butany J, Rao V. Antibody-mediated rejection: an evolving entity in heart transplantation. Journal of transplantation. 2012;2012:210210. doi: 10.1155/2012/210210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cuadrado A, San Segundo D, Lopez-Hoyos M, Crespo J, Fabrega E. Clinical significance of donor-specific human leukocyte antigen antibodies in liver transplantation. World journal of gastroenterology. 2015;21:11016–11026. doi: 10.3748/wjg.v21.i39.11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wiebe C, Gibson IW, Blydt-Hansen TD, Karpinski M, Ho J, Storsley LJ, Goldberg A, Birk PE, Rush DN, Nickerson PW. Evolution and clinical pathologic correlations of de novo donor-specific HLA antibody post kidney transplant. Am J Transplant. 2012;12:1157–1167. doi: 10.1111/j.1600-6143.2012.04013.x. [DOI] [PubMed] [Google Scholar]
- 7.Lund LH, Edwards LB, Kucheryavaya AY, Benden C, Christie JD, Dipchand AI, Dobbels F, Goldfarb SB, Levvey BJ, Meiser B, Yusen RD, Stehlik J, H. International Society of. Lung T. The registry of the International Society for Heart and Lung Transplantation: thirty-first official adult heart transplant report--2014; focus theme: retransplantation. J Heart Lung Transplant. 2014;33:996–1008. doi: 10.1016/j.healun.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 8.Viglietti D, Loupy A, Vernerey D, Bentlejewski C, Gosset C, Aubert O, Duong van Huyen JP, Jouven X, Legendre C, Glotz D, Zeevi A, Lefaucheur C. Value of Donor-Specific Anti-HLA Antibody Monitoring and Characterization for Risk Stratification of Kidney Allograft Loss. J Am Soc Nephrol. 2017;28:702–715. doi: 10.1681/ASN.2016030368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiebe C, Gibson IW, Blydt-Hansen TD, Pochinco D, Birk PE, Ho J, Karpinski M, Goldberg A, Storsley L, Rush DN, Nickerson PW. Rates and determinants of progression to graft failure in kidney allograft recipients with de novo donor-specific antibody. Am J Transplant. 2015;15:2921–2930. doi: 10.1111/ajt.13347. [DOI] [PubMed] [Google Scholar]
- 10.Atkinson C, Southwood M, Pitman R, Phillpotts C, Wallwork J, Goddard M. Angiogenesis occurs within the intimal proliferation that characterizes transplant coronary artery vasculopathy. J Heart Lung Transplant. 2005;24:551–558. doi: 10.1016/j.healun.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 11.Demetris AJ, Bellamy C, Hubscher SG, O'Leary J, Randhawa PS, Feng S, Neil D, Colvin RB, McCaughan G, Fung JJ, Del Bello A, Reinholt FP, Haga H, Adeyi O, Czaja AJ, Schiano T, Fiel MI, Smith ML, Sebagh M, Tanigawa RY, Yilmaz F, Alexander G, Baiocchi L, Balasubramanian M, Batal I, Bhan AK, Bucuvalas J, Cerski CTS, Charlotte F, de Vera ME, ElMonayeri M, Fontes P, Furth EE, Gouw ASH, Hafezi-Bakhtiari S, Hart J, Honsova E, Ismail W, Itoh T, Jhala NC, Khettry U, Klintmalm GB, Knechtle S, Koshiba T, Kozlowski T, Lassman CR, Lerut J, Levitsky J, Licini L, Liotta R, Mazariegos G, Minervini MI, Misdraji J, Mohanakumar T, Molne J, Nasser I, Neuberger J, O'Neil M, Pappo O, Petrovic L, Ruiz P, Sagol O, Sanchez Fueyo A, Sasatomi E, Shaked A, Shiller M, Shimizu T, Sis B, Sonzogni A, Stevenson HL, Thung SN, Tisone G, Tsamandas AC, Wernerson A, Wu T, Zeevi A, Zen Y. 2016 Comprehensive Update of the Banff Working Group on Liver Allograft Pathology: Introduction of Antibody-Mediated Rejection. Am J Transplant. 2016;16:2816–2835. doi: 10.1111/ajt.13909. [DOI] [PubMed] [Google Scholar]
- 12.DerHovanessian A, Todd JL, Zhang A, Li N, Mayalall A, Finlen Copeland CA, Shino M, Pavlisko EN, Wallace WD, Gregson A, Ross DJ, Saggar R, Lynch JP, 3rd, Belperio J, Snyder LD, Palmer SM, Weigt SS. Validation and Refinement of Chronic Lung Allograft Dysfunction Phenotypes in Bilateral and Single Lung Recipients. Annals of the American Thoracic Society. 2016;13:627–635. doi: 10.1513/AnnalsATS.201510-719OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Libby P, Zhao DX. Allograft arteriosclerosis and immune-driven angiogenesis. Circulation. 2003;107:1237–1239. doi: 10.1161/01.cir.0000059744.64373.08. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka H, Sukhova GK, Swanson SJ, Cybulsky MI, Schoen FJ, Libby P. Endothelial and smooth muscle cells express leukocyte adhesion molecules heterogeneously during acute rejection of rabbit cardiac allografts. The American journal of pathology. 1994;144:938–951. [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao XM, Hu Y, Miller GG, Mitchell RN, Libby P. Association of thrombospondin-1 and cardiac allograft vasculopathy in human cardiac allografts. Circulation. 2001;103:525–531. doi: 10.1161/01.cir.103.4.525. [DOI] [PubMed] [Google Scholar]
- 16.Bruneau S, Woda CB, Daly KP, Boneschansker L, Jain NG, Kochupurakkal N, Contreras AG, Seto T, Briscoe DM. Key Features of the Intragraft Microenvironment that Determine Long-Term Survival Following Transplantation. Frontiers in immunology. 2012;3:54. doi: 10.3389/fimmu.2012.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valenzuela NM, Reed EF. Antibodies to HLA Molecules Mimic Agonistic Stimulation to Trigger Vascular Cell Changes and Induce Allograft Injury. Current transplantation reports. 2015;2:222–232. doi: 10.1007/s40472-015-0065-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loupy A, Lefaucheur C, Vernerey D, Prugger C, Duong van Huyen JP, Mooney N, Suberbielle C, Fremeaux-Bacchi V, Mejean A, Desgrandchamps F, Anglicheau D, Nochy D, Charron D, Empana JP, Delahousse M, Legendre C, Glotz D, Hill GS, Zeevi A, Jouven X. Complement-binding anti-HLA antibodies and kidney-allograft survival. N Engl J Med. 2013;369:1215–1226. doi: 10.1056/NEJMoa1302506. [DOI] [PubMed] [Google Scholar]
- 19.Sis B, Campbell PM, Mueller T, Hunter C, Cockfield SM, Cruz J, Meng C, Wishart D, Solez K, Halloran PF. Transplant glomerulopathy, late antibody-mediated rejection and the ABCD tetrad in kidney allograft biopsies for cause. Am J Transplant. 2007;7:1743–1752. doi: 10.1111/j.1600-6143.2007.01836.x. [DOI] [PubMed] [Google Scholar]
- 20.Gloor JM, Sethi S, Stegall MD, Park WD, Moore SB, DeGoey S, Griffin MD, Larson TS, Cosio FG. Transplant glomerulopathy: subclinical incidence and association with alloantibody. Am J Transplant. 2007;7:2124–2132. doi: 10.1111/j.1600-6143.2007.01895.x. [DOI] [PubMed] [Google Scholar]
- 21.Haas M. Pathology of C4d-negative antibody-mediated rejection in renal allografts. Current opinion in organ transplantation. 2013;18:319–326. doi: 10.1097/MOT.0b013e32835d4daf. [DOI] [PubMed] [Google Scholar]
- 22.Loupy A, Cazes A, Guillemain R, Amrein C, Hedjoudje A, Tible M, Pezzella V, Fabiani JN, Suberbielle C, Nochy D, Hill GS, Empana JP, Jouven X, Bruneval P, Duong Van Huyen JP. Very late heart transplant rejection is associated with microvascular injury, complement deposition and progression to cardiac allograft vasculopathy. Am J Transplant. 2011;11:1478–1487. doi: 10.1111/j.1600-6143.2011.03563.x. [DOI] [PubMed] [Google Scholar]
- 23.Sis B, Jhangri GS, Bunnag S, Allanach K, Kaplan B, Halloran PF. Endothelial gene expression in kidney transplants with alloantibody indicates antibody-mediated damage despite lack of C4d staining. Am J Transplant. 2009;9:2312–2323. doi: 10.1111/j.1600-6143.2009.02761.x. [DOI] [PubMed] [Google Scholar]
- 24.Thomas KA, Valenzuela NM, Reed EF. The perfect storm: HLA antibodies, complement, FcgammaRs, and endothelium in transplant rejection. Trends in molecular medicine. 2015;21:319–329. doi: 10.1016/j.molmed.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reed EF. Mechanisms of action and effects of antibodies on the cells of the allograft. Human immunology. 2012;73:1211–1212. doi: 10.1016/j.humimm.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 26.Valenzuela NM, McNamara JT, Reed EF. Antibody-mediated graft injury: complement-dependent and complement-independent mechanisms. Current opinion in organ transplantation. 2014;19:33–40. doi: 10.1097/MOT.0000000000000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin YP, Fishbein MC, Said JW, Jindra PT, Rajalingam R, Rozengurt E, Reed EF. Anti-HLA class I antibody-mediated activation of the PI3K/Akt signaling pathway and induction of Bcl-2 and Bcl-xL expression in endothelial cells. Human immunology. 2004;65:291–302. doi: 10.1016/j.humimm.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 28.Jin YP, Korin Y, Zhang X, Jindra PT, Rozengurt E, Reed FE. RNA Interference Elucidates the Role of Focal Adhesion Kinase in HLA Class I-Mediated Focal Adhesion Complex Formation and Proliferation in Human Endothelial Cells. J Immunol. 2007;178:7911–7922. doi: 10.4049/jimmunol.178.12.7911. [DOI] [PubMed] [Google Scholar]
- 29.Jin YP, Singh RP, Du ZY, Rajasekaran AK, Rozengurt E, Reed EF. Ligation of HLA class I molecules on endothelial cells induces phosphorylation of Src, paxillin and focal adhesion kinase in an actin dependent manner. J Immunol. 2002;168:5415–5423. doi: 10.4049/jimmunol.168.11.5415. [DOI] [PubMed] [Google Scholar]
- 30.Coupel S, Leboeuf F, Boulday G, Soulillou JP, Charreau B. RhoA activation mediates phosphatidylinositol 3-kinase-dependent proliferation of human vascular endothelial cells: an alloimmune mechanism of chronic allograft nephropathy. J Am Soc Nephrol. 2004;15:2429–2439. doi: 10.1097/01.ASN.0000138237.42675.45. [DOI] [PubMed] [Google Scholar]
- 31.Jindra PT, Jin YP, Rozengurt E, Reed EF. HLA class I antibody-mediated endothelial cell proliferation via the mTOR pathway. J Immunol. 2008;180:2357–2366. doi: 10.4049/jimmunol.180.4.2357. [DOI] [PubMed] [Google Scholar]
- 32.Bian H, Reed EF. Alloantibody-Mediated Class I Signal Transduction in Endothelial Cells and Smooth Muscle Cells: Enhancement by IFN-gamma and TNF-alpha. J Immunol. 1999;163:1010–1018. [PubMed] [Google Scholar]
- 33.Jin YP, Valenzuela NM, Ziegler ME, Rozengurt E, Reed EF. Everolimus Inhibits Anti-HLA I Antibody-Mediated Endothelial Cell Signaling, Migration and Proliferation More Potently Than Sirolimus. Am J Transplant. 2014;14:806–819. doi: 10.1111/ajt.12669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lepin EJ, Zhang Q, Zhang X, Jindra PT, Hong LS, Ayele P, Peralta MV, Gjertson DW, Kobashigawa JA, Wallace WD, Fishbein MC, Reed EF. Phosphorylated S6 ribosomal protein: a novel biomarker of antibody-mediated rejection in heart allografts. Am J Transplant. 2006;6:1560–1571. doi: 10.1111/j.1600-6143.2006.01355.x. [DOI] [PubMed] [Google Scholar]
- 35.Jindra PT, Jin YP, Jacamo R, Rozengurt E, Reed EF. MHC class I and integrin ligation induce ERK activation via an mTORC2-dependent pathway. Biochem Biophys Res Commun. 2008;369:781–787. doi: 10.1016/j.bbrc.2008.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hassan GS, Mourad W. An unexpected role for MHC class II. Nature immunology. 2011;12:375–376. doi: 10.1038/ni.2023. [DOI] [PubMed] [Google Scholar]
- 37.Liu X, Zhan Z, Li D, Xu L, Ma F, Zhang P, Yao H, Cao X. Intracellular MHC class II molecules promote TLR-triggered innate immune responses by maintaining activation of the kinase Btk. Nature immunology. 2011;12:416–424. doi: 10.1038/ni.2015. [DOI] [PubMed] [Google Scholar]
- 38.Al-Daccak R, Mooney N, Charron D. MHC class II signaling in antigen-presenting cells. Curr Opin Immunol. 2004;16:108–113. doi: 10.1016/j.coi.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 39.Clerkin KJ, Farr MA, Restaino SW, Zorn E, Latif F, Vasilescu ER, Marboe CC, Colombo PC, Mancini DM. Donor-specific anti-HLA antibodies with antibody-mediated rejection and long-term outcomes following heart transplantation. J Heart Lung Transplant. 2017;36:540–545. doi: 10.1016/j.healun.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeNicola MM, Weigt SS, Belperio JA, Reed EF, Ross DJ, Wallace WD. Pathologic findings in lung allografts with anti-HLA antibodies. J Heart Lung Transplant. 2013;32:326–332. doi: 10.1016/j.healun.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Leary JG, Demetris AJ, Friedman LS, Gebel HM, Halloran PF, Kirk AD, Knechtle SJ, McDiarmid SV, Shaked A, Terasaki PI, Tinckam KJ, Tomlanovich SJ, Wood KJ, Woodle ES, Zachary AA, Klintmalm GB. The role of donor-specific HLA alloantibodies in liver transplantation. Am J Transplant. 2014;14:779–787. doi: 10.1111/ajt.12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wiebe C, Pochinco D, Blydt-Hansen TD, Ho J, Birk PE, Karpinski M, Goldberg A, Storsley LJ, Gibson IW, Rush DN, Nickerson PW. Class II HLA epitope matching-A strategy to minimize de novo donor-specific antibody development and improve outcomes. Am J Transplant. 2013;13:3114–3122. doi: 10.1111/ajt.12478. [DOI] [PubMed] [Google Scholar]
- 43.Wozniak LJ, Hickey MJ, Venick RS, Vargas JH, Farmer DG, Busuttil RW, McDiarmid SV, Reed EF. Donor-specific HLA Antibodies Are Associated With Late Allograft Dysfunction After Pediatric Liver Transplantation. Transplantation. 2015;99:1416–1422. doi: 10.1097/TP.0000000000000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pascher A, Klupp J. Biologics in the treatment of transplant rejection and ischemia/reperfusion injury: new applications for TNFalpha inhibitors? BioDrugs: clinical immunotherapeutics, biopharmaceuticals and gene therapy. 2005;19:211–231. doi: 10.2165/00063030-200519040-00002. [DOI] [PubMed] [Google Scholar]
- 45.Zika E, Ting JP. Epigenetic control of MHC-II: interplay between CIITA and histone-modifying enzymes. Curr Opin Immunol. 2005;17:58–64. doi: 10.1016/j.coi.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 46.Yeh M, Leitinger N, de Martin R, Onai N, Matsushima K, Vora DK, Berliner JA, Reddy ST. Increased transcription of IL-8 in endothelial cells is differentially regulated by TNF-alpha and oxidized phospholipids. Arterioscler Thromb Vasc Biol. 2001;21:1585–1591. doi: 10.1161/hq1001.097027. [DOI] [PubMed] [Google Scholar]
- 47.Ellis EL, Delbruck M. The Growth of Bacteriophage. The Journal of general physiology. 1939;22:365–384. doi: 10.1085/jgp.22.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, Rozengurt E, Reed EF. HLA class I molecules partner with integrin beta4 to stimulate endothelial cell proliferation and migration. Sci Signal. 2010;3:ra85. doi: 10.1126/scisignal.2001158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nature reviews Molecular cell biology. 2014;15:155–162. doi: 10.1038/nrm3757. [DOI] [PubMed] [Google Scholar]
- 50.Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith J, Rozengurt E. Different patterns of Akt and ERK feedback activation in response to rapamycin, active-site mTOR inhibitors and metformin in pancreatic cancer cells. PloS one. 2013;8:e57289. doi: 10.1371/journal.pone.0057289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jamieson S, Flanagan JU, Kolekar S, Buchanan C, Kendall JD, Lee WJ, Rewcastle GW, Denny WA, Singh R, Dickson J, Baguley BC, Shepherd PR. A drug targeting only p110alpha can block phosphoinositide 3-kinase signalling and tumour growth in certain cell types. The Biochemical journal. 2011;438:53–62. doi: 10.1042/BJ20110502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ni Y, Sinnett-Smith J, Young SH, Rozengurt E. PKD1 mediates negative feedback of PI3K/Akt activation in response to G protein-coupled receptors. PloS one. 2013;8:e73149. doi: 10.1371/journal.pone.0073149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B) Science. 1999;286:1741–1744. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]
- 54.Romano D, Nguyen LK, Matallanas D, Halasz M, Doherty C, Kholodenko BN, Kolch W. Protein interaction switches coordinate Raf-1 and MST2/Hippo signalling. Nature cell biology. 2014;16:673–684. doi: 10.1038/ncb2986. [DOI] [PubMed] [Google Scholar]
- 55.Soares HP, Ming M, Mellon M, Young SH, Han L, Sinnet-Smith J, Rozengurt E. Dual PI3K/mTOR Inhibitors Induce Rapid Overactivation of the MEK/ERK Pathway in Human Pancreatic Cancer Cells through Suppression of mTORC2. Molecular cancer therapeutics. 2015;14:1014–1023. doi: 10.1158/1535-7163.MCT-14-0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Damrongwatanasuk R, Fradley MG. Cardiovascular Complications of Targeted Therapies for Chronic Myeloid Leukemia. Current treatment options in cardiovascular medicine. 2017;19:24. doi: 10.1007/s11936-017-0524-8. [DOI] [PubMed] [Google Scholar]
- 57.Guest SK, Ribas R, Pancholi S, Nikitorowicz-Buniak J, Simigdala N, Dowsett M, Johnston SR, Martin LA. Src Is a Potential Therapeutic Target in Endocrine-Resistant Breast Cancer Exhibiting Low Estrogen Receptor-Mediated Transactivation. PloS one. 2016;11:e0157397. doi: 10.1371/journal.pone.0157397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taflin C, Favier B, Charron D, Glotz D, Mooney N. Study of the allogeneic response induced by endothelial cells expressing HLA class II after lentiviral transduction. Methods in molecular biology. 2013;960:461–472. doi: 10.1007/978-1-62703-218-6_34. [DOI] [PubMed] [Google Scholar]
- 59.Meissner M, Whiteside TL, Kaufmann R, Seliger B. CIITA versus IFN-gamma induced MHC class II expression in head and neck cancer cells. Archives of dermatological research. 2009;301:189–193. doi: 10.1007/s00403-008-0922-6. [DOI] [PubMed] [Google Scholar]
- 60.Galvani S, Auge N, Calise D, Thiers JC, Canivet C, Kamar N, Rostaing L, Abbal M, Sallusto F, Salvayre R, Bohler T, Zou Y, Stastny P, Negre-Salvayre A, Thomsen M. HLA class I antibodies provoke graft arteriosclerosis in human arteries transplanted into SCID/beige mice. Am J Transplant. 2009;9:2607–2614. doi: 10.1111/j.1600-6143.2009.02804.x. [DOI] [PubMed] [Google Scholar]
- 61.Orth SR, Odoni G, Karkoszka H, Ogata H, Viedt C, Amann K, Ferrari P, Ritz E. Combination treatment with an ET(A)-receptor blocker and an ACE inhibitor is not superior to the respective monotherapies in attenuating chronic transplant vasculopathy in different aorta allotransplantation rat models. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2003;18:62–69. doi: 10.1093/ndt/18.1.62. [DOI] [PubMed] [Google Scholar]
- 62.Bayliss J, Maguire JA, Bailey M, Leet A, Kaye D, Richardson M, Bergin PJ, Dowling J, Thomson NM, Stein AN. Increased vascular endothelial growth factor mRNA in endomyocardial biopsies from allografts demonstrating severe acute rejection: a longitudinal study. Transpl Immunol. 2008;18:264–274. doi: 10.1016/j.trim.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 63.Rozengurt E, Soares HP, Sinnet-Smith J. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Molecular cancer therapeutics. 2014;13:2477–2488. doi: 10.1158/1535-7163.MCT-14-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deng Y, Atri D, Eichmann A, Simons M. Endothelial ERK signaling controls lymphatic fate specification. J Clin Invest. 2013;123:1202–1215. doi: 10.1172/JCI63034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem. 2010;285:14071–14077. doi: 10.1074/jbc.R109.094003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Laplante M, Sabatini DM. mTOR signaling at a glance. Journal of cell science. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ali SM, Sabatini DM. Structure of S6 kinase 1 determines whether raptor-mTOR or rictor-mTOR phosphorylates its hydrophobic motif site. J Biol Chem. 2005;280:19445–19448. doi: 10.1074/jbc.C500125200. [DOI] [PubMed] [Google Scholar]
- 68.Armengol G, Rojo F, Castellvi J, Iglesias C, Cuatrecasas M, Pons B, Baselga J, Ramony Cajal S. 4E-binding protein 1: a key molecular “funnel factor” in human cancer with clinical implications. Cancer research. 2007;67:7551–7555. doi: 10.1158/0008-5472.CAN-07-0881. [DOI] [PubMed] [Google Scholar]
- 69.Long X, Muller F, Avruch J. TOR action in mammalian cells and in Caenorhabditis elegans. Current topics in microbiology and immunology. 2004;279:115–138. doi: 10.1007/978-3-642-18930-2_8. [DOI] [PubMed] [Google Scholar]
- 70.Um SH, D'Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell metabolism. 2006;3:393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 71.Inoki K, Guan KL. Complexity of the TOR signaling network. Trends in cell biology. 2006;16:206–212. doi: 10.1016/j.tcb.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 72.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 73.Matsuoka T, Tabata H, Matsushita S. Monocytes are differentially activated through HLA-DR, -DQ, and -DP molecules via mitogen-activated protein kinases. J Immunol. 2001;166:2202–2208. doi: 10.4049/jimmunol.166.4.2202. [DOI] [PubMed] [Google Scholar]
- 74.Stein R, Gupta P, Chen X, Cardillo TM, Furman RR, Chen S, Chang CH, Goldenberg DM. Therapy of B-cell malignancies by anti-HLA-DR humanized monoclonal antibody, IMMU-114, is mediated through hyperactivation of ERK and JNK MAP kinase signaling pathways. Blood. 2010;115:5180–5190. doi: 10.1182/blood-2009-06-228288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Haylett RS, Koch N, Rink L. MHC class II molecules activate NFAT and the ERK group of MAPK through distinct signaling pathways in B cells. European journal of immunology. 2009;39:1947–1955. doi: 10.1002/eji.200838992. [DOI] [PubMed] [Google Scholar]
- 76.Bertho N, Blancheteau VM, Setterblad N, Laupeze B, Lord JM, Drenou B, Amiot L, Charron DJ, Fauchet R, Mooney N. MHC class II-mediated apoptosis of mature dendritic cells proceeds by activation of the protein kinase C-delta isoenzyme. International immunology. 2002;14:935–942. doi: 10.1093/intimm/dxf058. [DOI] [PubMed] [Google Scholar]
- 77.Kissner TL, Ruthel G, Alam S, Ulrich RG, Fernandez S, Saikh KU. Activation of MyD88 signaling upon staphylococcal enterotoxin binding to MHC class II molecules. PloS one. 2011;6:e15985. doi: 10.1371/journal.pone.0015985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.