

Abstract
The ability of Mycobacterium tuberculosis (Mtb) to block host antimicrobial responses in infected cells provides a key mechanism for disease pathogenesis. The immune system has evolved to overcome this blockade to restrict the infection, but it is not clear whether two key innate cytokines IL-12/IL-18 involved in host defense can enhance anti-mycobacterial mechanisms. Here, we demonstrated that the combination of IL-12 and IL-18 triggered an antimicrobial response against mycobacteria in infected macrophages (THP-1 and human primary monocytes-derived macrophages) and pulmonary epithelial A549 cells. The inhibition of intracellular bacterial growth required both p38-MAPK and STAT4 pathways, the vitamin D receptor (VDR), VDR-derived antimicrobial peptide cathelicidin and autophagy, but not caspase-mediated apoptosis. Finally, the ability of IL-12+IL-18 to activate an innate antimicrobial response in human primary macrophages was dependent on the autonomous production of IFN-γ and the CAMP/autophagy pathway. Together, these data suggest that the IL-12+IL-18 co-signaling can trigger antimicrobial protein cathelicidin and autophagy, resulting in froth inhibition of intracellular mycobacteria in both macrophages and lung epithelial cells.
Keywords: IL-12+IL-18 co-signaling, IFN-γ, VDR, CAMP, autophagy, mycobacteria
Introduction
As part of the first line of defense against Mycobacterium tuberculosis (Mtb), the innate immune response involves macrophages and lung epithelial cells (1, 2). Within macrophages, Mtb can counteract host defense via multiple strategies resulting in its survival. In addition, Mtb infects pulmonary epithelial cells resulting in the production of pro-inflammatory cytokines (TNF-α, etc.), chemokines (CXCL8, CXCL10, CCL5, and CCL2), NO, and antimicrobial peptides (β-defensin-2, cathelicidin), but the bacteria are not eliminated (3-5). Despite the development of an adaptive T cell response, the infection can progress. Mtb is estimated to have infected one-third of population around the world and due to its ability to evade the human immune response, kills over 4,000 people each day. (http://www.who.int/tb/publications/global_report/en/).
A better understanding of the host-Mtb interaction is required to develop improved intervention strategies to block transmission of Mtb and reactivation of disease in the already-infected population. A successful immune response against Mtb involves the distinct and overlapping contributions of the innate and adaptive immune systems, including the production of cytokines by the innate immune response which instruct the adaptive T cell response (6, 7). For example, two key cytokines produced upon activation of the innate immune response by mycobacterial infection are interleukin-12 (IL-12) and IL-18 (8-15), which synergize to directly activate the adaptive T cell response towards Th1 cell differentiation (16). There is evidence that the combination of IL-12 and IL-18 can directly activate the innate immune system, by inducing IFN-γ production in NK cells (17) and macrophages (18-20). Yet it is not known whether IL-12 plus IL-18 directly triggers an antimicrobial response against Mtb in infected cells. To address this gap, we investigated whether IL-12 and IL-18 cooperate to directly activate a microbicidal pathway against intracellular mycobacteria in infected monocytes/macrophages as well as pulmonary epithelial cells.
Our initial studies indicated that IL-12 and IL-18 alone had no effect on the viability of intracellular mycobacteria, but that only co-treatment of infected macrophages with both IL-12+IL-18 together inhibited bacterial growth. Concomitantly, mRNAs encoding for the antimicrobial peptides CAMP and DEFB4A were induced by IL-12+IL-18 treatment, as well as protein levels of CAMP. The inhibition of intracellular mycobacteria growth in A549 and THP-1 cells mediated by IL-12+IL-18 was blocked by lentiviral transduction vectors knocking down the expression of VDR. Moreover, inhibition of the p38-MAPK or STAT4 signaling pathway as well as autophagy in BCG-infected IL-12+IL-18-treated cells abrogated the IL-12+IL-18 inhibition of intracellular mycobacteria growth. Furthermore, IL-12+IL8 treatment induces IFN-γ-dependent killing of intracellular mycobacteria in human primary macrophages. These data support the view that IL-12 and IL-18 cooperate to trigger an innate immune response which via the autonomous production of IFN-γ to inhibit the growth of intracellular mycobacteria in human macrophages.
Materials and Methods
Ethics statement
The protocols for use of human blood samples for in vitro experimental procedures were evaluated and approved by the institutional review boards for human donors’ research and institutional biosafety committees at Institut Pasteur of Shanghai and the University of Illinois–Chicago College of Medicine. All studies were consistent with guidelines of Office for Human Research Protections. All subjects were adults and anonymized, signed written informed consents.
Cells and Reagents
The human monocyte cell line, THP-1 and the human lung adenocarcinoma epithelial cell line, A549, were grown in RPMI 1640 supplemented with L-glutamine (2 mM), sodium pyruvate (1 mM) and 10% heat-inactivated fetal bovine serum [(FBS), all from Gibco)], Prior to infection, THP-1 cells were treated with 50 ng/ml Phorbol 12-myristate 13-actate (PMA, Sigma-Aldrich) for 48 h to differentiate into macrophages, then washed three times with pre-warmed PBS and maintained in antibiotic-free media at 37°C for further use. 293T cells (purchased from Cellbank, Shanghai), used to produce lentivirus particles, were maintained in the medium consisting of DMEM supplemented with high glucose (4.5 g/L), L-glutamine (2 mM), sodium pyruvate (1 mM) and 10% heat-inactivated fetal bovine serum(FBS). (all from Gibco) All cell lines currently in use were routinely tested for Mycoplasma infection using a 16s-based PCR routinely, and new cultures were established monthly from frozen stocks.
Human peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Ficoll-Paque PLUS medium(GE) from buffy coats prepared from the peripheral blood of healthy uninfected donors (from Blood Center of Shanghai Changhai hospital). Adherent-monocytes were enriched from PBMC by adherence on plastic culture plate for 2 hours. Non-adherent cells were removed via vigorous washing using pre-warmed PBS three times. Human monocytes-derived macrophages (hMDM) were differentiated from adherent-monocytes under the medium containing RPMI1640, supplemented with L-glutamine (2 mM), sodium pyruvate (1 mM), 10% heat-inactivated FBS and 50 ng/ml human M-CSF (Novoprotein) for 7 days.
Human IL-12 was purchased from Miltenyi Biotech (130-096-704); Human IL-18 was purchased from Biovison (4179-25). Both cytokines were used at 50 ng/ml for stimulation. Human TNF-α were purchased from Invitrogen(10602HNAE25), used at 50ng/ml. Human IFNβ was purchased from Peprotech(300-02BC), used at 100U/ml. Neutralizing anti-human IL-18 mAb(Clone 125-2H) and isotype control (Clone 11711), anti-human IL-12(AF-219-NA) and its isotype control (AB-108), anti-human TNF-α (Clone 28401) and its isotype control (Clone MAB002), anti-human IFN-γ (Clone 25718) and its isotype control (Clone MAB003) were all purchased from R&D. MAPK inhibitors (p38, SB203580, MEK1/2, U0126-EtOH), NF-κB inhibitor (PDTC), autophagy inhibitors (3-MA, Wortmannin), apoptosis inhibitor(zVAD.fmk) were used at 10 μM. All inhibitors were purchased from Beyotime.
Bacteria culture, infection of cells and measurement of intracellular mycobacterial growth
Mycobacterium bovis Bacillus Calmette-Guerin (BCG), Mycobacterium smegmatis and M. tuberculosis H37Rv were grown in Difco Middlebrook 7H9 broth medium (Becton Dickinson) with 10% oleic acid-albumin-dextrose-catalase (OADC) Enrichment (Becton Dickinson), 0.05% (v/v) Tween 80 and 0.2% (v/v) glycerol at 37°C as reported (21). Basically, cells were infected with BCG at a multiplicity-of-infection (MOI) of 10 bacilli to 1 cell overnight. A549 cells were infected with M. smeg at a MOI of 1 bacilli to 1 cell for 4 h. Human monocyte-derived macrophage (hMDM) cells were infected with H37Rv at a MOI of 4 for 4 hours. After infection, extracellular non-internalized bacilli were removed by washing with pre-warmed PBS four times. Then, 5×104 mycobacteria-infected THP-1, A549, or hMDM cells were cultured in multiple wells with 50 ng/ml of each cytokine or combinations in the presence or absence of 5 ug/ml neutralization Abs and their isotype controls, or inhibitors (inhibitors were pre-incubated with cells 60 mins, then cytokines were added) in 200ul media without antibiotic at 96-well-plate for 3 days. Then, wells were aspirated and the infected cells were lysed in 200ul of sterile PBS with 0.067%SDS. A 10-fold serial dilution was performed for quantitative culturing. 100ul of aliquots were platted in triplicate on Middlebrook 7H10 agar plates supplied with 10% OADC for 2-3 weeks until colonies were large enough to be counted. Mycobacteria viability were quantified via counting CFU. The percentage survival index represented as intracellular bacteria survival was calculated as follows: Survival Index = 100 × CFU of treatment/CFU of media.
Quantification of gene expression
Total RNA was isolated from cells using Direct-zol™ RNA MiniPrep kit (Zymo) based on spin-columns method. Contaminating genomic DNA was removed with DNase I in columns according to manufacturer’s protocols(Zymo). Then, RNA was reverse-transcribed into cDNA using mixed oligod-dT18 and random hexamers primers with HiScript II Q RT SuperMix for qPCR (Vanzyme). Real-time quantitative PCR (RT-qPCR) reactions were performed in triplicated in 384-well plates on an Applied Biosystems RT-qPCR System PRISM 7900HT using the THUNDERBIRD qPCR Mix (Toyobo). Expression values were normalized to those of the housekeeping gene EF1A. Oligonucleotides used for amplification were listed on table 1 and synthesized from Shanghai Sangon Biotech.
Table. 1.
The sequences of primers for RT-qPCR or RT-PCR used in this study.
| Gene Symbol | Forward | Reverse |
|---|---|---|
| DEFB4A | ATCAGCCATGAGGGTCTTGT | GAGACCACAGGTGCCAATTT |
| CAMP | AGGATTGTGACTTCAAGAAGGACG | GTTTATTTCTCAGAGCCCAGAAGC |
| IL-18R1 | GTTGAGTTGAATGACACAGG | TCCACTGCAACATGGTTAAG |
| IL-18Rbeta | CTGGACAGAACTCACAGCTC | TCAAAGGCTCTAAACCACAG |
| STAT4 | CAGTGAAAGCCATCTCGGAGGA | TGTAGTCTCGCAGGATGTCAGC |
| IFNGR1 | CATCACGTCATACCAGCCATTT | CATCACGTCATACCAGCCATTT |
| IFNGR2 | CTCCATTCTGCCTGGGTGACAA | CGTGGAGGTATCAGCGATGTCA |
| VDR | CTGACCCTGGAGACTTTGAC | TTCCTCTGCACTTCCTCATC |
| EF1A | GATTACAGGGACATCTCAGGCTG | TATCTCTTCTGGCTGTAGGGTGG |
Generation of LV-shRNA, pCDH-GFP-LC3 constructs, Lentivirus Preparation and Infection
The short hairpin RNA (shRNA) lentiviral construct (synthesized by Sangon) targeting VDR (CTCCTGCCTACTCACGATAAA), STAT4 (GCGAGACTACAAAGTTATTAT), CAMP (GCTTCGTGCTATAGATGGCAT) or control were cloned into pLKO.1 lentiviral vectors using restriction enzymes AgeI and EcoRI (NEB). The GFP-LC3 was cloned from pEGFP-LC3 (human, Addgene plasmid # 24920) and then subjected into pCDH-Puro backbone to create pCDH-GFP-LC3 construct. 293T cells were transfected with the lentivirus constructs, pMD2G and pxPAS2 vectors using HG-Trans293 transfection reagent (Yeasen) according to manufacturer’s protocol. After 24 hours, media was changed and fresh media was added. At day 4 and day 5, the cell suspension was collected, pooled and filtered through 0.45 μm PVDF unit (Merck/Millipore). Then, the virus particles were concentrated using GML-PC lentivirus concentration kit (Genomeditech) according to manufacturer’s protocol. A549 cells were treated with filtered viral supernatant and 8ug/ml polybrene (Yeasen). Then, virus was aspirated and fresh media was added. For THP-1, cells were infected by a spin-transduction through 800g for 2 hours at 32 degrees. Selection with puromycin (Yeasen) was started at 48 hours after viral transduction, and the selection concentrations for A549 and THP-1 cells is 2 and 0.5 ug/ml, respectively.
Autophagy Analysis
PMA-differentiated GFP-LC3-THP-1 cells were infected with BCG at MOI 10:1 overnight. Cells then treated with media or IL-12+IL-18 for 12 hours. For the quantitation of autophagy, the percentages of GFP-LC3-positive autophagic vacuoles in GFP-LC3-THP-1 cells were evaluated using fluorescence microscopy as described in (22).
Western blotting
Cells were lysed by incubation in RIPA lysis buffer supplemented with 1mMPMSF (Yeasen)on ice for 5 min. Then, cell lysates were separated by SDS/PAGE gel electrophoresis and transferred to PVDF membranes (Merck/Millipore). After blocking with 5% BSA in Tris-buffered saline plus 0.1% Tween-20 (TBST) for 2 hours, the membrane was incubated with antibody against STAT4(BBIAB, Sangon Biotech), VDR(Abcam), LC3 (Abcam) or GAPDH (BBIAB, Sangon Biotech) overnight at 4 °C, followed by incubation with respective secondary antibodies for 1 hours. Protein bands were checked using ECL detection solution (Yeasen). In independent western blot assays, cells for evaluating each of targeted proteins were treated in duplicate or triplicate wells in 6-well plate in each of experiments. Samples harvested from each well were run on separated SDS-PAGE gels, followed by transblotting and specific immune detection.
ELISA
The amounts of IFN-γ, TNF-α and IL-32 in cell supernatant were detected using human cytokines ELISA kit (C608 for Human IFN-γ; C609 for Human TNF-α; C687 for Human IL-32α) according to the manufacturer’s instructions.
Statistical analysis
Statistical analysis was performed with GraphPad Prism 6.0. All data are expressed as mean ± SEM. Differences between groups were firstly assessed by nonparametric t-test or ANOVA, the latter followed by the Dunnett’s test or Tukey’s multiple comparison test indicated in each figure. P values is adjusted for multiple comparisons (family-wise significance and confidence levels are 0.05 and 95% confidence interval).
Results
IL-12+ IL-18 co-treatment in mycobacterium-infected cells from monocytic and lung-epithelial linages led to inhibition of intracellular mycobacterial growth
To address whether IL-12 and/or IL-18 directly affected intracellular mycobacterial infection in monocytes/macrophages and lung epithelial cells, we used an in vitro human infection model (23, 24). Experiments were initiated in the biosafety level-2(BSL2) lab using less virulent mycobacteria, and then performed employing virulent Mtb H37Rv strain for confirmation in the BSL3 lab. Mycobacteria-infected cells were treated for 72 hours with media, recombinant human IL-12 alone, IL-18 alone or IL-12 plus IL-18 (IL-12+IL-18), and then the cells were lysed to release viable intracellular bacteria for measuring CFU counts in 7H10 plate. Thus, A549 cells, which usually serve as a model of studying host response of human type II alveolar epithelial cells to intracellular pathogens (25), were infected with M. smeg and M. bovis BCG, respectively. To select an optimal combination of cytokines required to limit intracellular bacteria, does response experiments were performed and a combination of 50 ng/ml cytokine each was shown a reduced recovered BCG and Mtb burden (Supplemental Fig. S1A-B). While IL18RAP mRNA and protein were expressed in both infected and uninfected A549 cells (Supplemental Fig. S2), only co-treatment with IL-12+IL-18, but not IL-12 or IL-18 alone, significantly reduced the growth of M. smeg in A549 cells (Fig. 1A). Approximately 40% inhibition of intracellular M. smeg viability was observed as compared to medium control (P< 0.0001, Fig. 1A). In the setting of BCG-infected A549 cells, IL-12+IL-18 also inhibited the intracellular BCG growth (Fig. 1B).
FIGURE 1. IL-12+ IL-18 co-treatment in mycobacterium-infected cells from monocytic and lung-epithelial linages led to reproducibly inhibition of intracellular mycobacterial growth.
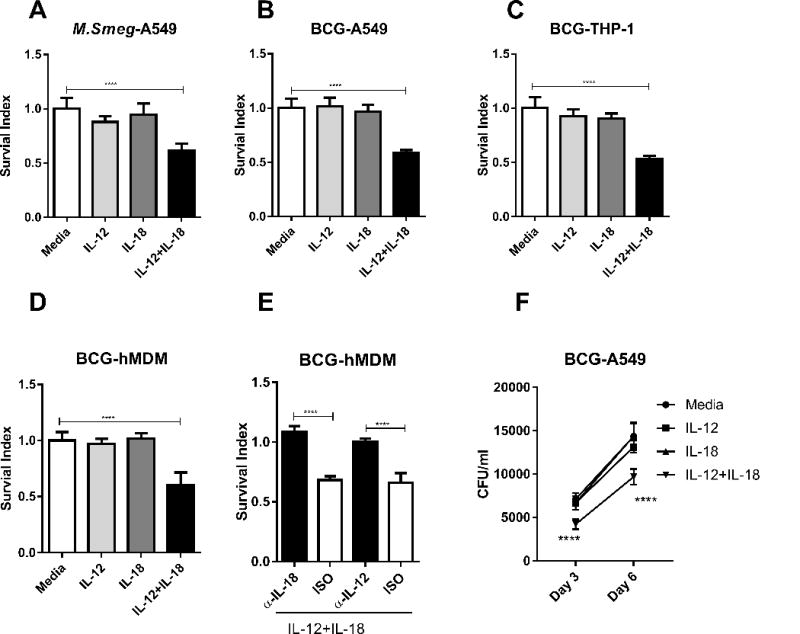
(A-B) Graph data of Survival Index (=100 × CFU of treatment/CFU of media only) show the IL-12+IL-18-induced inhibition of intracellular growth of M. smeg or M. bovis BCG in A549. A549 cells were infected with M. smeg or BCG, and then cultured for 3 days with 50ng/ml recombinant human IL-12, IL-18, their combination, or culture media alone as indicated in x-axis. (C) Mean Survival Index for BCG in THP-1 cells cultured with different treatments as indicated in x-axis. THP-1 cells were firstly treated with 50 ng/ml PMA for differentiated to be adherent. Then, differentiated and adherent THP-1 cells were infected with BCG. BCG-infected THP-1 cells were cultured for 3 days with 50ng/ml recombinant human IL-12, IL-18, their combination, or culture media alone. (D-E) Mean Survival Index for BCG in human monocyte-derived macrophages (hMDM) treated as above. The adherent monocytes enriched from PBMC were allowed to differentiate into human primary macrophages under stimulation of 50 ng/ml M-CSF in culture. HMDM cells were infected with BCG and then cultured for 3 days with 50ng/ml recombinant human IL-12, IL-18, their combination, culture media alone, in the absence or presence of antibody to IL-12 or IL-18 and isotype controls. (F) IL-12+Il-18 co-signaling decreased the number of intracellular BCG in A549 cells at both day 3 and day 6. A549 cells were infected with BCG, and then cultured for 3-6 days with 50ng/ml recombinant human IL-12, IL-18, their combination. Data in A-C are pooled from 5 independent experiments; data in D-E pooled from 4 independent experiments using cells from 20 healthy donors; data in F pooled from 8 replicates in 2 independent experiments. P values are calculated through ANOVA then Dunnett’s test compared with control condition: treated with culture media (for A-D); ANOVA then Tukey’s multiple comparison (for E); repeated ANOVA then Dunnett’s test compared with control condition: treated with culture media (for F), **** p< 0.0001.
We then tested whether co-treatment with IL-12+IL-18 inhibited the growth of mycobacteria not only in pulmonary epithelial cells, but also in human macrophages. We first employed PMA-matured THP-1 cells, which are often used as a model to study immune response of human macrophages (26). Co-treatment of BCG-infected THP-1 macrophage cells with IL-12+IL-18 significantly decreased the growth of intracellular mycobacteria, with approximately 50% inhibition compared to controls (Fig. 1C). We next examined whether the combination of IL-12+IL-18 triggered growth-inhibition could also be detected in human monocyte-derived macrophages (hMDM). Similarly, IL-12+IL-18 co-treatment significantly reduced the growth of intracellular BCG in hMDM, with a mean inhibition of ~40% compared to control treatment (Fig. 1D).
To validate the specificity of IL-12+IL-18 growth inhibition of intracellular mycobacteria, we performed blocking experiments using neutralizing antibodies. In the presence of IL-12+IL-18, blockade of IL-12 or IL-18 signaling by neutralizing anti-IL-12 or anti-IL-18 antibodies significantly reduced the intracellular growth of M. smeg and BCG in hMDM, as compared with isotype controls (Fig. 1E). This was consistent with the finding that the IL-12+IL-18 treatment up-regulated IL12A, IL12B and IL18 in BCG-infected hMDM (Supplemental Fig. S1C). In the absence of exogenous IL-12+IL-18, however, neutralization of endogenous IL-12 and IL-18 in BCG-infected hMDM cultures did not affect the intracellular mycobacteria growth (Supplemental Fig. S1D). While anti-IL12 or anti-IL18 neutralizing antibodies were unable to efficiently penetrate membrane into cells for blocking endogenous IL-12/IL-18, we cannot exclude the possibility that concentrations of endogenous IL-12 and IL-18 induced by mycobacterial infection were not high enough to inhibit extremely high levels of mycobacterial infection in cultures. Similar blocking effects by neutralizing anti-IL-12 or anti-IL-18 Ab were observed in other three systems involving IL-12+IL-18 co-treatment of M. smeg or BCG in A549 epithelial cells and THP-1 monocytic cells (Supplemental Fig. S1E-G).
We also sought to examine whether the IL-12+IL-18-mediated intracellular inhibition of mycobacteria could also be detected at day 6 after the co-culture. As seen in the Fig.1F, the IL-12+IL-18 co-treatment significantly restricted intracellular BCG growth in A549 cells not only at day 3 but also at day 6. Taken together, these data suggest that the simultaneous activation of IL-12 and IL-18 signaling pathways results in potent growth inhibition of intracellular mycobacteria in both human monocytic and lung-epithelial linages of cells.
Co-treatment of human monocyte-derived macrophages with IL-12+IL-18 consistently inhibited intracellular growth of virulent Mtb H37Rv
We then sought to determine whether co-treatment of monocytes/macrophages with IL-12+IL-18 could also inhibit the intracellular growth of virulent Mtb bacilli as we demonstrated in the setting of M. smeg and BCG infection. To address this, we performed the growth inhibition experiments using the virulent Mtb H37Rv strain for infection of hMDM under BSL-3 conditions. Similarly, Mtb H37Rv-infected hMDM were cultured with media or IL-12+IL-18 for 3 days and then subjected to measurement of CFU counts on 7H11 plates. Consistent with findings with M. smeg and BCG, co-treatment of Mtb-infected hMDM with IL-12+IL-18 also led to significant and reproducible inhibition of intracellular growth of H37Rv bacilli at day 3 (Fig. 2A, inhibition rate 39%, p=0.0296). Moreover, IL-12+IL-18 treatment still shown the suppression effect at day 6 (Fig. 2B). These results provided further support implicating that IL-12+IL-18 could restrict intracellular growth of mycobacteria including virulent Mtb.
FIGURE 2. IL-12+IL-18 co-treatment in human monocyte-derived macrophages consistently inhibited intracellular growth of virulent Mtb H37Rv.
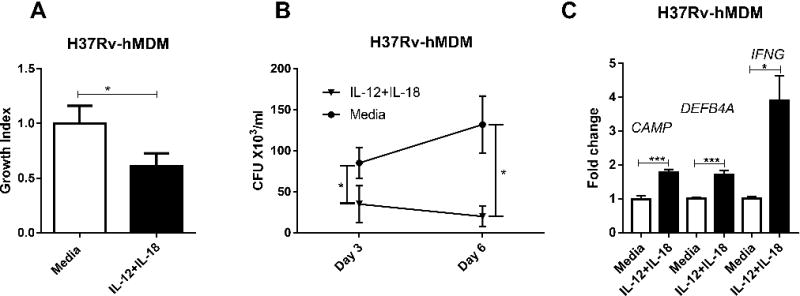
(A)Shown are mean Survival index for intracellular growth inhibition of Mtb H37Rv strain in hMDM. Mtb-infected hMDM cells were treated with IL-12+IL-18 or media for 3 days. Viable bacteria recovered from infected cells were quantified by counting CFU. Data are pooled from two independent experiments using cells from 8 healthy uninfected donors. P values are calculated through non-parametric t-test, * p < 0.05. (B) The IL-12+IL-18 co-signaling decreased the number of intracellular Mtb H37Rv bacilli in hMDM cells at both day 3 and day 6. hMDM were infected with H37Rv, and then cultured for 3-6 days in the presence of 50 ng/ml recombinant human IL-12+IL-18 or media. Data are pooled from two independent experiments from 8 uninfected donors. P values are calculated through non-parametric t-test, * p < 0.05. Individual cytokines alone were not found to inhibit the growth of BCG (Fig.1), thus were not evaluated here. Attenuated BCG may be similarly inhibited as does Mtb in IL12+IL18-treated hMDM cells (Figs.1D-1E). Presumably, at day 6 BCG may be restricted similarly to Mtb in IL12+IL18-stimulated hMDM cells. (C) shows mean fold changes in expression levels for CAMP, DEFB4A and IFNG in H37Rv-infected hMDM, respectively, in the presence of media or IL-12+IL-18. Data are derived from 2 independent experiments using hMDM derived from 8 healthy uninfected donors. P values are calculated through T test compared with control condition: treated with culture media, * p<0.05, *** p< 0.001.
VDR activation of anti-microbial peptides appeared to be required for the IL-12+IL-18 co-signaling-mediated growth inhibition of intracellular mycobacteria
We then conducted studies to explore the mechanism(s) by which co-treatment of infected cells with IL-12+IL-18 restricts intracellular mycobacterial growth. Since co-treatment with IL-12+IL-18 similarly inhibited Mtb and BCG growth in host cells, we employed BCG infection for the in-depth mechanistic manipulations in a BSL2 lab, rather than those involving Mtb in the complicated BSL3 condition. We sought to determine which of anti-mycobacterium effector functions were activated by IL-12+IL-18 during infection of target cells. Because mycobacteria can activate the expression of antimicrobial defense peptides in both lung epithelial cells and human macrophages (27-29), we rationalized that treatment with both IL-12+IL-18 would enhance expression of these peptides resulting in growth inhibition of intracellular mycobacteria. To address this, BCG-infected cells were treated with IL12+IL-18 together, individually or media control, and assessed by RT-qPCR for the expression of vitamin D receptor (VDR) downstream genes CAMP and DEFB4A, encoding cathelicidin and β-defensin 2, respectively, antimicrobial peptides capable of killing intracellular mycobacteria (29, 30). These experiments revealed that co-treatment with IL-12+IL-18 significantly increased expressions of CAMP by up to ~7 fold after BCG infection of A549, THP-1 and hMDM cells, respectively (Fig. 3A). Similarly, IL-12+IL-18 co-signaling also increased DEFB4A expression by up to ~8 fold (Fig. 3B). In H37Rv-infected hMDM cells, IL-12+IL-18 treatment still led to enhanced expression of CAMP and DEFB4A (Fig. 2C). This result is confirmed by the enhanced expression of CAMP in IL-12+IL-18 treated in BCG infected A549 and THP-1 cells at protein levels measured by western blot (Fig. 3C). Furthermore, when the expression of CAMP was impaired by knocking down (Fig. 3D-E), IL-12+IL-18 treatment failed to control the growth of intracellular BCG, comparing to media group (Fig. 3F-G). These results suggest that the IL-12+IL-18 co-signaling led to enhanced expression of genes encoding anti-mycobacterial peptide, such as CAMP and DEFB4A, during BCG infection of monocytic and lung-epithelial cell linages, which contributed to IL-12+IL-18 mediated inhibitory effect on intracellular mycobacteria growth.
FIGURE 3. CAMP appeared to be required for the IL-12+IL-18-mediated growth inhibition of intracellular mycobacteria.
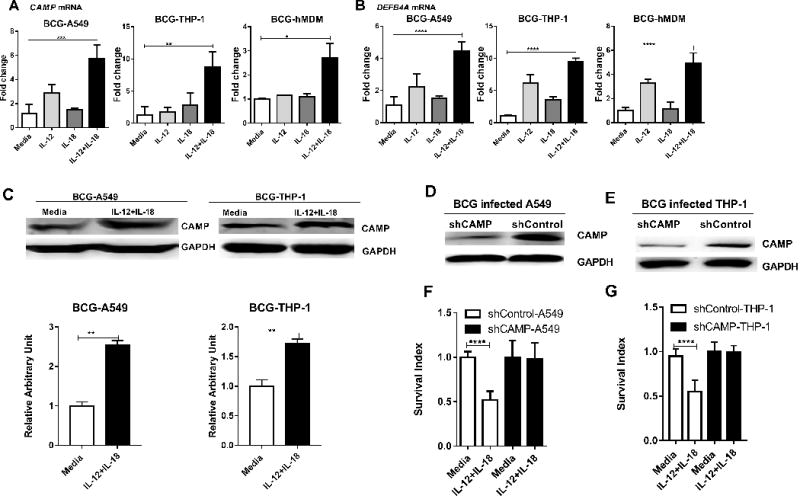
(A-B) shows mean fold changes in expression levels for CAMP(A), DEFB4A (B) in BCG-infected A549, THP-1 cells and hMDM, respectively, in the presence of media, IL-12, IL-18 or IL-12+IL-18 for 24 hours. Data are derived from 4 independent experiments using A549 or THP-1 cells, and 3 independent experiments using hMDM from 12 healthy uninfected donors. P values are calculated through ANOVA then Dunnett’s test compared with control condition: treated with culture media, * p<0.05, ** p<0.01, *** p < 0.001, **** p < 0.0001. (C) IL-12+IL-18 co-signaling led to enhanced expression of CAMP at protein levels in BCG-infected A549 (Left, Lane 1-2) and THP-1 (Right, Lane 3-4) cells. The upper panel are representative western blot analysis from 4 replicates, the lower panel are quantitative analysis of CAMP protein levels by densitometry. P values are calculated through T test compared with control condition: treated with culture media, ** p< 0.01. (D-E) shows representative western blot data demonstrating the shRNA knock-down of CAMP in A549 and THP-1 cells, respectively, compared to the shRNA-Control. (F-G) Bar graph shows that the shRNA knock-down of CAMP reverses the growth restriction of intracellular mycobacteria during IL-12+IL-18 treatment of BCG-infected A549 and THP-1 cells compared to shRNA control. Data are derived from 8 replicates in 2 independent experiments. P values are calculated through T test, **** p< 0.001.
Since VDR signaling is required for induction of CAMP and DEFB4A and mediates control of intracellular pathogens (31, 32), we hypothesized that VDR activation of cathelicidin and β-defensin 2 might be required for the IL-12+IL-18-induced growth restriction of intracellular mycobacteria. Initially, we found that the IL-12+IL-18 co-signaling significantly increased expression of VDR by 6.4±0.6 and 4.9±0.87 fold, respectively, during BCG infection of A549 cells and hMDM, though not in THP-1 cells (Fig. 4A). To prove that VDR activation of anti-mycobacterial peptides was required for the IL-12+IL-18 triggering of the antimycobacterial response, we developed the stably-transduced A549 and THP-1 cell lines displaying VDR knock-down (Fig. 4B, E). Notably, knockdown of the VDR in BCG-infected shVDR-A549 cells diminished the ability of co-treatment with IL-12+IL-18 to up-regulate CAMP and DEFB4A genes, when compared with the control shControl-A549 cells (Fig. 4C). The CAMP protein expression was also reduced by shRNA knock down of VDR in BCG-infected A549 and THP-1 cells treated with IL-12+IL-18 (Fig. 4G). More importantly, shRNA knockdown of the VDR in BCG-infected A549 and -THP-1 cells abrogated the ability of IL-12+IL-18 co-treatment to restrict the growth of intracellular mycobacteria (Fig. 4D, F). Taken together, these data suggest that VDR activation is required for the IL-12+IL-18 induced restriction of intracellular mycobacterial growth in both macrophages and lung epithelial cells.
FIGURE 4. VDR activation of anti-microbial peptides appeared to be required for the IL-12+IL-18-mediated growth inhibition of intracellular mycobacteria.
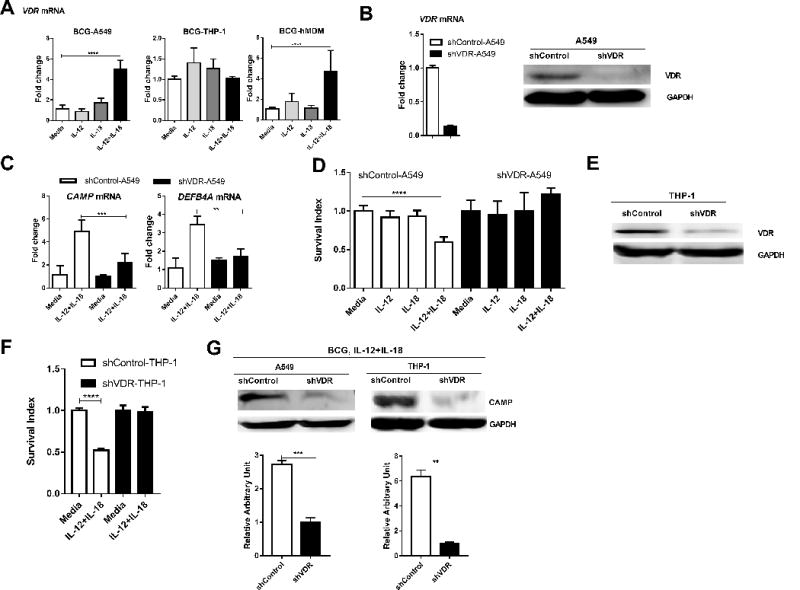
(A) shows mean fold changes in expression levels for VDR in BCG-infected A549, THP-1 cells and hMDM, respectively, in the presence of media, IL-12, IL-18 or IL-12+IL-18. Data are derived from 4 independent experiments using A549 or THP-1 cells, and 3 independent experiments using hMDM from 12 healthy uninfected donors. P values are calculated through ANOVA then Dunnett’s test compared with control condition: treated with culture media, * p<0.05, ** p<0.01, *** p < 0.001, **** p < 0.0001. (B) shows representative data demonstrating the knock-down for the VDR at both mRNA (left, RT-qPCR) and protein (right, western blot) levels in A549 cells stably transduced with the lentivirus construct LV-shVDR in comparisons with the control. Stably transduced cell lines were selected by 2 ug/ml puromycin treatment of cultures. (C) compares fold changes in expression levels of CAMP and DEFB4A between BCG-infected shVDR- and shControl-transduced A549 cells in the presence of media or IL-12+IL-18 for 24 hours. Data were generated by RT-qPCR, and pooled from three independent experiments. P values are calculated through T test, ** p<0.01, *** p < 0.001. (D) shows changes in mean Survival indexes for BCG bacilli in BCG-infected shVDR- and shControl-transduced A549 cells in the presence of media, IL-12, IL-18 or IL-12+IL-18. Data are derived from four independent experiments. Note that VDR knock-down led to reversion of IL-12+IL-18-mediated inhibition of BCG growth. P values are calculated through ANOVA then Dunnett’s test compared with control condition: treated with culture media, **** p < 0.0001. (E) shows representative western blot data demonstrating the VDR knock-down in THP-1 cells in comparisons with the shRNA control. (F) Bar graph shows that shRNA knock-down of VDR reverses the growth restriction of intracellular mycobacteria during IL-12+IL-18 treatment of BCG-infected THP-1 cells when compared with shRNA control. Data are derived from 8 replicates in 2 independent experiments. P values are calculated through T test, **** p< 0.0001. (G) Representative western blot analysis shows that shRNA knock-down of VDR abrogated the ability of the IL-12+IL-18 treatment to induce the CAMP expression in BCG-infected A549 and -THP-1 cells, respectively. The upper panel are representative western blot analysis from 4 replicates, the lower panel are quantitative analysis of CAMP protein levels by densitometry. P values are calculated through T test compared with control condition: treated with culture media, ** p< 0.01, *** p< 0.001.
Both p38-MAPK and STAT4 pathways are involved in the IL-12+IL-18 co-signaling that led to inhibition of intracellular mycobacteria
Next, studies of T cells and NK cells showed that IL-18 could activate NF-κB and MAPK-associated PI3K, MEK, and p38 signaling pathways (33, 34). We therefore investigated which of these pathways in monocyte was required for the IL-12+IL-18 co-signaling-induced the growth inhibition of intracellular mycobacteria. Thus, BCG-infected A549 and hMDM cells were incubated with individual well-documented inhibitors for MAPK and NF-κB pathways, respectively, in the presence of IL-12+IL-18 co-treatment. Notably, inhibitors of NF-κB pathway (PDTC) and MEK-MAPK (U0126-EtOH) each did not apparently alter the IL-12+IL-18-induced restriction of intracellular BCG growth (Fig. 5A-B). In contrast, the p38-MAPK inhibitor (SB203580) significantly blocked IL-12+IL-18 co-signaling-induced the growth restriction of mycobacteria in both BCG-infected hMDM and A549 cells (Fig. 5A-B). Consistently, we found that the IL-12+IL-18 co-signaling could modulate the expression of IL-18 receptors in hMDM as seen in T cells and NK cells (14, 33, 34), since co-treatment with IL-12+IL-18 significantly increased the expression of IL18R1 and IL18RAP by 5.7±2.1 and 5.2±2.6 fold, respectively, in mycobacteria-infected hMDM (Fig. 5C). Furthermore, the treatment with p38-MAPK inhibitor (SB203580) significantly inhibited IL-12+IL-18 co-signaling-induced CAMP at both mRNA and protein levels in BCG-infected hMDM and THP-1 cells (Fig. 5D-5E). These results suggest that the combined action of IL-12+IL-18 that resulted in growth inhibition of intracellular mycobacteria mainly occurred via the p38-MAPK axis signaling, but not via the NF-κB or MEK-MAPK pathway.
FIGURE 5.
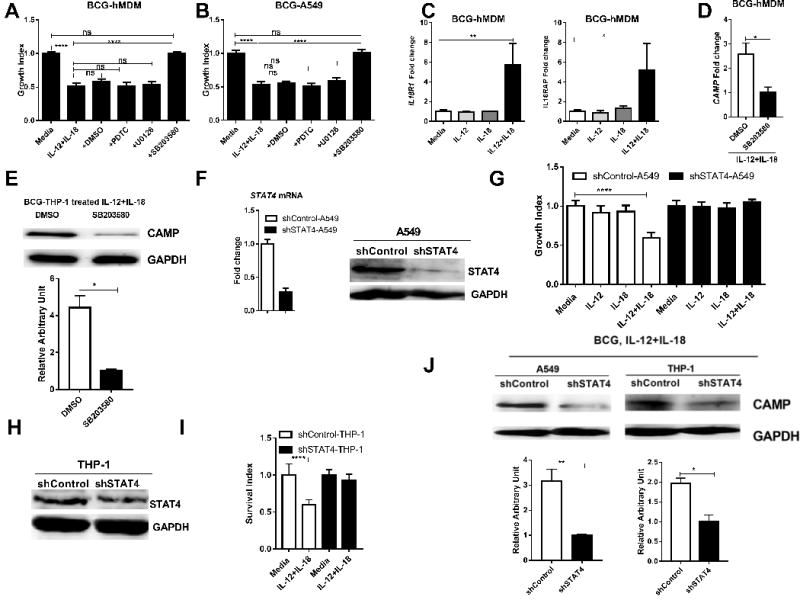
Both p38-MAPK and STAT4 pathways appeared to be the selected IL-12+IL-18 co-signaling that led to inhibition of intracellular mycobacteria.
(A-B) show changes in mean Survival index of BCG bacilli in IL-12+IL-18-cotreated A549 cells(A) and hMDM (B) in the absence and presence of chemical inhibitors PDTC (10uM), U0126 (10uM), SB203580 (10uM) and DMSO in the 3-day culture. Data were pooled from 3 independent experiments using A549 cells (A) and 5 different experiments using hMDM (B) derived from 15 healthy uninfected donors. P values are calculated through ANOVA then Tukey’s multiple comparisons test, **** p < 0.0001. ns, not significant. (C) shows graph data of mean fold changes in expression levels for IL-18 receptor genes IL18R1 and IL18RAP in BCG-infected hMDM incubated for 24 hours with media, IL-12, IL-18 or IL-12+IL-18. Fold changes were calculated by expression levels of cytokine treatment versus those of media control. Data were generated using RT-qPCR and pooled from 3 independent experiments using hMDM from 12 healthy uninfected donors. P values are calculated through ANOVA then Dunnett’s test compared with control condition: treated with culture media, * p<0.05, ** p<0.01. (D-E) The inhibition of p38 renders IL-12+IL-18 co-signaling fails to induce enhanced expression of CAMP at mRNA levels in BCG-hMDM cells (D) and at protein levels in BCG-THP-1 cells (E). Data are derived from 3 independent assays using 9 healthy uninfected donors for D. The upper panel in E is a represented image from 4 replicates, the lower panel is pooled data for optical density analysis. P values are calculated using T test, * p< 0.05. (F) shows representative data demonstrating the knock-down for the transcription factor STAT4 in A549 cells stably transduced with the lentivirus construct LV-shSTAT4 in comparisons with the control. (G) compares mean Survival indexes for BCG bacilli in shSTAT4- and shControl-transduced A549 cells in the presence of media, IL-12, IL-18 or IL-12+IL-18. Note the reversion of IL-12+IL-18-mediated inhibition in the setting of STAT4 knock-down. Data were derived from 4 independent experiments. P values are calculated through ANOVA then Dunnett’s test compared with control condition: treated with culture media, **** p < 0.0001. (H). Representative western blot data shows that the shRNA knock-down of STAT4 in THP-1 cells in comparisons with the shRNA control. (I). Bar graph shows that the shRNA knock-down of STAT4 reverses the growth restriction of intracellular mycobacteria during IL-12+IL-18 treatment of BCG-infected THP-1 cells, compared to the shRNA control. Data are derived from 8 replicates in 2 independent experiments. P values are calculated through T test, **** p< 0.001. (J) Representative western blot shows shRNA knock-down of STAT4 reduced the CAMP protein expression during the IL-12+IL-18 treatment of BCG-infected A549 and THP-1 cells, respectively. The upper panel are representative western blot analysis from 4 replicates, the lower panel are quantitative analysis of CAMP protein levels by densitometry. P values are calculated through T test compared with control condition: treated with culture media, * p< 0.05, ** p< 0.01.
Since studies of T cells and NK cells identified STAT4 as a major down-stream molecule for IL-12 signaling (35), we presumed that the IL-12/STAT4 pathway in monocytic/lung-epithelial linages of cells might contribute to the combined activity of IL-12+IL-18 resulting in the growth inhibition of intracellular mycobacteria. To test this, we produced lentivirus shRNA knock-down constructs and developed stable A549 and THP-1 cell lines displaying STAT4 knock-down, with the shControl serving as control. The STAT4 knock-down was confirmed in these transduced A549 and THP-1 cells (Fig. 5F, H). Clearly, knocking down STAT4 expression in both A549 and THP-1 cells reversed the IL-12+IL-18-induced growth inhibition of intracellular mycobacteria (Fig. 5G, I). Notably, the reversing effect by the STAT4 knock down corelated with the reduced ability of IL-12+IL-18 to up-regulate CAMP protein, when compared with the control (Fig. 5J). Taken together, these data suggest that both p38-MAPK and STAT4 pathways are required for the combined activity of IL-12+IL-18 in the restriction of intracellular mycobacterial growth in both monocytes and pulmonary epithelial cells.
Autophagy, but not caspase-mediated apoptosis, was involved in the IL-12+IL-18 co-signaling inhibition of intracellular mycobacteria growth
The VDR activation of anti-microbial peptides has been shown to induce autophagy and mycobactericidal activity (36). Thus, we sought to examine whether autophagy was involved in the co-signaling induced by IL-12+IL-18 that triggered restriction of intracellular mycobacterial growth. Here, we assessed an increase in the LC3II: LC3I ratio (LC3II/LC3I) or LC3II convention as a parameter to examine autophagic flux in BCG-infected target cells treated with IL-12+IL-18 or control. We found that the IL-12+IL-18 co-signaling in BCG-infected A549 cells led to an increase in the LC3II/LC3I ratio, i.e. autophagy flux (Fig. 6A), beyond the background autophagy induced by BCG infection of A549 cells in the absence of cytokines (37, 38). In parallel, we developed a stable THP-1 cell line expressing GFP-LC3 using lentivirus vector to support our results in western blot assays. Consistently, the IL-12+IL-18 treatment remarkably formed fluorescence LC3 aggregates/microdots of the autophagy machinery in GFP-LC3-transduced, BCG-infected THP-1 cells (Fig. 6B). Since p38, STAT4 and VDR were indispensable for IL-12+IL-18-induced antimicrobial response, we sought to determine whether autophagy was involved in connecting p38/STAT4/VDR activation and growth inhibition of intracellular mycobacteria. To this end, we examined whether the conversion of LC3I into LC3II was altered by p38 inhibition, STAT4-, VDR-, or CAMP-knockdown during IL-12+IL-18 treatment of BCG-infected THP-1 cells. Treatment with p38 inhibitor SB203580 reduced IL-12+IL-18-induced LC3II convention, compared to the control DMSO (Fig. 6C). This was consistent with the decreased formation of LC3II by two well-documented autophagy inhibitors, wortmannin and 3-MA (39, 40) (Fig. 6C). Moreover, the knock-down of CAMP by shRNA significantly reduced the conversion of LC3I into LC3II during the IL-12+IL-18 treatment of BCG-infected THP-1 cells (Fig. 6D), suggesting a critical role of CAMP in the IL-12+IL-18 activation of autophagy and BCG growth inhibition in THP-1 cells. Since we already showed that CAMP was up-regulated upstream by VDR (Fig.4G) and STAT4 (Fig.5J), we sought to investigate if shRNA knock-down of VDR or STAT4 would impact autophagy. As expected, knocking down VDR or STAT4 reduced the formation of LC3II during the IL-12+IL-18 treatment of BCG-infected THP-1 cells (Fig.6 E). These results suggest the IL-12+IL-18 co-signaling of p38/STAT4 can activate VDR-derived CAMP and autophagy process.
FIGURE 6. Autophagy, but not caspase-mediated apoptosis, was involved in the IL-12+IL-18-induced inhibition of intracellular mycobacteria growth.
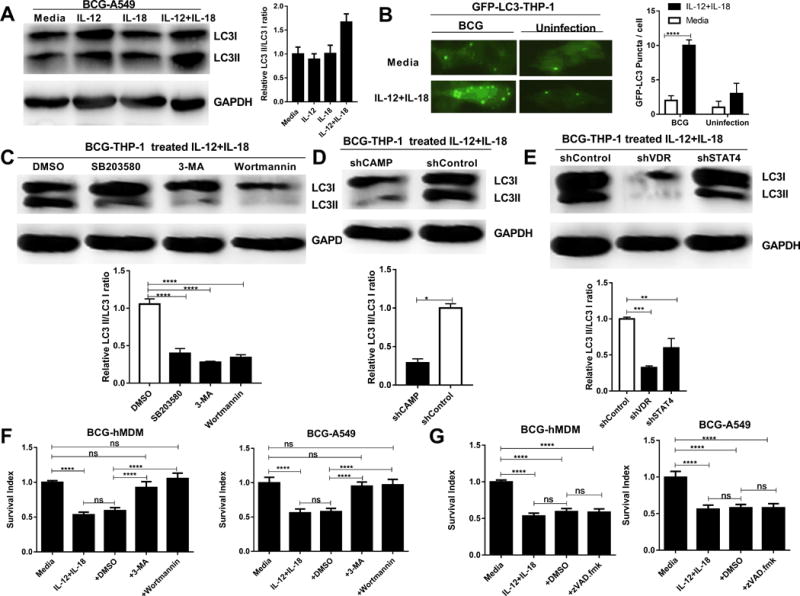
(A) shows representative western blot image data (Left) displaying autophagy-related proteins LC3II and LC3I in lysates from BCG-infected hMDM treated for 6 hours with media, IL-12, IL-18 or IL-12+IL-18, and bar plot of the ratio of the intensities of the LC3-II versus LC3-I bands from the blot (Right, n=3). (B) On left is representative fluorescence microscopic image showing that GFP-LC3 puncta or fluorescence GFP-LC3 aggregates of the autophagy machinery were formed after the IL-12+IL-18 treatment of GFP-LC3-transduced, BCG-infected THP-1 cells. Shown are high-power images of individual cells treated with IL-12+IL-18 or media control in the presence or absence of BCG infection (original magnification x400). On right is bar graph showing comparative quantitation of GFP-LC3 puncta per cell under the microscopy. n ≥ 25 cells for each group in 3 independent experiments. Low background of fluorescence machinery dots is seen in GFP-LC3-THP-1 cells treated only with media or IL-12+IL-18 alone. (C-E) The upper panel: Representative western blot data show that the conversion of LC3II (autophagy flux) was reduced by p38 inhibitor SB203580/autophagy inhibitors 3-MA and wortmannin (C), shRNA knock-downs of CAMP, VDR and STAT4 (D-E) during the IL-12+IL-18 treatment of BCG-infected THP-1 cells from 4 replicates; The lower panel: Quantitative analysis of relative LC3II/LC3I ratios by densitometry. P values are calculated through ANOVA then Dunnett’s test compared with control condition: treated with DMSO, **** p < 0.0001 (for C); T test compared with shControl-THP-1 cells, * p<0.05 (for D); ANOVA then Dunnett’s test compared with shControl-THP-1 cells, ** p < 0.01, *** p < 0.001 (for C). (F) shows that a 3-day treatment with autophagy-blocking reagent 3-MA (10 uM) or Wortmannin (10 uM) could abrogate the IL-12+IL-18-mediated inhibition of intracellular BCG in BCG-infected hMDM or A549 cells when compared with DMSO control. (G) shows that a 3-day treatment with apoptosis-blocking reagent zVAD.fmk (10 uM) was not able to abrogate the IL-12+IL-18-mediated inhibition of intracellular BCG in BCG-infected hMDM or A549 cells. In the absence of IL12+IL18, autophagy inhibitors 3-MA and wortmannin, but not zVAD.fmk, have reverse impact on mycobacterial growth (Supplementary Fig. S3B). Data (F and G) are pooled from 4 independent experiments using A549 cells, and 3 independent experiments using hMDM from 12 healthy uninfected donors. P values are calculated through ANOVA then Tukey’s multiple comparisons test, **** P < 0.0001, ns, no significance.
We then questioned whether the autophagy induced by the IL-12+IL-18 co-signaling indeed contributed to the detectable inhibition of intracellular mycobacterial growth. To address this, wortmannin and 3-MA were used for blocking autophagy flux in the context of IL-12+IL-18-mediated growth restriction of intracellular mycobacteria. In parallel, the apoptosis inhibitor zVAD.fmk was also evaluated, as it inhibits caspase-mediated apoptosis (41). Notably, addition of 3-MA or wortmannin to BCG-infected A549 and THP-1 cultures significantly blocked the IL-12+IL-18 co-signaling restriction of intracellular mycobacterial growth, as compared to DMSO control (Fig. 6F). In contrast, the apoptosis inhibitor zVAD.fmk did not have significant blocking activity in BCG-infected A549 and hMDM cells (Fig. 6G). Taken together, these data together implicate that the IL-12+IL-18 co-signaling of p38 and STAT4 activates CAMP and autophagy and leads to the growth inhibition of intracellular mycobacteria.
Activation of the IFN-γ-IL-32 pathway was required for the IL-12+IL-18 co-signaling-mediated growth inhibition of intracellular mycobacteria
Finally, we sought to explore the possibility that the combined action of IL-12+IL-18 activates IFN-γ and/or TNF-α, resulting in induction of downstream genes that mediate growth inhibition of intracellular mycobacteria (31, 40, 42-44). Human MDM, infected or uninfected with BCG, were treated with media, IL-12, IL-18 or IL-12+IL-18 for three days, then culture supernatants were assessed for IFN-γ and TNF-α. Treatment of hMDM by IL-12 and/or IL-18 without BCG infection did not significantly induce secretion of IFN-γ and TNF-α in culture supernatant (data not shown). Similarly, in BCG infected hMDM, IL-18 alone did not induce IFN-γ release, but IL-12 did so moderately in BCG infected macrophages. Co-treatment of BCG-infected hMDM with IL-12+IL-18 led to secretion of significantly higher levels of IFN-γ than the addition of IL-12 alone (Fig. 7A, 444±15 vs 275±21 pg/ml), suggesting additional innate signaling from mycobacteria was required for induction of IFN-γ. In contrast, there was no significant changes in TNF-α secretion (Fig. 7B). Consistently, co-treatment with IL-12+IL-18 up-regulated expression of IFN-γ receptors IFNGR1 and IFNGR2 on the target cells at mRNA level (Fig. S3C). In addition, IL-12+IL-18 together, while facilitating IFN-γ production, led to the induction of significantly higher levels of IL-32 than then treatment with IL-18 alone (Fig. 7C), implicating a possible IL-12+IL-18→IFN-γ→ IL-32 activation axis (31, 42).
FIGURE 7. Activation of the IFN-γ-depended antimycobacterial pathway was required for the IL-12+IL-18 co-signaling-mediated growth inhibition of intracellular mycobacteria in human macrophage.
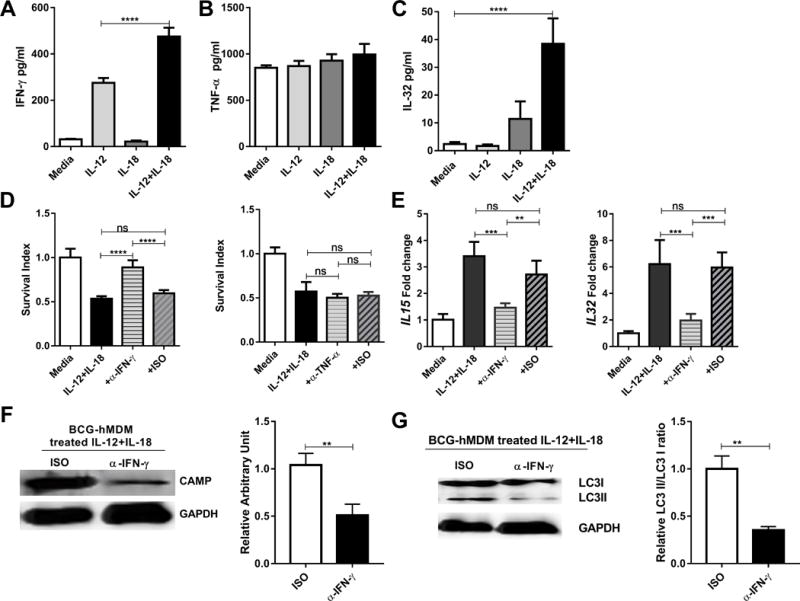
(A-C) show mean cytokine protein concentrations for IFN-γ (A), TNF-α (B) and IL-32 (C) in culture supernatants from BCG-infected hMDM in the presence of media, IL-12, IL-18 or IL-12+IL-18. Culture conditions were the same as those in Fig.1D. Cytokine proteins were measured by ELISA. Data were pooled from three independent experiments using hMDM from 12 healthy uninfected donors. P values are calculated through ANOVA then Tukey’s multiple comparisons test, **** p < 0.0001. (D) shows that IFN-γ blockade (left) but not TNF-α blockade (right) impacts mean survival indexes for BCG bacilli in IL-12+IL-18-treated hMDM. BCG-infected hMDM were treated with media or IL-12+IL-18 in the presence of neutralizing anti-IFN-γ, anti-TNF-α Ab and their isotype controls (5 ug/ml for each), respectively, and then measured for CFU counts and calculated for survival indexes as described above. In the absence of IL-12+IL-18, anti-IFN-γ or anti-TNF-α antibody does not lead to changes in intracellular mycobacterial growth (Supplementary Fig.S3A). Data are derived from 12 healthy donors in 3 independent experiments using hMDM from 12 healthy uninfected donors. P values are calculated through ANOVA then Tukey’s multiple comparisons test, **** p < 0.0001, ns, not significant. (E) shows that IFN-γ blockade-induced abrogation of intracellular BCG growth coincided with decreased expressions of IL-15 and IL-32 in BCG-infected hMDM in comparisons with the IL-12+IL-18 alone without anti-IFN-γ or isotype control. The expression levels of IL15 and IL32 mRNA in BCG infected hMDM were quantified by RT-qPCR. Data are derived from 3 independent experiments using hMDM from 12 healthy uninfected donors. P values are calculated through ANOVA then Tukey’s multiple comparisons test, *** p < 0.001, ** p < 0.01, ns, not significant. (F) The left panel: Representative western blot shows that anti-IFN-γ neutralizing Ab, not isotype control (ISO), reduced the CAMP expression during the IL-12+IL-18 treatment of BCG-infected hMDM cells; the right panel: quantitative analysis of CAMP protein levels by densitometry. Data are derived from 6 replicates. P values are calculated through T test compared with treated with isotype control, ** p<0.01. (G) The left panel: Representative western blot shows that anti-IFN-γ neutralizing Ab, not isotype control (ISO), reduced the conversion of LC3II during the IL-12+IL-18 treatment of BCG-infected hMDM; the right panel: quantitative analysis of relative LC3II/LC3I ratios by densitometry. Data are derived from 6 replicates. P values are calculated through T test compared with treated with isotype control, * p<0.05.
To authenticate that IL-12+IL-18 activation of the IFN-γ pathway contributed to inhibition of intracellular mycobacteria, we blocked IFN-γ or TNF-α with neutralizing monoclonal antibodies. The addition of neutralizing anti-IFN-γ Ab, but not anti-TNF-α Ab, significantly abrogated the IL-12+IL-18-mediated growth inhibition of intracellular mycobacteria in BCG-infected hMDM (Fig. 7D). Consistently, the blockade of IFN-γ also significantly reduced the IL-12+IL-18-mediated induction of IL-15 and IL-32 (Fig. 7E). Moreover, the neutralization of IFN-γ in the IL-12+IL-18 treated BCG infected hMDM resulted in reduced expression of CAMP and less extend of autophagy (Fig. 7 F, G). These data suggested that activation of the IL-12+IL-18 →IFN-γ → IL-15/IL-32 → CAMP → autophagy pathway was required for the IL-12+IL-18-induced growth inhibition of intracellular mycobacteria. Of note, the IL-12+IL-18-activation of the IL-12+IL-18 →IFN-γ → IL-15 → IL-32 pathway did not appear to be negated by type I interferon IFN-β, previously shown to inhibit IFN-γ induced antimicrobial capability (45, 46). The addition of IFN-β enhanced intracellular BCG growth in the infected hMDM in the absence of IL-12+IL-18 treatment, but not in the IL-12+IL-18-co-treated cells (Supplemental Fig. S4).
Discussion
Activation of the innate immune response triggers the release of cytokines which induce inflammation and instruct the adaptive T cell response. Two key innate cytokines induced by mycobacteria and involved in host defense are IL-12 (9-12, 15) and IL-18 (13-15, 47, 48). These cytokines act individually on NK and T cells (10, 16, 47, 49, 50), but together, IL-12 and IL-18 provide a potent stimulus for innate instruction of the adaptive T cell response towards Th1 cell differentiation (16). The current study provides evidence that together, IL-12 and IL-18 trigger anti-mycobacterial responses against intracellular pathogens in innate cells, including macrophage and pulmonary epithelial cells, involving the co-signaling of p38-MAPK and STAT4, activating of VDR-derived CAMP or β-defensin 2, and autophagy. Moreover, the ability of IL-12+IL-18 to induce production of IFN-γ by macrophages was required for upregulation of the antimicrobial activity against Mtb, identifying a common link between the innate and adaptive immune response. Our data provide new insight into the combined role of IL-12+IL-18 in host defense with clinical relevance in the context that polymorphisms of IL-12 and IL-18 are reported in patients with TB (51, 52).
The ability of macrophages to upregulate antimicrobial peptide expression provides one mechanism of innate host defense against intracellular pathogens (29). BCG infection also triggers the activation of microbicidal peptides in A549 epithelial cell line through multiple mechanisms (27, 28). Our data suggest that IL-12+IL-18 treatment indeed induces the up-regulation of the antimicrobial peptide genes, CAMP and DEFB4A. Particularly, the increased patterns of CAMP and autophagy were observed in both lung epithelial cells and human macrophages, suggesting a common mechanism of the IL-12+IL-18-induced antimicrobial response in these two different cell types. Consistent with multiple studies, the increased expression of antimicrobial peptide genes might represent the down-stream effector function of the IL-12+IL-18-induced growth inhibition of intracellular mycobacteria. (32, 53, 54). Consistent with the demonstration that both these antimicrobial peptide genes are regulated by a transcription factor, VDR (30, 36, 54), we found that blockade of the VDR signaling by transduction with shRNA lentivirus affected the IL-12+IL-18-induced microbicidal activity. Furthermore, the requirement of the VDR partly explains the vital role of autophagy in the clearance of intracellular bacteria induced by the combined action of IL-12+IL-18 (55).
Both IL-12 and IL-18 cooperate to activate co-signaling pathways and inhibit the intracellular growth of mycobacteria in target host cells, as the anti-mycobacterial effect cannot be induced by either alone or in the presence of anti-IL-12 or anti-IL-18 neutralizing Abs to the co-treatment culture. Together, IL-12+IL-18 lead to signaling via the p38-MAPK and STAT4 pathways which are required for inhibiting the growth of intracellular mycobacteria, since small molecule inhibition of p38-MAPK or knockdown of STAT4 clearly reduced the antimicrobial effect. Although blocking the p38-MAPK pathway strongly reduced the IL-12+IL-18-induced anti-mycobacterial activity, blocking the MEK-MAPK and NF-κB pathways had no effect. The discrepant requirement of specific MAPK pathways provides new insights into host intracellular pathway-network regulating intracellular pathogens survival (36).
The IL-12+IL-18-induced growth restriction of intracellular bacteria is dependent on VDR-derived CAMP and autophagy. The IL12+IL18 up-regulation of VDR mRNA in THP-1 cells is not as striking as that in A549 or hMDM cells, presumably due to complex cell-type factors. However, knock down of VDR protein by shRNA manipulation consistently abrogates the IL12+IL18-induced activation of antimicrobial peptides and reversed the IL12+IL18-mediated inhibition of BCG growth in both THP-1 and A549 cells. These results suggest that the VDR is required for the IL12+IL18-induced activation of a downstream antimicrobial pathway in both cell types. Autophagy has been shown to facilitate the delivery of antimicrobial peptides to the intracellular compartments containing the pathogen (56). The data in our study using chemical inhibitors emphasize the significant role of autophagy, which is consistent with multiple reports (31, 38, 57-59). Although, caspase-mediated apoptosis has been associated with the suppression of intracellular bacterial growth (36, 60, 61), it has been suggested by other groups that apoptosis may facilitate the spread of mycobacteria during infection in vivo in zebrafish and in a mouse model (62, 63). Our data suggests that IL-12+IL-18-induced antimycobacterial activity does not require the induction of apoptosis in BCG-infected hMDM and A549 cells, indicating that autophagy but not apoptosis contributes to lower bacillary load. Also, this data partly explains the unnecessary role of endogenous production of TNF-α, which is known to induce apoptosis (36).
It is well known that the production of IFN-γ by the adaptive T cell response is necessary for immune protection against TB (64-66). In addition, it has been shown that IFN-γ is produced by cells of the innate immune system in response to IL-12+IL-18, including NK cells (17) and murine macrophages (19, 20), as well as in response to mycobacterial infection in human macrophages (18). A surprising finding of the present study was that the ability of IL-12+IL-18 to induce in macrophages the autonomous production of IFN-γ, a cytokine typically associated with the adaptive T cell response, was required for generation of an innate antimicrobial response. Here, we found that treatment of macrophages with both IL-12 and IL-18 induced the induction of IL-15 and IL-32, activation of the VDR leading to upregulation of antimicrobial peptides known to inhibit the growth of intracellular mycobacteria, the identical pathway induced by exogenous treatment with IFN-γ (31). Evidence for this vital role of IFN-γ was demonstrated by experiments in which neutralizing IFN-γ blockade abrogated the IL-12+IL-18-induced production of IL-15 and IL-32, as well as the antimycobacterial response. These data provide new insight into the mechanism of innate antimicrobial activity against Mtb in human macrophage, delineated as IL-12+IL-18 →IFN-γ → IL-15/IL-32 →antimicrobial peptides and autophagy. Furthermore, the innate and adaptive immune response have both evolved to have distinct functions, yet both have retained the ability to produce IFN-γ to activate the vitamin D-dependent antimicrobial pathway (31). It is noteworthy that the IL-12+IL-18 activation of IFN-γ was not inhibited by IFN-β. This finding suggests that the combined action of IL-12+IL-18 can override the immunosuppressive action of Type I IFNs, which are upregulated as part of the pathogenesis of TB and have been shown to negatively regulate host immune defense against bacterial infection (36, 46, 67). One mechanism by which Type I IFNs downregulate IFN-γ-induced immune responses is via the production of IL-10 (46).
The IL-12+IL-18-induced restriction of mycobacteria in pulmonary epithelial cells does not involve the production of IFN-γ or IL-15 as seen in the setting of mycobacteria-infected macrophages. However, the restriction of intracellular mycobacterial growth in lung epithelial cells still requires both VDR-derived antimicrobial peptides and autophagy, suggesting the existence of an alternate pathway for induction of these host defense responses. Importantly, IL-12+IL-18 co-signaling of p38/STAT4 can activate the VDR, VDR-derived CAMP, and autophagy, leading to inhibition of intracellular mycobacterial growth in both macrophage and pulmonary epithelial cell lineages.
IL-12+IL-18 co-signaling does not apparently up-regulate TNF-α production, and neutralizing anti-TNF-α Ab blockade fails to reduce or abrogate the IL-12+IL-18-mediated growth inhibition of intracellular mycobacteria. Several points might help to explain the lack of a role for TNF-α in the IL-12+IL-18 restriction of intracellular mycobacteria. First, an absence of changes in low pg-scale TNF-α baseline after IL-12+IL-18 treatment implicates that low-level TNF-α does not contribute to the intrinsic function of the IL-12+IL-18-induced growth inhibition of mycobacteria. Virtually, we found that only 50 ng/ml or higher concentrations of exogenous TNF-α were able to reveal TNF-α-mediated inhibition of intracellular mycobacteria (data not shown). Second, in vitro blockade of TNF-α signal might not be efficient enough to uncover up-regulation of intracellular mycobacteria growth. In fact, intracellular mycobacterial growth could not be enhanced by three well-documented TNF-α blockade drugs capable of reactivating latent TB (68). Third, in vitro TNF-α mediated growth inhibition of intracellular mycobacteria appears to require collaboration of granzyme A or other factors produced by T cells, and such cooperative TNF-α inhibition is independent of autophagy (40). In contrast, the IL-12+IL-18–induced inhibition involves VDR induction of cathelicidin, β-defensin 2 and autophagy, without the need for granzyme A.
Whether the IL-12+IL-18-induced anti-microbial pathway is active in vivo appears to be complex and beyond the scope of the current study. Natural Mtb infection of humans certainly induces production of IL-12/IL-18 (10, 14), and it will be interesting to determine whether IL-12/IL-18 rapidly produced upon Mtb exposure can help control infection or even sterilizing immunity. In this natural setting, a much lower concentration of IL-12+IL-18 may only be required for Mtb growth restriction.
Thus, treatment of both macrophage and lung epithelial cells with IL-12+IL-18 induced an antimicrobial response, providing evidence for a mechanism involving the p38-MAPK and STAT4 signaling pathways, the production of antimicrobial peptides cathelicidin as well as enhanced induction of autophagy. Furthermore, IL-12+IL-18 treatment led to the production of IFN-γ, and induction of IL32 and IL15 in human macrophages. Defining this novel mechanism may also provide innovative treatment strategies for blocking further transmission of Mtb and reactivation of TB in the already-infected population.
Supplementary Material
Acknowledgments
We thank other members in Chen Lab for technical support.
This work was supported by the following research grants: The National Key Research and Development Program of China (2016YFA0502204, to Z.W.C.), the National Institutes of Health R01 grants (OD015092/RR13601, HL64560 and HL129887, all to Z.W.C.), and the National Program Project grant (2013ZX10003009-002, to Z.W.C.)
Footnotes
Conflict of Interest Statement
All authors declare no financial or commercial conflict of interest.
References
- 1.Wickremasinghe MI, Thomas LH, Friedland JS. Pulmonary epithelial cells are a source of IL-8 in the response to Mycobacterium tuberculosis: essential role of IL-1 from infected monocytes in a NF-kappa B-dependent network. J Immunol. 1999;163:3936–3947. [PubMed] [Google Scholar]
- 2.Diamond G, Legarda D, Ryan LK. The innate immune response of the respiratory epithelium. Immunol Rev. 2000;173:27–38. doi: 10.1034/j.1600-065x.2000.917304.x. [DOI] [PubMed] [Google Scholar]
- 3.Roy S, Sharma S, Sharma M, Aggarwal R, Bose M. Induction of nitric oxide release from the human alveolar epithelial cell line A549: an in vitro correlate of innate immune response to Mycobacterium tuberculosis. Immunology. 2004;112:471–480. doi: 10.1046/j.1365-2567.2004.01905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chuquimia OD, Petursdottir DH, Rahman MJ, Hartl K, Singh M, Fernandez C. The role of alveolar epithelial cells in initiating and shaping pulmonary immune responses: communication between innate and adaptive immune systems. PloS one. 2012;7:e32125. doi: 10.1371/journal.pone.0032125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chuquimia OD, Petursdottir DH, Periolo N, Fernandez C. Alveolar epithelial cells are critical in protection of the respiratory tract by secretion of factors able to modulate the activity of pulmonary macrophages and directly control bacterial growth. Infect Immun. 2013;81:381–389. doi: 10.1128/IAI.00950-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper AM. Cell-mediated immune responses in tuberculosis. Annu Rev Immunol. 2009;27:393–422. doi: 10.1146/annurev.immunol.021908.132703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stenger S, Modlin RL. T cell mediated immunity to Mycobacterium tuberculosis. Curr Opin Microbiol. 1999;2:89–93. doi: 10.1016/s1369-5274(99)80015-0. [DOI] [PubMed] [Google Scholar]
- 8.Megyeri K, Buzas K, Miczak A, Buzas E, Kovacs L, Seprenyi G, Falus A, Mandi Y. The role of histamine in the intracellular survival of Mycobacterium bovis BCG. Microbes Infect. 2006;8:1035–1044. doi: 10.1016/j.micinf.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 9.Henderson RA, Watkins SC, Flynn JL. Activation of human dendritic cells following infection with Mycobacterium tuberculosis. J Immunol. 1997;159:635–643. [PubMed] [Google Scholar]
- 10.Zhang M, Gately MK, Wang E, Gong J, Wolf SF, Lu S, Modlin RL, Barnes PF. Interleukin 12 at the site of disease in tuberculosis. J Clin Invest. 1994;93:1733–1739. doi: 10.1172/JCI117157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sieling PA, Wang XH, Gately MK, Oliveros JL, McHugh T, Barnes PF, Wolf SF, Golkar L, Yamamura M, Yogi Y, Uyemura K, Rea TH, Modlin RL. IL-12 regulates T helper type 1 cytokine responses in human infectious disease. J Immunol. 1994;153:3639–3647. [PubMed] [Google Scholar]
- 12.Munk ME, Mayer P, Anding P, Feldmann K, Kaufmann SH. Increased numbers of interleukin-12-producing cells in human tuberculosis. Infect Immun. 1996;64:1078–1080. doi: 10.1128/iai.64.3.1078-1080.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fulton SA, Johnsen JM, Wolf SF, Sieburth DS, Boom WH. Interleukin-12 production by human monocytes infected with Mycobacterium tuberculosis: role of phagocytosis. Infect Immun. 1996;64:2523–2531. doi: 10.1128/iai.64.7.2523-2531.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vankayalapati R, Wizel B, Weis SE, Samten B, Girard WM, Barnes PF. Production of interleukin-18 in human tuberculosis. J Infect Dis. 2000;182:234–239. doi: 10.1086/315656. [DOI] [PubMed] [Google Scholar]
- 15.Giacomini E, Iona E, Ferroni L, Miettinen M, Fattorini L, Orefici G, Julkunen I, Coccia EM. Infection of human macrophages and dendritic cells with Mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T cell response. J Immunol. 2001;166:7033–7041. doi: 10.4049/jimmunol.166.12.7033. [DOI] [PubMed] [Google Scholar]
- 16.Tominaga K, Yoshimoto T, Torigoe K, Kurimoto M, Matsui K, Hada T, Okamura H, Nakanishi K. IL-12 synergizes with IL-18 or IL-1beta for IFN-gamma production from human T cells. Int Immunol. 2000;12:151–160. doi: 10.1093/intimm/12.2.151. [DOI] [PubMed] [Google Scholar]
- 17.Fehniger TA, Shah MH, Turner MJ, VanDeusen JB, Whitman SP, Cooper MA, Suzuki K, Wechser M, Goodsaid F, Caligiuri MA. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12: implications for the innate immune response. J Immunol. 1999;162:4511–4520. [PubMed] [Google Scholar]
- 18.Fenton MJ, Vermeulen MW, Kim S, Burdick M, Strieter RM, Kornfeld H. Induction of gamma interferon production in human alveolar macrophages by Mycobacterium tuberculosis. Infect Immun. 1997;65:5149–5156. doi: 10.1128/iai.65.12.5149-5156.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munder M, Mallo M, Eichmann K, Modolell M. Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: A novel pathway of autocrine macrophage activation. J Exp Med. 1998;187:2103–2108. doi: 10.1084/jem.187.12.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schindler H, Lutz MB, Rollinghoff M, Bogdan C. The production of IFN-gamma by IL-12/IL-18-activated macrophages requires STAT4 signaling and is inhibited by IL-4. J Immunol. 2001;166:3075–3082. doi: 10.4049/jimmunol.166.5.3075. [DOI] [PubMed] [Google Scholar]
- 21.Shen H, Wang F, Zeng G, Shen L, Cheng H, Huang D, Wang R, Rong L, Chen ZW. Bis-biguanide dihydrochloride inhibits intracellular replication of M. tuberculosis and controls infection in mice. Sci Rep. 2016;6:32725. doi: 10.1038/srep32725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeng G, Chen CY, Huang D, Yao S, Wang RC, Chen ZW. Membrane-bound IL-22 after de novo production in tuberculosis and anti-Mycobacterium tuberculosis effector function of IL-22+ CD4+ T cells. J Immunol. 2011;187:190–199. doi: 10.4049/jimmunol.1004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu Y, Chen J, Liao H, Zhang Y, Wang H, Li S, Luo Y, Fang D, Li G, Zhou B, Shen L, Chen CY, Huang D, Cai J, Cao K, Jiang L, Zeng G, Chen ZW. Tim-3-expressing CD4+ and CD8+ T cells in human tuberculosis (TB) exhibit polarized effector memory phenotypes and stronger anti-TB effector functions. PLoS Pathog. 2012;8:e1002984. doi: 10.1371/journal.ppat.1002984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma M, Sharma S, Roy S, Varma S, Bose M. Pulmonary epithelial cells are a source of interferon-gamma in response to Mycobacterium tuberculosis infection. Immunol Cell Biol. 2007;85:229–237. doi: 10.1038/sj.icb.7100037. [DOI] [PubMed] [Google Scholar]
- 26.Stokes RW, Doxsee D. The receptor-mediated uptake, survival, replication, and drug sensitivity of Mycobacterium tuberculosis within the macrophage-like cell line THP-1: a comparison with human monocyte-derived macrophages. Cell Immunol. 1999;197:1–9. doi: 10.1006/cimm.1999.1554. [DOI] [PubMed] [Google Scholar]
- 27.Mendez-Samperio P, Miranda E, Trejo A. Expression and secretion of cathelicidin LL-37 in human epithelial cells after infection by Mycobacterium bovis Bacillus Calmette-Guerin. Clin Vaccine Immunol. 2008;15:1450–1455. doi: 10.1128/CVI.00178-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mendez-Samperio P, Miranda E, Trejo A. Regulation of human beta-defensin-2 by Mycobacterium bovis bacillus Calmette-Guerin (BCG): involvement of PKC, JNK, and PI3K in human lung epithelial cell line (A549) Peptides. 2008;29:1657–1663. doi: 10.1016/j.peptides.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 29.Shin DM, Jo EK. Antimicrobial Peptides in Innate Immunity against Mycobacteria. Immune Netw. 2011;11:245–252. doi: 10.4110/in.2011.11.5.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu PT, Schenk M, Walker VP, Dempsey PW, Kanchanapoomi M, Wheelwright M, Vazirnia A, Zhang X, Steinmeyer A, Zugel U, Hollis BW, Cheng G, Modlin RL. Convergence of IL-1beta and VDR activation pathways in human TLR2/1-induced antimicrobial responses. PloS one. 2009;4:e5810. doi: 10.1371/journal.pone.0005810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fabri M, Stenger S, Shin DM, Yuk JM, Liu PT, Realegeno S, Lee HM, Krutzik SR, Schenk M, Sieling PA, Teles R, Montoya D, Iyer SS, Bruns H, Lewinsohn DM, Hollis BW, Hewison M, Adams JS, Steinmeyer A, Zugel U, Cheng G, Jo EK, Bloom BR, Modlin RL. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med. 2011;3:104ra102. doi: 10.1126/scitranslmed.3003045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bloom BR, Modlin RL. Mechanisms of Defense against Intracellular Pathogens Mediated by Human Macrophages. Microbiol Spectr. 2016;4 doi: 10.1128/microbiolspec.MCHD-0006-2015. [DOI] [PubMed] [Google Scholar]
- 33.Lee JK, Kim SH, Lewis EC, Azam T, Reznikov LL, Dinarello CA. Differences in signaling pathways by IL-1beta and IL-18. Proc Natl Acad Sci U S A. 2004;101:8815–8820. doi: 10.1073/pnas.0402800101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chandrasekar B, Mummidi S, Valente AJ, Patel DN, Bailey SR, Freeman GL, Hatano M, Tokuhisa T, Jensen LE. The pro-atherogenic cytokine interleukin-18 induces CXCL16 expression in rat aortic smooth muscle cells via MyD88, interleukin-1 receptor-associated kinase, tumor necrosis factor receptor-associated factor 6, c-Src, phosphatidylinositol 3-kinase, Akt, c-Jun N-terminal kinase, and activator protein-1 signaling. J Biol Chem. 2005;280:26263–26277. doi: 10.1074/jbc.M502586200. [DOI] [PubMed] [Google Scholar]
- 35.Bacon CM, Petricoin EF, 3rd, Ortaldo JR, Rees RC, Larner AC, Johnston JA, O’Shea JJ. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proc Natl Acad Sci U S A. 1995;92:7307–7311. doi: 10.1073/pnas.92.16.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu G, Wang J, Gao GF, Liu CH. Insights into battles between Mycobacterium tuberculosis and macrophages. Protein Cell. 2014;5:728–736. doi: 10.1007/s13238-014-0077-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo XG, Ji TX, Xia Y, Ma YY. Autophagy protects type II alveolar epithelial cells from Mycobacterium tuberculosis infection. Biochem Biophys Res Commun. 2013;432:308–313. doi: 10.1016/j.bbrc.2013.01.111. [DOI] [PubMed] [Google Scholar]
- 38.Fine KL, Metcalfe MG, White E, Virji M, Karls RK, Quinn FD. Involvement of the autophagy pathway in trafficking of Mycobacterium tuberculosis bacilli through cultured human type II epithelial cells. Cell Microbiol. 2012;14:1402–1414. doi: 10.1111/j.1462-5822.2012.01804.x. [DOI] [PubMed] [Google Scholar]
- 39.Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–246. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- 40.Spencer CT, Abate G, Sakala IG, Xia M, Truscott SM, Eickhoff CS, Linn R, Blazevic A, Metkar SS, Peng G, Froelich CJ, Hoft DF. Granzyme A produced by gamma(9)delta(2) T cells induces human macrophages to inhibit growth of an intracellular pathogen. PLoS Pathog. 2013;9:e1003119. doi: 10.1371/journal.ppat.1003119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu H, Fearnhead HO, Cohen GM. An ICE-like protease is a common mediator of apoptosis induced by diverse stimuli in human monocytic THP.1 cells. FEBS Lett. 1995;374:303–308. doi: 10.1016/0014-5793(95)01116-v. [DOI] [PubMed] [Google Scholar]
- 42.Bai X, Dinarello CA, Chan ED. The role of interleukin-32 against tuberculosis. Cytokine. 2015;76:585–587. doi: 10.1016/j.cyto.2015.06.013. [DOI] [PubMed] [Google Scholar]
- 43.Douvas GS, Looker DL, Vatter AE, Crowle AJ. Gamma interferon activates human macrophages to become tumoricidal and leishmanicidal but enhances replication of macrophage-associated mycobacteria. Infect Immun. 1985;50:1–8. doi: 10.1128/iai.50.1.1-8.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar D, Nath L, Kamal MA, Varshney A, Jain A, Singh S, Rao KV. Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell. 2010;140:731–743. doi: 10.1016/j.cell.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 45.Bouchonnet F, Boechat N, Bonay M, Hance AJ. Alpha/beta interferon impairs the ability of human macrophages to control growth of Mycobacterium bovis BCG. Infect Immun. 2002;70:3020–3025. doi: 10.1128/IAI.70.6.3020-3025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teles RM, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, Komisopoulou E, Kelly-Scumpia K, Chun R, Iyer SS, Sarno EN, Rea TH, Hewison M, Adams JS, Popper SJ, Relman DA, Stenger S, Bloom BR, Cheng G, Modlin RL. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science. 2013;339:1448–1453. doi: 10.1126/science.1233665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia VE, Uyemura K, Sieling PA, Ochoa MT, Morita CT, Okamura H, Kurimoto M, Rea TH, Modlin RL. IL-18 promotes type 1 cytokine production from NK cells and T cells in human intracellular infection. J Immunol. 1999;162:6114–6121. [PubMed] [Google Scholar]
- 48.Sugawara I, Yamada H, Kaneko H, Mizuno S, Takeda K, Akira S. Role of interleukin-18 (IL-18) in mycobacterial infection in IL-18-gene-disrupted mice. Infect Immun. 1999;67:2585–2589. doi: 10.1128/iai.67.5.2585-2589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshimoto T, Takeda K, Tanaka T, Ohkusu K, Kashiwamura S, Okamura H, Akira S, Nakanishi K. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: synergism with IL-18 for IFN-gamma production. J Immunol. 1998;161:3400–3407. [PubMed] [Google Scholar]
- 50.Fantuzzi L, Puddu P, Varano B, Del Corno M, Belardelli F, Gessani S. IFN-alpha and IL-18 exert opposite regulatory effects on the IL-12 receptor expression and IL-12-induced IFN-gamma production in mouse macrophages: novel pathways in the regulation of the inflammatory response of macrophages. J Leukoc Biol. 2000;68:707–714. [PubMed] [Google Scholar]
- 51.Thada S, Ponnana M, Sivangala R, Joshi L, Alasandagutti M, Ansari MS, Schumann RR, Valluri V, Gaddam S. Polymorphisms of IFN-gamma (+874A/T) and IL-12 (+1188A/C) in tuberculosis patients and their household contacts in Hyderabad, India. Hum Immunol. 2016;77:559–565. doi: 10.1016/j.humimm.2016.04.016. [DOI] [PubMed] [Google Scholar]
- 52.Han M, Yue J, Lian YY, Zhao YL, Wang HX, Liu LR. Relationship between single nucleotide polymorphism of interleukin-18 and susceptibility to pulmonary tuberculosis in the Chinese Han population. Microbiol Immunol. 2011;55:388–393. doi: 10.1111/j.1348-0421.2011.00332.x. [DOI] [PubMed] [Google Scholar]
- 53.Montoya D, Inkeles MS, Liu PT, Realegeno S, Teles RM, Vaidya P, Munoz MA, Schenk M, Swindell WR, Chun R, Zavala K, Hewison M, Adams JS, Horvath S, Pellegrini M, Bloom BR, Modlin RL. IL-32 is a molecular marker of a host defense network in human tuberculosis. Sci Transl Med. 2014;6:250ra114. doi: 10.1126/scitranslmed.3009546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krutzik SR, Hewison M, Liu PT, Robles JA, Stenger S, Adams JS, Modlin RL. IL-15 links TLR2/1-induced macrophage differentiation to the vitamin D-dependent antimicrobial pathway. J Immunol. 2008;181:7115–7120. doi: 10.4049/jimmunol.181.10.7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin DM, Yuk JM, Lee HM, Lee SH, Son JW, Harding CV, Kim JM, Modlin RL, Jo EK. Mycobacterial lipoprotein activates autophagy via TLR2/1/CD14 and a functional vitamin D receptor signalling. Cell Microbiol. 2010;12:1648–1665. doi: 10.1111/j.1462-5822.2010.01497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alonso S, Pethe K, Russell DG, Purdy GE. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc Natl Acad Sci U S A. 2007;104:6031–6036. doi: 10.1073/pnas.0700036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. 2012;150:803–815. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 59.Klug-Micu GM, Stenger S, Sommer A, Liu PT, Krutzik SR, Modlin RL, Fabri M. CD40 ligand and interferon-gamma induce an antimicrobial response against Mycobacterium tuberculosis in human monocytes. Immunology. 2013;139:121–128. doi: 10.1111/imm.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jayaraman P, Sada-Ovalle I, Nishimura T, Anderson AC, Kuchroo VK, Remold HG, Behar SM. IL-1beta promotes antimicrobial immunity in macrophages by regulating TNFR signaling and caspase-3 activation. J Immunol. 2013;190:4196–4204. doi: 10.4049/jimmunol.1202688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen M, Divangahi M, Gan H, Shin DS, Hong S, Lee DM, Serhan CN, Behar SM, Remold HG. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J Exp Med. 2008;205:2791–2801. doi: 10.1084/jem.20080767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davis JM, Ramakrishnan L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell. 2009;136:37–49. doi: 10.1016/j.cell.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blomgran R, Desvignes L, Briken V, Ernst JD. Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe. 2012;11:81–90. doi: 10.1016/j.chom.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin Immunol. 2014;26:454–470. doi: 10.1016/j.smim.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Du Q, Xie J, Kim HJ, Ma X. Type I interferon: the mediator of bacterial infection-induced necroptosis. Cell Mol Immunol. 2013;10:4–6. doi: 10.1038/cmi.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saliu OY, Sofer C, Stein DS, Schwander SK, Wallis RS. Tumor-necrosis-factor blockers: differential effects on mycobacterial immunity. J Infect Dis. 2006;194:486–492. doi: 10.1086/505430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.