

Abstract
Rationale
Heart failure is characterized by electrical remodeling that contributes to arrhythmic risk. The unfolded protein response (UPR) is active in heart failure and can decrease protein levels by increasing mRNA decay, accelerating protein degradation, and inhibiting protein translation.
Objective
Therefore, we investigated whether the UPR downregulated cardiac ion channels that may contribute to arrhythmogenic electrical remodeling.
Methods
Human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) were used to study cardiac ion channels. Action potentials (APs) and ion channel currents were measured by patch clamp recording. The mRNA and protein levels of channels and the UPR effectors were determined by quantitative RT-PCR and Western blotting. Tunicamycin (TM, 50 ng/mL and 5 μg/mL), GSK2606414 (GSK, 300 nmol/L), and 4μ8C (5 μmol/L) were utilized to activate the UPR, inhibit protein kinase-like ER kinase (PERK) and inositol-requiring protein-1 (IRE1), respectively.
Results
TM-induced activation of the UPR caused significant prolongation of the AP duration (APD) and a reduction of the maximum upstroke velocity (dV/dtmax) of the AP phase 0 in both acute (20-24 h) and chronic treatment (6 days). These changes were explained by reductions in the sodium, L-type calcium, the transient outward and rapidly/slowly activating delayed rectifier potassium currents. Nav1.5, Cav1.2, Kv4.3, and KvLQT1 channels showed concomitant reductions in mRNA and protein levels under activated UPR. Inhibition of PERK or IRE1 shortened the APD and reinstated dV/dtmax. The PERK branch regulated Nav1.5, Kv4.3, hERG, and KvLQT1. The IRE1 branch regulated Nav1.5, hERG, KvLQT1, and Cav1.2.
Conclusions
Activated UPR downregulates all major cardiac ion currents and results in electrical remodeling in hiPSC-CMs. Both PERK and IRE1 branches downregulate Nav1.5, hERG, and KvLQT1. The PERK branch specifically downregulates Kv4.3, while the IRE1 branch downregulates Cav1.2. Therefore, the UPR contributed to electrical remodeling, and targeting the UPR might be anti-arrhythmic.
Keywords: PERK, IRE1, heart failure, hiPSC-derived cardiomyocytes
Introduction
The cardiac action potential (AP) is a complicated event that relies upon highly regulated active and passive ion transport through Na+, K+, Ca2+ and other channels and transporters. The cardiac Na+ channel (Nav1.5) governs phase 0 of depolarization of the AP. The K+ and Ca2+ channels determine the characteristic plateau of phase 2. K+ channels are also responsible for the phases 3 and 4 (the resting membrane potential) of the AP. Any disturbance of these ion channels can alter the delicate balance between depolarizing and repolarizing ionic currents, leading to slow conduction and prolongation of the AP duration (APD) that are observed in human heart failure and animal cardiomyopathic models. For example, Nav1.5 protein and the macroscopic Na+ current (INa) are reduced and contribute to a decreased upstroke velocity (dV/dtmax) of the AP phase 0 in cardiomyopathy [1-5]. Animal model studies reveal reductions in K+ currents including the transient outward (Ito, conducted by rapidly inactivating K+ channel Kv4.3), inward rectifying (IK1, conducted by Kir2.1), and slow delayed rectifying (IKs, conducted by slowly inactivating K+ channel KvLQT1) potassium currents that are responsible for prolonged APD, afterdepolarizations, heterogeneous repolarization patterns and ventricular arrhythmias (Table S1) [6-12]. Nevertheless, the reasons for these changes are unknown, and reversing these changes could represent a new anti-arrhythmic therapy.
The endoplasmic reticulum (ER) is the location for protein translation, folding and assembling before trafficking to the plasma membrane. Recently, we observed that human heart failure is associated with activation of the ER unfolded protein response (UPR), a mechanism that responds to ER protein overload [5]. When under ER stress, glucose-regulated protein/78 kDa (Grp78) dissociates from the three main UPR sensors, protein kinase-like ER kinase (PERK), inositol-requiring protein-1 (IRE1), and activating transcription factor-6α (ATF6α), leading to their activation. Activation of the UPR sensors initiates complicated signal transduction (Fig. 1) to increase protein folding capacity by increasing UPR genes expression and translation (such as chaperone proteins Grp78, Grp94, calreticulin, and GADD34). To reduce ER protein burden, the UPR enhances mRNA decay, inhibits protein translation, and accelerates protein degradation mainly through the PERK and IRE1 branches. The signaling cascades of the three branches have been investigated mainly in neural degeneration diseases and diabetes mellitus [13-16], although a wide variety of cardiovascular diseases have been associated with the UPR activation, such as ischemia/reperfusion, myocardial infarction, hypertension, and heart failure (reviewed in [17-23]). When Grp78 dissociates, PERK and IRE1 oligomerize, become phosphorylated, and induce activation of downstream effectors: phosphorylation of eukaryotic initiation factor 2α (eIF2α) and X-box binding protein 1 (XBP1) splicing, respectively. Phosphorylated eIF2α (p-eIF2α) inhibits ribosomal-mRNA interactions, leading to subsequent mRNA degradation and nascent protein translation reduction. Phosphorylation of eIF2α also enhances gene expression of activating transcription factor 4 (ATF4), which in turn increases the gene expression of chaperone proteins. Spliced XBP1 (sXBP1) degrades mRNA, upregulates gene expression of ER chaperones, and enhances ER-associated protein degradation. The activated form of ATF6α, ATF6N (cleaved N-terminus of ATF6α), translocates to the nucleus to enhance the gene expression of UPR targets such as ER chaperones.
Fig. 1.
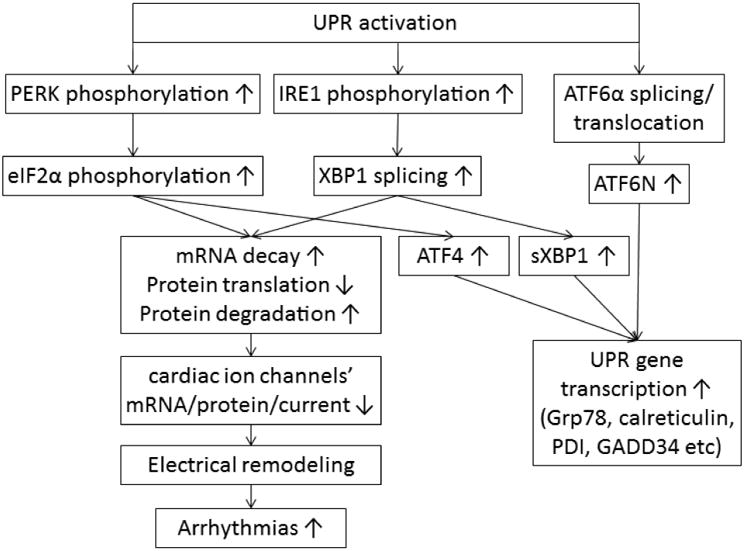
The scheme of the UPR branches signaling cascades and functions and our hypothesis of UPR downregulation on cardiac ion channels, which could induce electrical remodeling and cause increased arrhythmic risk.
Our group has reported that PERK is activated in human heart failure and decreases expression of the Nav1.5 α subunit encoded by SCN5A. This phenomenon contributes significantly to cardiac INa reduction.
This UPR effect is not specific to Nav1.5 and does not affect all ion channels [5]. In this study, we used human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) to investigate whether and how activated UPR regulates major cardiac ion channels and contributes to arrhythmogenic electrical remodeling similar to that observed in heart failure patients. We focused on the PERK and IRE1 branches, since the ATF6α branch is not known to alter mRNA decay, protein translation, or protein degradation.
Materials and Methods
Compounds and reagents were purchased from Sigma (St. Louis, MO) except as stated otherwise. Detailed methods are in the “Supplementary Materials.”
Cell culture
Episomally reprogrammed human induced pluripotent stem cells (ThermoFisher Scientific, Watham, MA) were cultured in fully chemically defined conditions on truncated recombinant human vitronectin (VTN-N; Life Technologies, Carlsbad, CA) in Essential 8 medium (Life Technologies) as colonies. Cells were replated onto Matrigel growth factor reduced basement membrane matrix (ThermoFisher Scientific) for chemically defined cardiac differentiation into hiPSC-CMs, according to previously published methods [24-27]. Although we have considerable experience with human embryonic stem cells and hiPSCs [5,28-33], we recognized that variation in the key reagents can affect results. Therefore, we took several precautions to ensure reliability and repeatability of the cell preparations (see “Supplementary Materials”). After day 30 of differentiation, hiPSC-CMs were considered “mature” and were cultured in maintenance medium (RPMI 1640, B27, 1% pen/strep) for real-time PCR quantification and Western blot of protein [33]. For patch clamp recording, mature cells (30-60 days differentiation) were replated at low density to have single cells on 0.1% gelatin coated coverslips [28]. We also used iCell® Cardiomyocytes2 (Cellular Dynamics, Madison, WI) for patch clamp recording and obtained similar results for action potentials and channel currents.
Activation of the UPR and inhibition of PERK and IRE1
hiPSC-CMs were treated with tunicamycin (TM, 5 μg/mL) [34-36] to activate the UPR. GSK2606414 (GSK, 300 nmol/L, MilliporeSigma, Burlington, MA) or 4-methyl umbelliferone 8-carbalde-hyde (4μ8C, 5 μmol/L, Millipore Sigma) was applied to cells to inhibit the PERK and IRE1 branches, respectively. GSK is an orally available, potent, and selective inhibitor for PERK phosphorylation with IC50 of 0.4 nmol/L [37]. The selective inhibitor for IRE1 RNase activity, 4μ8C, has an IC50 of 62 nmol/L [38]. Isoproterenol (ISO, 1 μmol/L) [39,40] was also used to treat hiPSC-CMs to compare with TM for the extent of UPR activation. All treatments were in a 95% O2/5% CO2 incubator at 37°C for 20-24 h. For chronic UPR effect, we treated hiPSC-CMs with TM at 50 ng/mL [41-44], GSK at 300 nmol/L, and 4μ8C at 5 μmol/L for 6 days, together or respectively and measured action potentials.
Electrophysiology
Channel currents were measured using the whole-cell patch clamp technique in the voltage clamp mode at room temperature, as we have done previously [1,45]. Nifedipine (10 μmol/L) was used to block ICaL when measuring K+ currents [46]. IKr and IKs in hiPSC-CMs were small, and we did not measure them seperately. Because hiPSC-CMs have been reported to express very low levels of Kir2.1 [47,48], we therefore did not measure IK1. APs were recorded under current clamp mode. Standard pulse protocols were used to determine the current-voltage, steady state availability, steady state inactivation, and other gating behaviors appropriate to the channels studied [1,46,49,50]. All electrophysiological measurements were carried out with an Axopatch 200B amplifier driven by a pCLAMP system (Digidata A/D and D/A boards and pCLAMP 9.2, Axon Instruments, Burlingame, CA). Data were analyzed with Clampfit.
Real-Time PCR quantification of mRNA levels
hiPSC-CMs were collected from three wells of 12-well plates for each group as three individual samples for total RNA measurements. Total RNA was isolated from hiPSC-CMs using the RNeasy Mini Kit (Qiagen, Valencia, CA) following the manufacturer's instructions. Primers for the target genes were made by Invitrogen (Thermo Fisher Scientific). The primers sequences are listed in Table S2.
Western blot of proteins
hiPSC-CMs were collected from three wells of 6-well plates for each group as three individual samples for Western blot. TGX Stain-Free™ precast gels (Bio-Rad, Hercules, CA) were used to confirm if the loading control GAPDH was altered under UPR activation by comparing GAPDH with the total protein levels. Protein levels of ion channels and the UPR sensors and effectors were measured by standard Western blot. Antibodies were purchased from Alomone Labs (Jerusalem, Israel), Cell Signaling Technology (Danvers, MA), Proteintech (Rosemont, IL), Santa Cruz Biotechnology (Dallas, TX), or ThermoFisher Scientific. Horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG secondary antibodies were used with dilution of 1:5000. GAPDH was used as a loading control.
Statistical analysis
Values are presented as mean ± SEM. The t test, one-way analysis of variance with post hoc tests of significance, the Tukey's honest significant difference test, and the Fisher exact test for 2 × 2 tables were used when appropriate, and a P value of < 0.05 was considered statistically significant (P=NS indicated no significant difference).
Results
TM and ISO induced UPR activation
UPR activation by TM (5 μg/mL) was confirmed by the elevated activated forms of the UPR effectors (Fig. 2A and Table S3), including Grp78 (1.8±0.3-fold increase over untreated), p-PERK/p-eIF2α/ATF4 for the PERK branch (2.1±0.3-, 2.8±0.2-, and 3.8±0.5-fold increases over untreated, respectively), p-IRE1/sXBP1 for the IRE1 branch (2.6±0.4- and 14.1±5.2-fold increases over untreated, respectively), and ATF6N for the ATF6 branch (1.7±0.2-fold increase over untreated) (P<0.05 vs. untreated for all). Fig. 2B presents representative protein bands of these UPR effectors with and without TM treatment. To evaluate the extent of TM-induced UPR activation, we also treated hiPSC-CMs with ISO (1 μmol/L for 24 h) [39,40], which induced similar UPR activation as shown in Fig. 2C and 2D. By comparing the GAPDH amount with the total protein amount, we verified that the loading control was not altered under UPR activation induced by either TM or ISO.
Fig. 2.
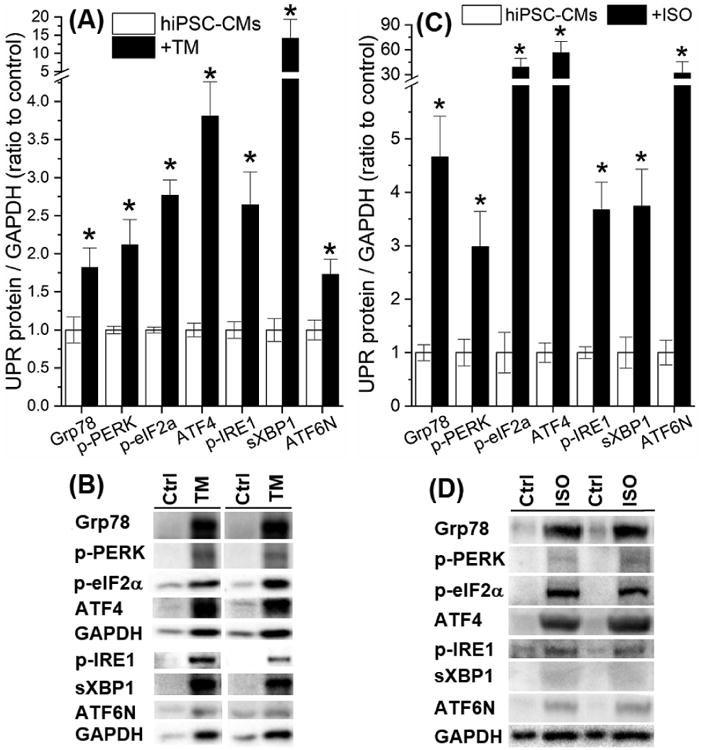
(A) TM (5 μg/mL, 20-24 h at 37 °-C) activated the UPR and elevated protein levels of the activated forms of UPR effectors. (B) The representative Western blot bands of the UPR effectors with or without TM treatment. (C) ISO (1 μmol/L, 24 h at 37 °-C) activated the UPR and elevated protein levels of the activated forms of UPR effects. (D) The representative Western blot bands of the UPR effectors with or without ISO treatment. *P<0.05 vs. the untreated control group.
Activated UPR altered APs and downregulated cardiac ion channels
As shown in Fig. 3A and 3B, TM-induced activation of UPR prolonged the APD and slowed down dV/dtmax. The APD at 90% of repolarization (APD90) was prolonged from 477±23 ms in untreated cells to 1575±154 ms by TM (P<0.05). The dV/dtmax was significantly decreased from 77±4 V/s in untreated myocytes to 66±1 V/s by TM (P<0.05). Therefore, activated UPR induced electrical remodeling similar to that seen in heart failure.[51]
Fig. 3.
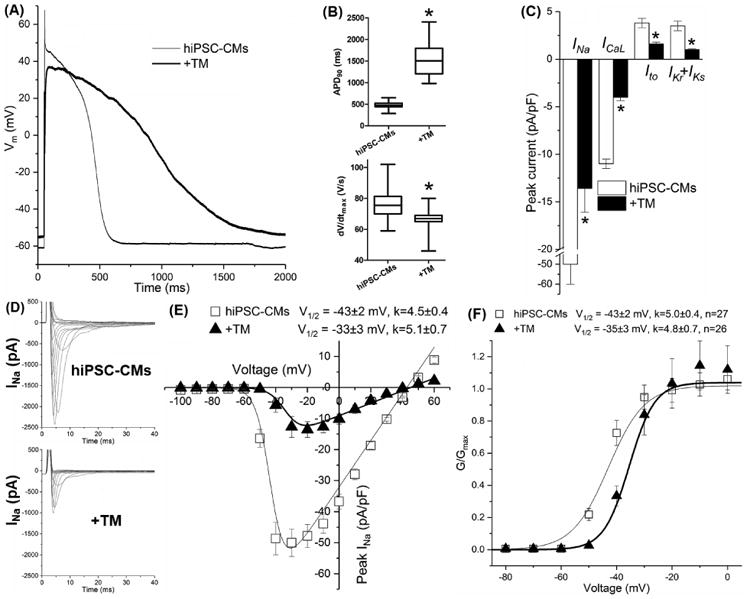
TM treatment altered (A) the morphology of the action potential with (B) prolonged action potential duration (APD90) and decreased dV/dtmax of hiPSC-CMs. TM treatment downregulated (C) all major cardiac ion channel currents and positively shifted (D) the V1/2 value of INa steady state activation. Up to 30 cells were used for data average for each group. *P<0.05 vs the untreated group.
All major cardiac ion channels' currents that contribute to the AP were reduced significantly by UPR activation, including INa, ICaL (conducted by L-type Ca2+ channel Cav1.2), and three prominent K+ currents Ito and IKr+IKs (conducted by hERG and KvLQT1, respectively). The peak currents were reduced to 27±7%, 36±7%, 42±14% and 29±9% of untreated cells, respectively (P<0.05 vs. untreated, Table 1 and Fig. 3C). These current reductions were consistent with the changes of the dV/dtmax and APD.
Table 1.
Whole-cell cardiac ion channel currents and the AP data of hiPSC-CMs were obtained with voltage- and current-clamp recording, respectively, with TM treatment (5 μg/mL, 20-24 h at 37 °C). Currents were measured at different testing membrane potential and normalized to the cell capacitance.
| Testing potential (mV) | Control | TM | ||||
|---|---|---|---|---|---|---|
| pA/pF | n | pA/pF | n | Ratio to control | ||
| INa | -20 | -50±10 | 27 | -13.6±2.5* | 26 | 27±7%* |
| ICaL | +10 | -11±1 | 20 | -4.0±0.7* | 21 | 36±7%* |
| Ito | +50 | 3.8±0.8 | 11 | 1.6±0.4* | 21 | 42±14%* |
| IKs+IKr | +50 | 3.5±0.9 | 11 | 1.0±0.2* | 21 | 29±9%* |
|
| ||||||
| APD90 (ms) | 477±23 | 30 | 1575±154* | 29 | ||
| dV/dtmax (V/s) | 77±4 | 30 | 66±1* | 29 | ||
P<0.05 vs untreated control cells; n, tested number of cells.
With evaluation of the electrophysiological properties of steady state activation (SSA) and inactivation of channel currents, we found that only the V1/2 of INa SSA were positively shifted from -43±2 mV of untreated cells to -33±3 mV by TM (Fig. 3E-3F, P<0.05), perhaps explaining some reduction of INa observed with UPR activation. Steady state inactivation of L-type Ca2+, transient outward K+, and rapid and slow delayed rectifying K+ currents was unaffected.
The mechanism of UPR effects on ion channels
UPR can reduce protein levels by activating mRNA decay, translation inhibition, or protein degradation. In order to understand at which levels the channels were regulated by the UPR, we evaluated the mRNA and protein levels of cardiac ion channels in TM-treated hiPSC-CMs. As shown in Fig. 4, significant concurrent reductions of mRNA and protein levels were observed with SCN5A-Nav1.5, CACNA1C-Cav1.2, KCND3-Kv4.3, KCNH2-hERG, and KCNQ1-KvLQT1 with TM treatment (see Table S4 for values), consistent with the reduction of corresponding currents. However, the mRNA and protein levels of KCNIP2-KChIP2 and KCNA4-Kv1.4 were not affected by TM treatment (Table S4). This indicated a certain specificity of UPR regulation of the cardiac ion channels. Fig. 4C shows representative Western blot protein bands of cardiac ion channels obtained with or without TM present.
Fig. 4.
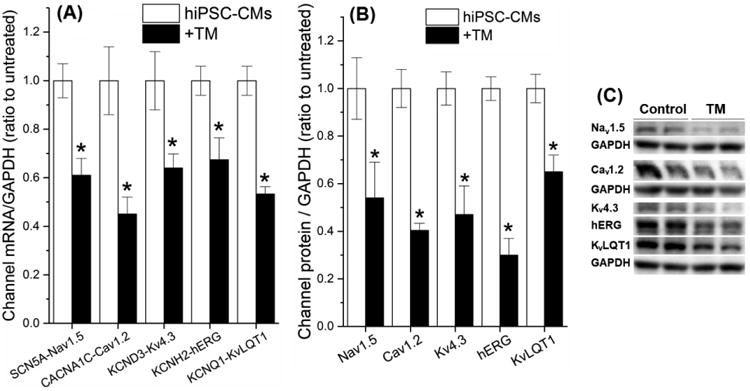
TM-induced UPR activation downregulated cardiac ion channels at the mRNA and protein levels. (A) The mRNA and (B) protein levels of cardiac ion channels of TM-treated hiPSC-CMs were measured by qRT-PCR and Western blot. (C) Representative Western blot bands of the ion channels were present with or without TM treatment. *P<0.05 vs. the untreated control group.
The role of PERK in electrical remodeling
We used a specific PERK inhibitor, GSK to investigate the role of the PERK branch in the UPR-dependent ion channel changes. As shown in Fig. 5, GSK suppressed activation of the PERK branch specifically by decreasing protein levels of p-PERK, p-eIF2α, and ATF4 (1.2±0.2-, 1.7±0.2-, and 2.6±0.4-fold changes of untreated, respectively, P<0.05 vs. the TM group, Table S3). These results were similar to those reported previously [13,37]. GSK did not alter TM-induced elevation of p-IRE1, sXBP1, or ATF6N, confirming GSK specificity for the PERK branch. GSK alone showed no significant effects on the protein level of UPR effectors. Fig. 5B shows representative protein bands of these UPR effectors.
Fig. 5.
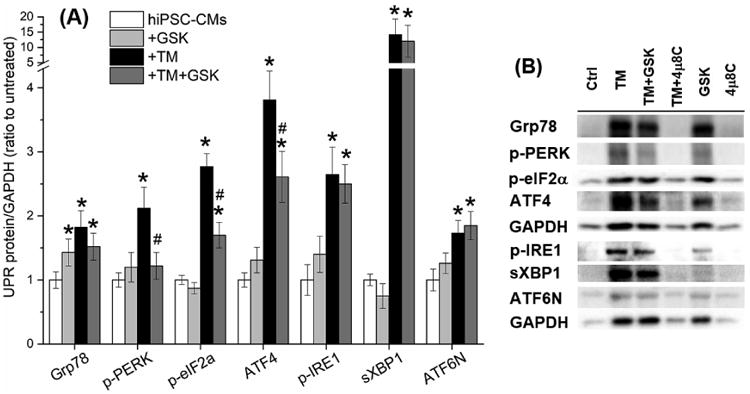
GSK specifically inhibited the PERK branch of the UPR. (A) GSK suppressed the elevation of the PERK branch protein levels induced by TM treatment, while showed no effect on the IRE1 and ATF6α branches. (B) Representative Western blot bands of the activated UPR effectors were present with or without TM, GSK, and 4μ8C treatment. *P<0.05 vs. the untreated control group; #P<0.05 vs. the TM group.
As shown in Fig. 6 and Table S5, GSK shortened TM-induced APD prolongation (APD90: 627±20 ms of the TM+GSK group, #P<0.05 vs. 1575±154 ms of the TM group, and *P<0.05 vs. 477±23 ms of the untreated group) and prevented completely the TM-induced reduction of dV/dtmax (79±3 V/s of the TM+GSK group, #P<0.05 vs. 66±1 V/s of the TM group, and P=NS vs. 77±4 V/s of the untreated group). As shown in Fig. 6C-6F and Table S5, GSK co-application partially recovered INa (56±13% of the untreated, P<0.05 vs. untreated or TM) and shifted the V1/2 of INa SSA to -38±3 mV, compared to -33±3 mV for the TM only group (P<0.05 vs. TM). GSK restored Ito completely (84±16% of the untreated, P<0.05 vs. TM and P=NS vs untreated) and IKr+IKs partially (54±27% of the untreated, P<0.05 vs. untreated or TM), while the TM-induced ICaL reduction was not affected (TM+GSK: 25±6% of the untreated, P<0.05 vs. the untreated, and P=NS vs. TM; Table S5). These changes help explain the partially corrected APD. The relation of dV/dtmax to peak INa is nonlinear [52], and the dV/dtmax overestimates Na+ channel availability. Therefore, fully recovered dV/dtmax may not indicate fully recovered INa, which is what we observed. GSK alone did not show significant effects on the currents or the AP.
Fig. 6.
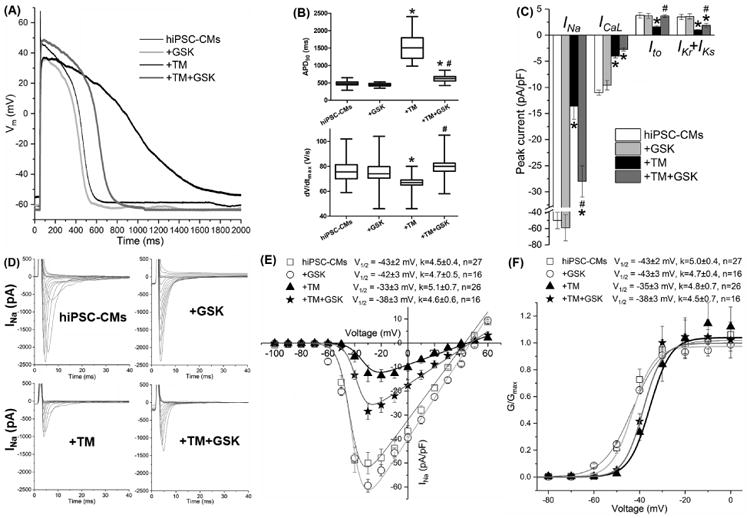
PERK inhibition by GSK restored selective channel currents partially, shortened the APD, and completely reinstated dV/dtmax that were altered by TM in hiPSC-CMs. (A) Representative traces of the action potentials obtained with or without TM and GSK treatment. Untreated hiPSC-CMs, black thin line; +GSK, light gray line; +TM, black bold line; +TM+GSK, dark gray bold line. (B) The APD90 and dV/dtmax values of the action potentials and (C) whole-cell cardiac ion channel currents of hiPSC-CMs were obtained with patch clamp recording with treatments of TM and GSK. Currents were measured at different testing membrane potentials (as listed in Table 1) and normalized to the cell capacitances. (D) The TM-induced positive shift of INa steady state activation V1/2 was partially reinstated by GSK. Up to 30 cells were used for data average for each group. *P<0.05 vs. the untreated group; #P<0.05 vs. the TM group.
The role of IRE1 in electrical remodeling
To distinguish the IRE1 effects on cardiac ion channels, we inhibited the IRE1 branch with a specific inhibitor, 4μ8C (5 μmol/L, 20-24 h at 37 °C) [38]. As shown in Fig. 7 and Table S3, 4μ8C inhibited TM-induced activation of the IRE1 branch by decreasing protein levels of p-IRE1 and sXBP1 (TM+4μ8C: 1.6±0.3- and 1.2±0.4-fold changes of the untreated cells, respectively, P<0.05 vs. the TM group, Table S3). Inhibition was also observed with Grp78 (0.9±0.3-fold increase with the TM+4μ8C vs. 1.8±0.3-fold increase with the TM group, P<0.05). These data confirmed the specificity of 4μ8C for the IRE1 branch. Fig. 7B shows representative protein bands of the UPR effectors.
Fig. 7.
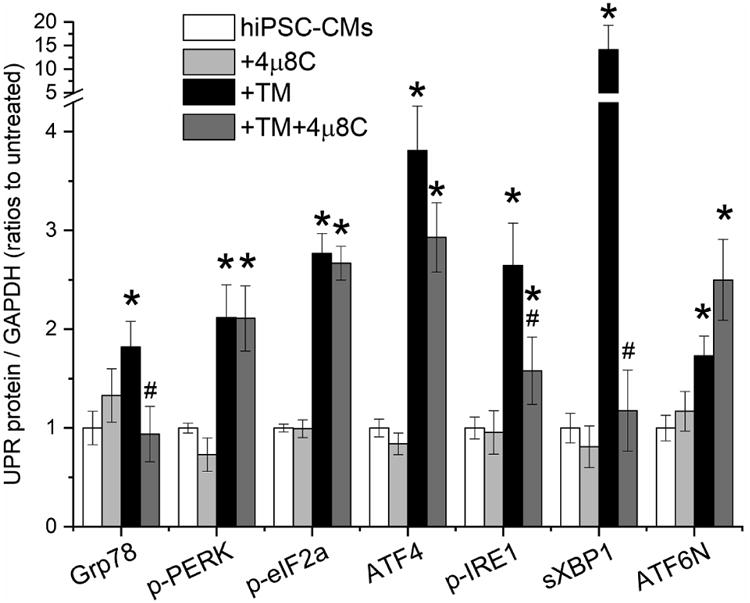
The IRE1 branch of the UPR was specifically inhibited by 4μ8C, which suppressed the elevation of the IRE1 branch protein levels of p-IRE1 and sXBP1 induced by TM treatment, while showed no significant effects on the PERK and ATF6α branches. *P<0.05 vs. the untreated control group; #P<0.05 vs. the TM group.
IRE1 inhibition by 4μ8C shortened TM-induced APD prolongation (Fig. 8 and Table S5, APD90: 1166±75 ms of the TM+4μ8C group, #P<0.05 vs. 1575±154 ms of the TM group, and *P<0.05 vs. 477±23 ms of the untreated group). TM-induced reduction in dV/dtmax was completely reversed by 4μ8C (74±5 V/s of the TM+4μ8C group, #P<0.05 vs. 66±1 V/s of the TM group; P=NS vs. 77±4 V/s of the untreated group). As shown in Fig. 8C and Table S5, 4μ8C restored INa completely (100±21% of the untreated, #P<0.05 vs. the TM group, P=NS vs. the untreated group), and ICaL and IKr+IKs partially (50±6% and 43±11% of the untreated, P<0.05 vs the untreated or TM group). As shown in Fig. 8E-8F, the TM-induced positive shift of V1/2 of INa SSA (-33±3 mV) was restored fully to -45±5 mV with TM+4μ8C (P<0.05 vs. the TM group and P=NS vs. the untreated group). IRE1 inhibition by 4μ8C alone prolonged the APD (APD90: 750±12 ms of the 4μ8C group, P<0.05 vs. 477±23 ms of the untreated group) and decreased ICaL and IKr+IKs (72±9% and 66±18% of the untreated group, P<0.05). This indicated that the IRE1 branch was important to maintain Cav1.2 and hERG/KvLQT1 channel function under control conditions.
Fig. 8.
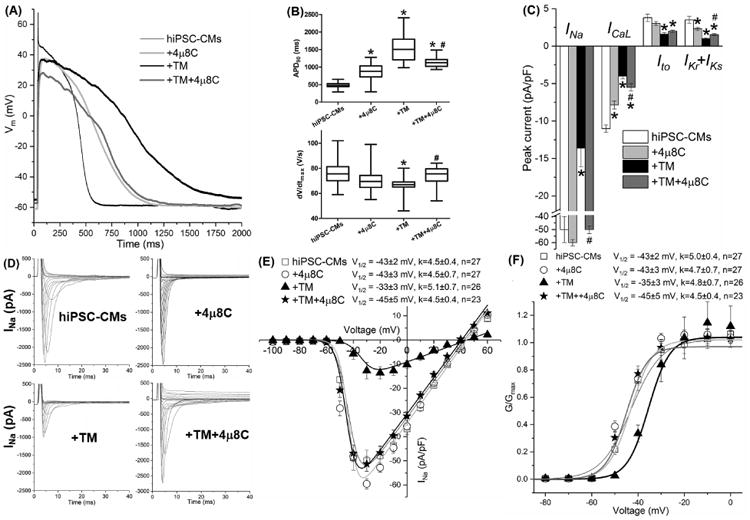
IRE1 inhibition by 4μ8C shorted the APD partially and restored dV/dtmax completely that were altered by TM in hiPSC-CMs. (A) Representative traces of the action potentials obtained with or without TM and 4μ8C treatment. Untreated hiPSC-CMs, black thin line; +4μ8C, light gray line; +TM, black bold line; +TM+4μ8C, dark gray bold line. (B) The APD90 and dV/dtmax values of the action potentials and (C) whole-cell cardiac ion channel currents of hiPSC-CMs were obtained with patch clamp recording with treatments of TM and 4μ8C. Currents were measured at different testing membrane potentials (as listed in Table 1) and normalized to the cell capacitances. (D) The TM-induced positive shift of INa steady state activation V1/2 was completely reinstated by 4μ8C. Up to 30 cells were used for data average for each group. *P<0.05 vs. the untreated control group; #P<0.05 vs. the TM group.
Comparison of the acute and chronic UPR effects on cardiomyocyte action potentials
To investigate the chronic effects of the UPR on cardiomyocytes, we treated hiPSC-CMs with TM (50 ng/mL) with or without GSK (300 nmol/L) or 4μ8C (5 μmol/L) for 6 days. As shown in Fig. 9A and 9B, chronic treatment of TM induced APD prolongation and dV/dtmax reduction, similar to the acute TM treatment (5 μg/mL, 20-24 h). GSK or 4μ8C shortened the APD in the chronic treatment (Fig. 9A), similar to their acute effects (Fig. 9C). GSK or 4μ8C increased dV/dtmax similarly in the chronic or acute settings (Fig. 9B or Fig. 9D, respectively).
Fig. 9.
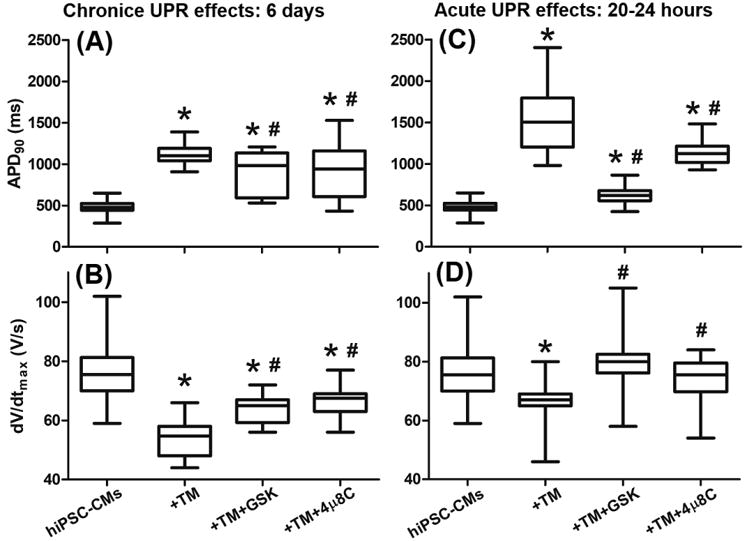
Comparison of the effects of chronic and acute UPR activation on the APD90 and dV/dtmax of hiPSC-CMs. Chronic UPR activation induced by TM (50 ng/mL for 6 days) resulted in (A) APD prolongation and (B) dV/dtmax reduction, which were partially reversed by co-application of GSK or 4μ8C for 6 days. These changes are similar to (C) and (D) the acute UPR effects on hiPSC-CMs. For each group, 27 to 32 cells were tested for data average. *P<0.05 vs. the untreated control group; #P<0.05 vs. the TM group.
Discussion
Short-term activation of the unfolded protein response (UPR) triggers adaptive adjustments and promotes cell survival, while long-term and severe UPR activation triggers cell apoptosis. Activated UPR has been reported in heart diseases such as ischemia/reperfusion [53-56], dilated cardiomyopathy [57], atherosclerosis [18,19,58,59], myocardial infarction [34,60-63], hypertension [64-67], diabetic cardiomyopathy [68-70], and heart failure [5,21,51,65,71-73] with increased expression of ER chaperones (Grp78 and calreticulin [57,65,74]) and effectors from all three UPR branches [5,57,63,73,74]. In this study, tunicamycin (TM) activated all three branches of the UPR in hiPSC-CMs in both acute (20-24 h) and chronic (6 days) treatment, and UPR activation resulted in reduced current levels of all major cardiac ion channels: Nav1.5, Cav1.2, Kv4.3, hERG, and KvLQT1. These changes explained UPR-mediated dV/dtmx reduction and APD prolongation. These observations are consistent with the ion channel changes seen in cardiomyopathy (Table S1). Since these electrical remodeling changes are thought to underlie part of the arrhythmic risk in cardiomyopathy, we further investigated which branches of the UPR regulated these changes.
The specific inhibitor of the PERK branch, GSK, partially reversed UPR-induced electrical remodeling. This incomplete effect may represent the fact that GSK showed only partial inhibition of the PERK branch. There was PERK-dependent regulation of Nav1.5, Kv4.3, hERG, and KvLQT1 but not Cav1.2 (Fig. 9). This suggests that PERK-mediated channel downregulation is specific for a certain set of channels. The determinants of this specificity are unknown currently. UPR induced a positive shift of the V1/2 of INa SSA that was reversed partially by GSK (Fig. 6D), suggesting that the UPR activation resulted in changes in post-translational modifications of the channel or possibly changes in associated subunits.
IRE1 inhibition with 4μ8C shortened the APD prolongation and restored the reduction of dV/dtmax. Inhibition of IRE1 showed specific regulation of Nav1.5, Cav1.2, hERG, and KvLQT1 but not of Kv4.3 (Fig. 9). These data imply some cross talk between branches of UPR. Application of 4μ8C alone decreased ICaL and IKr+IKs and prolonged the APD (Fig. 8), indicating that certain IRE1 activity may be necessary to maintain these channels under physiological conditions.
Limitations of this study include the fact that, although similar, TM may not recapitulate all the UPR changes seen in cardiomyopathy. Nevertheless, the TM-induced changes were similar to the electrical remodeling reported in various forms of heart disease. In this work, TM induced elevation of UPR effectors in hiPSC-CMs by ∼2- to 4-fold for phospho-PERK, phospho-eIF2α, ATF4, phospho-IRE1 and ATF6N, and ∼14-fold for sXBP1 (Fig. 2). In human HF, phospho-PERK and phospho-eIF2α have been reported to be increased by ∼2.4- and 4.5-fold, respectively [75]. Significant elevation of sXBP1 (5.2±0.5-fold) is reported in failing human heart tissue [74]. In ischemic mouse heart [63], phospho-PERK and phospho-eIF2α were increased ∼3- and 1.5-fold, respectively and ATF6N was significantly increased ∼9-to 12-fold as well. These elevations of UPR effectors in human and animal models are comparable to our observation with TM-treated hiPSC-CMs. We also used ISO to treat the cells and observed similar UPR activation (Fig. 2), suggesting that our observation would be relevant to human disease. While hiPSC-CMs do not fully recapitulate all aspects of adult myocyte electrophysiology, they do exhibit a human-like profile of ion channels, transporters, and currents, excepting Kir2.1 and IK1 which are below native cardiomyocyte levels.[76] Nevertheless, hiPSC-CMs represent an accepted model for drug screening for electrophysiological effects and are used for the first time in the current study to evaluate the UPR. Both IRE1 and PERK inhibition showed partial reversal of electrical remodeling, suggesting that ATF6 may play a role in electrical remodeling or that there are overlapping effects of the UPR branches. Finally, it is possible that the UPR effects are chamber specific, necessitating further studies to fully understand the role of UPR in arrhythmogenesis.
In summary, our study showed that activated UPR downregulated all major cardiac ion currents and resulted in electrical remodeling in hiPSC-CMs. Activated PERK and IRE1 branches showed distinct patterns of downregulation of cardiac ion channels. Electrical remodeling could be partially reversed by inhibition of either the PERK or IRE1 branch. Our study suggests that UPR inhibition may be anti-arrhythmic in cardiomyopathic states.
Supplementary Material
Fig. 10.
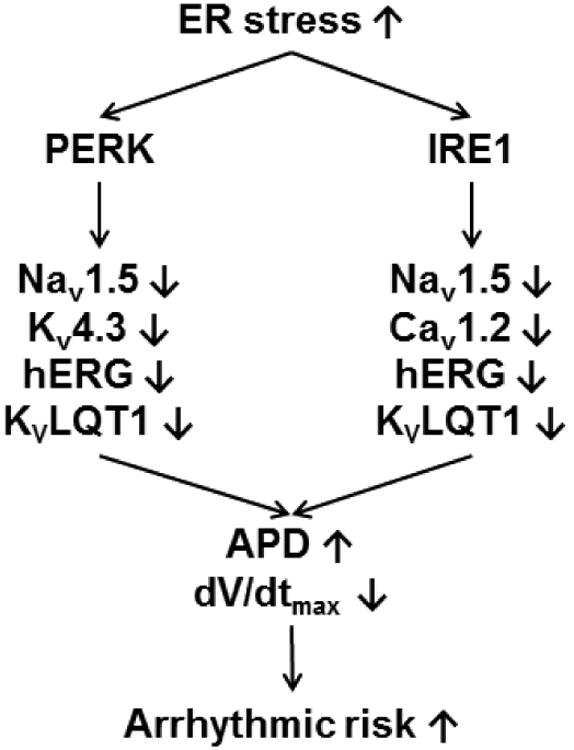
A summarized scheme of the UPR regulation on human cardiac ion channels. Activated UPR downregulates selective ion channels, leads to prolonged APD and reduced dV/dtmax, which can contribute to electrical remodeling and arrhythmias. The PERK branch downregulates Nav1.5, Kv4.3, hERG, and KvLQT1, while the IRE1 branch downregulates Nav1.5, Cav1.2, hERG, and KvLQT1.
Highlights.
The unfolded protein response mediates arrhythmic electrical remodeling
Activated unfolded protein response downregulates all major cardiac ion currents
Electrical remodeling results in action potential prolongation
Electrical remodeling can be attenuated by inhibiting the unfolded protein response
Acknowledgments
None.
Sources of funding: This work was supported by Rhode Island Foundation grant 20154145 (ML) and R01 HL104025 (SCD).
Footnotes
Disclosures: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liu M, Gu L, Sulkin MS, Liu H, Jeong EM, Greener I, Xie A, Efimov IR, Dudley SC., Jr Mitochondrial dysfunction causing cardiac sodium channel downregulation in cardiomyopathy. J Mol Cell Cardiol. 2013;54:25–34. doi: 10.1016/j.yjmcc.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pu J, Boyden PA. Alterations of Na+ currents in myocytes from epicardial border zone of the infarcted heart: a possible ionic mechanism for reduced excitability and postre polarization refractoriness. Circ Res. 1997;81:110–119. doi: 10.1161/01.res.81.1.110. [DOI] [PubMed] [Google Scholar]
- 3.Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–483. doi: 10.1016/j.yjmcc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Ufret-Vincenty CA, Baro DJ, Lederer WJ, Rockman HA, Quinones LE, Santana LF. Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. J Biol Chem. 2001;276:28197–28203. doi: 10.1074/jbc.M102548200. [DOI] [PubMed] [Google Scholar]
- 5.Gao G, Xie A, Zhang J, Herman AM, Jeong EM, Gu L, Liu M, Yang KC, Kamp TJ, Dudley SC. Unfolded protein response regulates cardiac sodium current in systolic human heart failure. Circ Arrhythm Electrophysiol. 2013;6:1018–24. doi: 10.1161/CIRCEP.113.000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valenzeno DP, Tarr M. Calcium as a modulator of photosensitized killing of H9c2 cardiac cells. Photochem Photobiol. 2001;74:605–610. doi: 10.1562/0031-8655(2001)074<0605:caamop>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 7.Sah R, Ramirez RJ, Oudit GY, Gidrewicz D, Trivieri MG, Zobel C, Backx PH. Regulation of cardiac excitation-contraction coupling by action potential repolarization: role of the transient outward potassium current (Ito) J Physiol. 2003;546:5–18. doi: 10.1113/jphysiol.2002.026468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosati B, McKinnon D. Regulation of ion channel expression. Circ Res. 2004;94:874–883. doi: 10.1161/01.RES.0000124921.81025.1F. [DOI] [PubMed] [Google Scholar]
- 9.Scholz EP, Welke F, Joss N, Seyler C, Zhang W, Scherer D, Völkers M, Bloehs R, Thomas D, Katus HA, Karle CA, Zitron E. Central role of PKCα in isoenzyme-selective regulation of cardiac transient outward current Ito and Kv4.3 channels. J Mol Cell Cardiol. 2011;51:722–729. doi: 10.1016/j.yjmcc.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 10.Janse MJ. Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovasc Res. 2004;61:208–217. doi: 10.1016/j.cardiores.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 11.Li GR, Lau CP, Ducharme A, Tardif JC, Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol. 2002;283:H1031–H1041. doi: 10.1152/ajpheart.00105.2002. [DOI] [PubMed] [Google Scholar]
- 12.Nuss HB, Kaab S, Kass DA, Tomaselli GF, Marban E. Cellular basis of ventricular arrhythmias and abnormal automaticity in heart failure. Am J Physiol. 1999;277:H80–H91. doi: 10.1152/ajpheart.1999.277.1.H80. [DOI] [PubMed] [Google Scholar]
- 13.Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, Mallucci GR. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med. 2013;5:206ra138–206ra138. doi: 10.1126/scitranslmed.3006767. [DOI] [PubMed] [Google Scholar]
- 14.Li Z, Zhang T, Dai H, Liu G, Wang H, Sun Y, Zhang Y, Ge Z. Involvement of endoplasmic reticulum stress in myocardial apoptosis of streptozocin-induced diabetic rats. J Clin Biochem Nutr. 2007;41:58–67. doi: 10.3164/jcbn.2007008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang P, Mc Grath B, Li Sa, Frank A, Zambito F, Reinert J, Gannon M, Ma K, Mc Naughton K, Cavener DR. The PERK eukaryotic initiation factor 2α kinase is required for the development of the skeletal systme, postnatal growth and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang L, Zhao D, Ren J, Yang J. Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1852:209–218. doi: 10.1016/j.bbadis.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 17.Glembotski CC. The role of the unfolded protein response in the heart. J Mol Cell Cardiol. 2008;44:453–459. doi: 10.1016/j.yjmcc.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res. 2010;107:1071–1082. doi: 10.1161/CIRCRESAHA.110.227819. [DOI] [PubMed] [Google Scholar]
- 19.Scull C, Tabas I. Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:2792–2797. doi: 10.1161/ATVBAHA.111.224881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hasty AH, Harrison DG. Endoplasmic reticulum stress and hypertension - a new paradigm? J Clin Invest. 2012;122:3859–3861. doi: 10.1172/JCI65173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu M, Dudley SC. Targeting the unfolded protein response in heart diseases. Expert Opin Ther Targets. 2014;18:719–723. doi: 10.1517/14728222.2014.918605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu M, Dudley SC., Jr Role for the unfolded protein response in heart disease and cardiac arrhythmias. Int J Mol Sci. 2016;17:52. doi: 10.3390/ijms17010052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang S, Binder P, Fang Q, Wang Z, Xiao W, Liu W, Wang X. Endoplasmic reticulum stress in the heart: insights into mechanisms and drug targets. Br J Pharmacol. 2017 doi: 10.1111/bph.13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, Plews JR, Abilez OJ, Cui B, Gold JD, Wu JC. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11:855–860. doi: 10.1038/nmeth.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lian X, Zhang J, Azarin S, Zhu K, Hazeltine L, Bao X, Hsiao C, Kamp TJ, Palecek SP. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat Protoc. 2013;8:162–175. doi: 10.1038/nprot.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y, Egashira T, Seki T, Muraoka N, Yamakawa H, Ohgino Y, Tanaka T, Yoichi M, Yuasa S, Murata M, Suematsu M, Fukuda K. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell. 2013;12:127–137. doi: 10.1016/j.stem.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 27.Rupert CE, Coulombe KLK. IGF1 and NRG1 enhance proliferation, metabolic maturity, and the force-frequency response in hESC-derived engineered cardiac tissues. Stem Cells Int. 2017;2017:7648409. doi: 10.1155/2017/7648409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao G, Xie A, Huang SC, Zhou A, Zhang J, Herman AM, Ghassemzadeh S, Jeong EM, Kasturirangan S, Raicu M, Sobieski MA, Bhat G, Tatooles A, Benz EJ, Kamp TJ, Dudley SC. Role of RBM25/LUC7L3 in abnormal cardiac sodium channel splicing regulation in human heart failure/clinical perspective. Circulation. 2011;124:1124–1131. doi: 10.1161/CIRCULATIONAHA.111.044495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang YM, Hartzell C, Narlow M, Dudley SC., Jr Stem cell-derived cardiomyocytes demonstrate arrhythmic potential. Circulation. 2002;106:1294–1299. doi: 10.1161/01.cir.0000027585.05868.67. [DOI] [PubMed] [Google Scholar]
- 30.Zhang YM, Shang L, Hartzell C, Narlow M, Cribbs L, Dudley SC. Characterization and regulation of T-type Ca2+ channels in embryonic stem cell-derived cardiomyocytes. Am J Physiol Heart Circ Physiol. 2003;285:H2770–H2779. doi: 10.1152/ajpheart.01114.2002. [DOI] [PubMed] [Google Scholar]
- 31.Kreutziger KL, Muskheli V, Johnson P, Braun K, Wight TN, Murry CE. Developing vasculature and stroma in engineered human myocardium. Tissue Eng Part A. 2011;17:1219–1228. doi: 10.1089/ten.tea.2010.0557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerbin KA, Yang X, Murry CE, Coulombe KLK. Enhanced electrical integration of engineered human myocardium via intramyocardial versus epicardial delivery in infarcted rat hearts. PLoS One. 2015;10:e0131446. doi: 10.1371/journal.pone.0131446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rupert CE, Chang HH, Coulombe KLK. Hypertrophy changes 3D shape of hiPSC-cardiomyocytes: implications for cellular maturation in regenerative medicine. Cell Mol Bioeng. 2017;10:54–62. doi: 10.1007/s12195-016-0462-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res. 2006;99:275–282. doi: 10.1161/01.RES.0000233317.70421.03. [DOI] [PubMed] [Google Scholar]
- 35.Hammadi M, Oulidi A, Gackière F, Katsogiannou M, Slomianny C, Roudbaraki M, Dewailly E, Delcourt P, Lepage G, Lotteau S, Ducreux S, Prevarskaya N, Van Coppenolle F. Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: involvement of GRP78. FASEB J. 2013;27:1600–1609. doi: 10.1096/fj.12-218875. [DOI] [PubMed] [Google Scholar]
- 36.Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KKH, Wilson C, Okreglak V, Ashkenazi A, Hann B, Nader K, Arkin MR, Renslo AR, Sonenberg N, Walter P, Ron D. Pharmacological brake-release of mRNA translation enhances cognitive memory. ELife. 2013;2:e00498. doi: 10.7554/eLife.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Axten JM, Medina J, Feng Y, Shu A, Romeril SP, Grant SW, Li WHH, Heerding DA, Minthorn E, Mencken T, Atkins C, Liu Q, Rabindran S, Kumar R, Hong X, Goetz A, Stanley T, Taylor JD, Sigethy SD, Tomberlin GH, Hassell AM, Kahler KM, Shewchuk LM, Gampe RT. Discovery of 7-Methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) J Med Chem. 2012;55:7193–7207. doi: 10.1021/jm300713s. [DOI] [PubMed] [Google Scholar]
- 38.Cross BCS, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D, Harding HP. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci USA. 2012;109:E869–E878. doi: 10.1073/pnas.1115623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu ZF, Zhang X, Qiao YX, Xu WQ, Ma CT, Gu HL, Zhou XM, Shi L, Cui CX, Xia D, Chen YG. Neuroglobin protects cardiomyocytes against apoptosis and cardiac hypertrophy induced by isoproterenol in rats. Int J Clin Exp Med. 2015;8:5351–5360. [PMC free article] [PubMed] [Google Scholar]
- 40.Lundy SD, Zhu WZ, Regnier M, Laflamme MA. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev. 2013;22:1991–2002. doi: 10.1089/scd.2012.0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamada J, Onuma H, Ochi F, Hirai H, Takemoto K, Miyoshi A, Matsushita M, Kadota Y, Ohashi J, Kawamura R, Takata Y, Nishida W, Hashida S, Ishii E, Osawa H. Endoplasmic reticulum stress induced by tunicamycin increases resistin messenger ribonucleic acid through the pancreatic endoplasmic reticulum eukaryotic initiation factor 2α kinase–activating transcription factor 4–CAAT/enhancer binding protein-α homologous protein pathway in THP-1 human monocytes. J Diabetes Investig. 2016;7:312–323. doi: 10.1111/jdi.12434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rutkowski D, Arnold S, Miller C, Wu J, Li J, Gunnison K, Mori K, Sadighi Akha A, Raden D, Kaufman R. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nickson P, Toth A, Erhardt P. PUMA is critical for neonatal cardiomyocyte apoptosis induced by endoplasmic reticulum stress. Cardiovasc Res. 2007;73:48–56. doi: 10.1016/j.cardiores.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu J, Rutkowski DT, Swathirajan J, Sauners T, Song B, Yau GDY, Kaufman RJ. ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13:351–364. doi: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 45.Liu M, Sanyal S, Gao G, Gurung IS, Zhu X, Gaconnet G, Kerchner LJ, Shang LL, Huang CLH, Grace A, London B, Dudley SC., Jr Cardiac Na+ current regulation by pyridine nucleotides. Circ Res. 2009;105:737–745. doi: 10.1161/CIRCRESAHA.109.197277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brunner M, Peng X, Liu G, Ren X, Ziv O, Choi B, Mathur R, Hajjiri M, Odening K, Steinberg E, Folco E, Pringa E, Centracchio J, Macharzina R, Donahay T, Schofield L, Rana N, Kirk M, Mitchell G, Poppas A, Zehender M, Koren G. Mechanisms of cardiac arrhythmias and sudden death in transfenic rabbits with long QT syndrome. J Clin Invest. 2008;118:2246–2259. doi: 10.1172/JCI33578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lieu DK, Fu JD, Chiamvimonvat N, Tung KC, McNerney GP, Huser T, Keller G, Kong CW, Li RA. Mechanism-based facilitated maturation of human pluripotent stem cell–derived cardiomyocytes. Circ Arrhythm Electrophysiol. 2013;6:191–201. doi: 10.1161/CIRCEP.111.973420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bett GCL, Kaplan AD, Lis A, Cimato TR, Tzanakakis ES, Zhou Q, Morales MJ, Rasmusson RL. Electronic “expression” of the inward rectifier in cardiocytes derived from human-induced pluripotent stem cells. Heart Rhythm. 2013;10:1903–1910. doi: 10.1016/j.hrthm.2013.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.El Gebeily G, Fiset C. 4-Hydroxytamoxifen inhibits K+ currents in mouse ventricular myocytes. Eur J Pharmacol. 2010;629:96–103. doi: 10.1016/j.ejphar.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 50.Liu GX, Choi BR, Ziv O, Li W, de Lange E, Qu Z, Koren G. Differential conditions for early after-depolarizations and triggered activity in cardiomyocytes derived from transgenic LQT1 and LQT2 rabbits. J Physiol. 2012;590:1171–1180. doi: 10.1113/jphysiol.2011.218164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nattel S, Maguy A, Le BS, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007;87:425–456. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- 52.Sheets MF, Hanck DA, Fozzard HA. Nonlinear relation between Vmax and INa in canine cardiac Purkinje cells. Circ Res. 1988;63:386–398. doi: 10.1161/01.res.63.2.386. [DOI] [PubMed] [Google Scholar]
- 53.Wei K, Liu L, Xie F, Hao X, Luo J, Min S. Nerve growth factor protects the ischemic heart via attenuation of the endoplasmic reticulum stress induced apoptosis by activation of phosphatidylinositol 3-kinase. Int J Med Sci. 2015;12:83–91. doi: 10.7150/ijms.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li C, Hu M, Wang Y, Lu H, Deng J, Yan X. Hydrogen sulfide preconditioning protects against myocardial ischemia/reperfusion injury in rats through inhibition of endo/sarcoplasmic reticulum stress. Int J Clin Exp Pathol. 2015;8:7740–7751. [PMC free article] [PubMed] [Google Scholar]
- 55.Hou JY, Liu Y, Liu L, Li XM. Protective effect of hyperoside on cardiac ischemia reperfusion injury through inhibition of ER stress and activation of Nrf2 signaling. Asian Pac J Trop Med. 2016;9:76–80. doi: 10.1016/j.apjtm.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 56.Tadimalla A, Belmont PJ, Thuerauf DJ, Glassy MS, Martindale JJ, Gude N, Sussman MA, Glembotski CC. Mesencephalic astrocyte-derived neurotrophic factor Is an ischemia-inducible secreted endoplasmic reticulum stress response protein in the heart. Circ Res. 2008;103:1249–1258. doi: 10.1161/CIRCRESAHA.108.180679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ortega A, Roselló-Lletí E, Tarazón E, Molina-Navarro MM, Martínez-Dolz L, González-Juanatey J, Lago F, Montoro-Mateos JD, Salvador A, Rivera M, Portolés M. Endoplasmic reticulum stress Induces different molecular structural alterations in human dilated and ischemic Cardiomyopathy. PLoS One. 2014;9:e107635. doi: 10.1371/journal.pone.0107635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107:839–850. doi: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou A, Tabas I. The UPR in atherosclerosis. Semin Immunopathol. 2013;35:321–332. doi: 10.1007/s00281-013-0372-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xin W, Li X, Lu X, Niu K, Cai J. Involvement of endoplasmic reticulum stress-associated apoptosis in a heart failure model induced by chronic myocardial ischemia. Int J Mol Med. 2011;27:503–509. doi: 10.3892/ijmm.2011.612. [DOI] [PubMed] [Google Scholar]
- 61.Liu M, Wang X, Wang C, Song D, Liu X, Shi D. Panax quinquefolium saponin attenuates ventricular remodeling after acute myocardial infarction by inhibiting chop-mediated apoptosis. Shock. 2013;40:339–344. doi: 10.1097/SHK.0b013e3182a3f9e5. [DOI] [PubMed] [Google Scholar]
- 62.Shi ZY, Liu Y, Dong L, Zhang B, Zhao M, Liu WX, Zhang X, Yin XH. Cortistatin improves cardiac function after acute myocardial infarction in rats by suppressing myocardial apoptosis and endoplasmic reticulum stress. J Cardiovasc Pharmacol Ther. 2016;22:83–93. doi: 10.1177/1074248416644988. [DOI] [PubMed] [Google Scholar]
- 63.Toko H, Takahashi H, Kayama Y, Okada S, Minamino T, Terasaki F, Kitaura Y, Komuro I. ATF6 is important under both pathological and physiological states in the heart. J Mol Cell Cardiol. 2010;49:113–120. doi: 10.1016/j.yjmcc.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 64.Santos C, Nabeebaccus A, Shah A, Camargo L, Filho S, Lopes L. Endoplasmic reticulum stress and Nox-mediated reactive oxygen species signaling in the peripheral vasculature: potential role in hypertension. Antioxid Redox Signal. 2014;20:121–134. doi: 10.1089/ars.2013.5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Okada Ki, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- 66.Dromparis P, Paulin R, Stenson TH, Haromy A, Sutendra G, Michelakis ED. Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation. 2013;127:115. doi: 10.1161/CIRCULATIONAHA.112.133413. [DOI] [PubMed] [Google Scholar]
- 67.Liu X, Kwak D, Lu Z, Xu X, Fassett J, Wang H, Wei Y, Cavener DR, Hu X, Hall J, Bache RJ, Chen Y. Endoplasmic reticulum stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK) protects against pressure overload-induced heart failure and lung remodeling. Hypertension. 2014;64:738–744. doi: 10.1161/HYPERTENSIONAHA.114.03811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Z, Zhang T, Dai H, Liu G, Wang H, Sun Y, Zhang Y, Ge Z. Endoplasmic reticulum stress is involved in myocardial apoptosis of streptozocin-induced diabetic rats. J Endocrinol. 2008;196:565–572. doi: 10.1677/JOE-07-0230. [DOI] [PubMed] [Google Scholar]
- 69.Liu ZW, Zhu HT, Chen KL, Dong X, Wei J, Qiu C, Xue JH. Protein kinase RNA-like endoplasmic reticulum kinase (PERK) signaling pathway plays a major role in reactive oxygen species (ROS)-mediated endoplasmic reticulum stress-induced apoptosis in diabetic cardiomyopathy. Cardiovasc Diabetol. 2013;12:158. doi: 10.1186/1475-2840-12-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guo R, Liu W, Liu B, Zhang B, Li W, Xu Y. SIRT1 suppresses cardiomyocyte apoptosis in diabetic cardiomyopathy: An insight into endoplasmic reticulum stress response mechanism. Int J Cardiol. 2015;191:36–45. doi: 10.1016/j.ijcard.2015.04.245. [DOI] [PubMed] [Google Scholar]
- 71.Dickhout JG, Carlisle RE, Austin RC. Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: endoplasmic reticulum stress as a mediator of pathogenesis. Circ Res. 2011;108:629–642. doi: 10.1161/CIRCRESAHA.110.226803. [DOI] [PubMed] [Google Scholar]
- 72.Liu Y, Wang J, Qi SY, Ru LS, Ding C, Wang HJ, Zhao JS, Li JJ, Li Ay, Wang DM. Reduced endoplasmic reticulum stress might alter the course of heart failure via caspase-12 and JNK pathways. Can J Cardiol. 2014;30:368–375. doi: 10.1016/j.cjca.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 73.Fu HY, Okada Ki, Liao Y, Tsukamoto O, Isomura T, Asai M, Sawada T, Okuda K, Asano Y, Sanada S, Asanuma H, Asakura M, Takashima S, Komuro I, Kitakaze M, Minamino T. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation. 2010;122:361–369. doi: 10.1161/CIRCULATIONAHA.109.917914. [DOI] [PubMed] [Google Scholar]
- 74.Dally S, Monceau V, Corvazier E, Bredoux R, Raies A, Bobe R, del Monte F, Enouf J. Compartmentalized expression of three novel sarco/endoplasmic reticulum Ca2+ATPase 3 isoforms including the switch to ER stress, SERCA3f, in non-failing and failing human heart. Cell Calcium. 2009;45:144–154. doi: 10.1016/j.ceca.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 75.Ni L, Zhou C, Duan Q, Lv J, Fu X, Xia Y, Wang DW. β-AR blockers suppresses ER stress in cardiac hypertrophy and heart failure. PLoS One. 2011;6:e27294. doi: 10.1371/journal.pone.0027294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barbuti A, Benzoni P, Campostrini G, Dell'Era P. Human derived cardiomyocytes: A decade of knowledge after the discovery of induced pluripotent stem cells. Dev Dyn. 2016;245:1145–1158. doi: 10.1002/dvdy.24455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.