


Abstract
The process of transcriptional elongation by RNA polymerase II (RNAPII) in a chromatin context involves a large number of crucial factors. Spn1 is a highly conserved protein encoded by an essential gene and is known to interact with RNAPII and the histone chaperone Spt6. Spn1 negatively regulates the ability of Spt6 to interact with nucleosomes, but the chromatin binding properties of Spn1 are largely unknown. Here, we demonstrate that full length Spn1 (amino acids 1–410) binds DNA, histones H3–H4, mononucleosomes and nucleosomal arrays, and has weak nucleosome assembly activity. The core domain of Spn1 (amino acids 141–305), which is necessary and sufficient in Saccharomyces cerevisiae for growth under ideal growth conditions, is unable to optimally interact with histones, nucleosomes and/or DNA and fails to assemble nucleosomes in vitro. Although competent for binding with Spt6 and RNAPII, the core domain derivative is not stably recruited to the CYC1 promoter, indicating chromatin interactions are an important aspect of normal Spn1 functions in vivo. Moreover, strong synthetic genetic interactions are observed with Spn1 mutants and deletions of histone chaperone genes. Taken together, these results indicate that Spn1 is a histone binding factor with histone chaperone functions.
INTRODUCTION
In eukaryotic cells, chromatin plays a significant role in the regulation of transcription by RNA polymerase II (RNAPII) (1). Chromatin obscures the underlying DNA and blocks the binding of proteins required for the formation of the preinitiation complex (PIC). It also physically impedes the progression of RNAPII during transcription elongation. Therefore, proper gene expression requires numerous accessory elongation factors that, in addition to regulating RNAPII activity, modulate the structure of chromatin to promote productive transcription (1,2). These transcription elongation factors associate with RNAPII and are found throughout the body of actively transcribed genes.
Spn1 is a highly conserved transcription elongation factor that associates with the histone chaperone Spt6, and also interacts with numerous elongation and chromatin-regulating factors (3–9). Spn1 is comprised of three domains: an acidic N-terminal region, a well conserved core domain (amino acids 141–305) and a basic C-terminal region (10,11). Iws1, the ortholog of Spn1 in higher eukaryotes, also associates with Spt6 (8,12) and has maintained the three domains found in yeast Spn1, although the N-terminal region of both mammalian and plant Iws1 is significantly longer than that of Saccharomyces cerevisiae Spn1. We, and others, have determined that the conserved core domain of Spn1 is structured and contains the binding site for Spt6 (7,11,13).
Mutations in Spt6 that abolish Spn1 binding are lethal, indicating the binding of Spn1 to Spt6 is essential for cellular viability (7). As a histone chaperone, Spt6 has been shown to bind DNA, histones, and nucleosomes and to assemble nucleosomes both in vitro and in vivo (3,7,14–16). The binding of Spn1 to Spt6 precludes Spt6 binding to nucleosomes and therefore, it has been proposed that Spn1 regulates Spt6 binding to nucleosomes (7). Interestingly, mutations in histones H2A and H2B suppress phenotypes seen with Spt6-F249K, an Spt6 mutant that has reduced binding to Spn1 (16). These H2A and H2B mutations are thought to destabilize histone interfaces within the nucleosome, and it was suggested that the destabilized nucleosomal structures activate a nucleosome monitoring system that circumvents the need for a Spn1–Spt6 associated function (16).
While the Spn1–Spt6 complex provides critical cellular functions, both proteins appear to have functions that are independent of the other. An analysis of elongation factor occupancies throughout the yeast genome found that the occupancy profiles for Spn1 and Spt6 differ, especially near the 3′-end of actively transcribed genes (17). In addition, Spn1 occupies the CYC1 promoter in the absence of Spt6 (18), presumably through its association with RNAPII although, it could also be interacting with other proteins at the promoter.
Spn1 and Spt6 are both required for the repression of SER3 transcription (19). Currently, it is thought that SER3 repression requires nucleosome occupancy over the SER3 promoter to occlude it from transcription factor binding (19). Spt6 is required for the reassembly of nucleosomes over the SER3 promoter that were disrupted by transcription of the SRG1 gene located immediately upstream of SER3 (19). How Spn1 is required for SER3 repression is not known. It is possible that Spn1 needs to be present for Spt6 to assemble nucleosomes or that Spn1 independently plays a role in the assembly of nucleosomes over the SER3 promoter. Spn1 mutants have Spt− phenotypes (6,10) which are correlated with reduced nucleosome assembly, further suggesting that Spn1 has functions associated with nucleosome assembly. However, to date there is no direct evidence that Spn1 functions in nucleosome binding or assembly.
To further our understanding of cellular functions of the conserved Spn1 protein, we investigated the properties of full length (wild type) Spn1 and compared those to the core domain of Spn1 (residues 141–305, Spn1141–305). We show that full-length Spn1 binds DNA, histones H3–H4, nucleosomes and nucleosomal arrays, and can assemble nucleosomes in vitro. The core domain of Spn1 is not sufficient for any of these functions. We also demonstrate that the N- and C-terminal domains of Spn1 are not required for association with RNAPII or Spt6 in vivo, but are required for Spn1 occupancy at the CYC1 promoter prior to activation. Finally, although Spn1141–305 is sufficient for yeast cell viability under optimal conditions, it produces significant synthetic growth defects when combined with elongation factor and histone chaperone deletions. Taken together, these results indicate that Spn1 is an important protein that functions in the regulation of chromatin architecture.
MATERIALS AND METHODS
See Supplemental Data for the experimental procedures used in preparing yeast strains, Spn1 protein expression and purification, and media preparation. A list of the strains and plasmids used in this manuscript may be found in Supplemental Table S1.
Sucrose gradient sedimentation assay
Wild type Spn1, Spn1141–305, Xenopus laevis histones H2A–H2B (5 μM) or 2.5 μM histones H3–H4 alone, or in combination as indicated were brought to 120 μl with the following buffer: 10 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM DTT. The protein samples were then incubated on ice for 30 min and 100 μl of each sample was layered onto 12 ml, 5–25% sucrose gradients (prepared in the same buffer as above). The remainder of each protein sample was set aside so that a portion (2 μl) could be used as an input control. The gradients were spun in a SW41Ti rotor (Beckman) at 28 000 RPM and 4°C for 18 h. The gradients were fractionated by pulling off 50 μl aliquots from the top and proceeding down the gradient. The sedimentation of the proteins through the gradients was analyzed by SDS-PAGE (15% gels or 4–12% precast polyacrylamide gels (BioRad Cat. # 3450124)) of 20 μl aliquots from every other fraction. Proteins were visualized with Sypro Ruby (Invitrogen) staining, scanned using a Trio imager (GE Healthcare Life Sciences)) and quantified using ImageJ software (http://rsbweb.nih.gov/ij/index.html). Protein band intensities cannot be directly compared between gels following staining with Sypro Ruby; therefore, the H3–H4 intensities in each gradient were normalized by setting samples with the lowest intensity to 0 and highest intensity to 100.
Size-exclusion chromatography binding assay
Purified recombinant proteins were mixed at equimolar concentrations (20 μM, H3–H4 was calculated as a (H3–H4)2 tetramer) and incubated for 15 min at 4°C. The protein mixture was applied to a 120 ml Superdex 200 16/60 column (GE Healthcare) in 20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 10% glycerol and 5 mM 2-mercaptoethanol. Fractions were collected and analyzed by SDS-PAGE (15% gel).
DNA and nucleosome binding assay
DNA (‘601’ sequence (20), 147 base pairs) was purified as described (21). X. laevis histones H2A, H2B, H3 and H4 were purified, refolded into octamers, and assembled into nucleosomes as described (21). DNA (601–147) or nucleosomes assembled from X. laevis octamers and 601–147 DNA were incubated at 2.5 μM with a 2- or 4-fold molar excess of wild type Spn1 or Spn1141–305 for 30 min at room temperature. The binding buffer was 50 mM Tris–HCl (pH 7.5), 50 mM NaCl, 2 mM arginine. The binding reactions were loaded onto a 5% polyacrylamide gel in 45 mM Tris (pH 8.3), 45 mM boric acid,1 mM EDTA. The samples were electrophoresed through the gel at 300 V and 4°C for 3 h. DNA and DNA/protein complexes were visualized in the gel by staining with ethidium bromide.
Nucleosomal array binding assays
Nucleosomal arrays were reconstituted as described in (22) using native chicken histone octamers and a DNA template comprised of 12 repeats of the 207 base pair 601 nucleosome positioning sequence (207–212 DNA) (20). Wild type Spn1 or Spn1141–305 was combined with nucleosomal arrays at a molar ratio of 0.5, 1.0 and 2.0 (protein to 207 repeat) in 10 mM Tris–HCl (pH 7.5), 0.25 mM EDTA, and 2.5 mM NaCl with varying concentrations of MgCl2. The samples were incubated at room temperature for 15 min and then their volume was brought to 400 μl with reaction buffer. Aliquots (10 μl) from each sample were removed and analyzed by EMSA using 1% agarose gels. The remainder of each sample was analyzed by sedimentation velocity analytical ultracentrifugation (SV-AUC) sedimentation velocity in a Beckman XL-I or XL-A ultracentrifuge in either a An50Ti or An60Ti rotor at 25°C. The resulting boundaries were analyzed using the improved van Holde–Weischet method as implemented in Ultrascan II (23).
Histones H3–H4 deposition assay
Asf11–168 was purified as previously described (24). Xenopus laevis histones H3C110A and H4T71C were purified and labeled with Alexa488 as previously described (25). Bovine Serum Albumin (BSA) was purchased from ThermoFisher Scientific (catalog #: 23209). A 79 base pair (bp) DNA fragment containing the 601 positioning sequence was prepared as previously described (26). Reconstituted tetrasomes were formed on the 79 bp DNA using labeled H3–H4 and a continuous salt gradient dialysis (21). Asf11–168, Spn1, Spn1141–305 and BSA (100 or 200 nM) were incubated with histones H3–H4 (200 nM) for 10 min at room temperature in 20 mM Tris–HCl (pH 7.5), 180 mM NaCl, and 1 mM DTT. The DNA was added to the reactions and the incubation continued an additional 15 min. The reactions were then electrophoresed through 5% polyacrylamide gels, the gels stained with ethidium bromide and the bands in the gel visualized using a Typhoon FLA 9500 (GE). Band intensities were quantified using ImageQuant TL. Tetrasome band intensities were normalized to the amount of tetrasome formed in the H3–H4 plus DNA reaction and the normalized values plotted using GraphPad.
Chromatin assembly assay
The DNA supercoiling assay was carried out as described (27). Wild type Spn1, Spn11–305, Spn1141–410 and Spn1141–305 were combined with X. laevis histone octamers (0.8 μM) to reach a final Spn1 to histone octamer molar ratio of 0.5:1 and 1:1. Nap1 was purified as described in (28) and combined with histone octamers to reach a final ratio of 1:1 and 2:1. The reactions were incubated at 37°C for 10 min. Relaxed pBR322 plasmid DNA was added to the reactions and the reactions incubated at 37°C for 1 h in a buffer containing 10 mM Tris–HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA and 100 μg/μl BSA. Eight units of wheat germ topoisomerase I (Promega) were then added to each reaction and the reactions incubated at 37°C for one additional hour. To terminate the reactions, Proteinase K and SDS were added to reach a final concentration of 0.2 mg/ml and 0.5%, respectively and the reactions incubated at 55°C for 30 min. The plasmid DNA was purified by phenol/chloroform extraction and ethanol precipitation. The final products were electrophoresed through a 1.2% agarose gel and the DNA visualized by SYBR Gold (Invitrogen) staining.
Circular dichroism spectroscopy
CD was performed using a Jasco-720 spectropolarimeter at 20°C. Wild type Spn1 and Spn1141–305 were extensively dialyzed against 10 mM NaH2PO4/Na2HP04 (pH 7.4) and 100 mM NaF buffer prior to obtaining measurements. Experiments were performed with protein concentrations ranging from 20 to 30 μM using cells with a pathlength of 0.01 or 0.02 cm. Each scan was obtained by averaging measurements taken from 260 nm down to 185 nm at 10 nm/min with a response time of 16 s. Each spectrum was baseline subtracted from a similar scan performed with dialysis buffer. The molar ellipticity [Θ] was obtained by normalization of the measured ellipticity (Θ, millidegree), where [Θ] = (Θ × 100)/(nlc), n is the number of residues, c is the total concentration (mM), and l is the cell path length (cm). The percentage of secondary structure was determined from each spectra using the CONTINLL, SELCON3 and CDSSTR methods within CDPro analytical software (29). The SDP42 basis set was used to deconvolute the CD spectrum. The data reported are from two independent biological replicates.
Sedimentation velocity analytical ultracentrifugation
SV-AUC experiments were performed using the Beckman XL-I or a Beckman XL-A analytical ultracentrifuge and the absorbance optical system as described (30). Wild type Spn1 and Spn1141–305 were extensively dialyzed against 20 mM Tris–HCl (pH 7.5) with 150, 300 or 500 mM NaCl buffer prior to analysis. Boundaries were analyzed to yield the integral distribution of sedimentation coefficients, g(s) according to the method of Demeler et al. (23,31) using Ultrascan (version 9.9). Sedimentation coefficients (s) were corrected to water at 20°C (s20,w). Modeling of hydrodynamic parameters (sedimentation coefficient, molecular weight, f/fo and RMSD values) was performed within Ultrascan.
Tandem affinity purification (TAP)
TAP tagged proteins were purified as described (32) with a few modifications. Yeast cells were grown in 2 L of YPD (1% yeast extract, 2% Bacto-Peptone and 2% glucose) to an optical density at 600 nm (OD600) of ∼1.0. Harvested cells were broken by grinding in liquid nitrogen with mortar and pestle and resuspended in lysis buffer (40 mM HEPES–KOH, pH 7.5, 10% glycerol, 150 mM NaCl, 0.1% Tween 20, 1 mM EDTA, 1 mM DTT and protease inhibiter cocktail). The suspension was sonicated 10 times for 1 min each, using a Branson Sonifier 450 with 2 min incubations on ice between each cycle. The cell lysate was clarified by centrifuging at 3200 rpm for 20 min at 4°C and the supernatant was subjected to purification using IgG Sepharose 6 Fast Flow (GE, Cat. #: 17-0969-01).
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assays were performed as described in (33,34). Specific details of the culturing of cells for the ChIP analysis, sample preparation and qPCR may be found in the Supplemental Data.
Phenotypic assays
To assess the growth of the Spn1141–305 and Spn1K192N strain under different conditions and to further assess the genetic interaction of SPN1 with DST1, RTF1, NAP1, RTT106, HIR1, HIR2, ASF1, CAC1, CAC3 and VPS75 the indicated strains were grown in YPD to an OD600 between 0.7 and 1. Ten-fold serial dilutions of the strains were prepared and spotted onto solid media and incubated as indicated.
RESULTS
Spn1 preferentially binds histones H3–H4
Spn1 interacts with numerous proteins that are associated with maintaining or modulating chromatin architecture including histone chaperones (Spt6), histone modifiers (Rtf1), nucleosome remodelers (Swi/Snf and INO80); and is recruited to chromatin in vivo (4–6,9,18,35). Therefore, we wondered whether Spn1 might have uncharacterized functions associated with chromatin interactions. The binding partner of Spn1, Spt6, binds both histones H2A–H2B and H3–H4, assembles nucleosomes and in the presence of Nhp6, binds nucleosomes (3,7,16). As a first step in evaluating whether Spn1 has chromatin associated functions, we investigated whether Spn1 can bind histones. Spn1 and histones H2A–H2B or H3–H4 do not enter polyacrylamide gels under non-denaturing conditions therefore, an Electrophoretic Mobility Shift Assay (EMSA) could not be used to evaluate Spn1 binding to histones. Therefore, we utilized a sucrose gradient sedimentation assay to investigate Spn1 binding to histones. Equimolar amounts of wild type Spn1, and histones H3–H4 or histones H2A–H2B, were incubated alone or in combination at 150 mM NaCl, and then sedimented through 5–25% sucrose gradients. Fractions from the gradients were collected and analyzed by denaturing polyacrylamide gel electrophoresis (SDS-PAGE). Spn1 and the histone proteins sedimented near the top of the gradients (fractions 1–31 out of 240 collected) (Supplemental Figure S1). We found that Spn1 binds histones H3–H4, as indicated by the significant shift in the H3–H4 peak from fractions 9 and 11 to fraction 19 when Spn1 is present (Figure 1A and Supplemental Figure S1A). Spn1 does not appear to bind histones H2A–H2B under these conditions, the H2A–H2B sedimentation peak only shifted slightly (two fractions) when Spn1 was present (Figure 1B and Supplemental Figure S1B).
Figure 1.

Spn1 preferentially binds histones H3–H4. The binding of Spn1 to histones H3–H4 or H2A–H2B was assessed by sucrose gradient sedimentation (A and B), and size exclusion chromatography (C and D). Histones H3–H4, Histones H2A–H2B, and Spn1 with H3–H4 or H2A–H2B were sedimented through 5–25% sucrose gradients or passed through a Superdex 200 column. Sedimentation of the proteins through the gradients was analyzed by SDS-PAGE for every other fraction. Results are shown for fractions 1–31 from the top of the gradient. The sedimentation profiles for (A) H3–H4 alone or H3–H4 with Spn1 and (B) H2A–H2B alone or H2A–H2B with Spn1. Overlays of the size exclusion chromatograms for (C) Spn1 alone and Spn1 with H3–H4 and (D) Spn1 alone and Spn1 with H2A–H2B.
Histones H2A–H2B and H3–H4 are highly charged proteins that tend to interact non-specifically with themselves and other proteins, frequently forming large aggregates. To validate the histone-binding activity of Spn1 observed using sucrose gradient sedimentation, we also assessed Spn1-histone complex formation by size exclusion chromatography (SEC). Spn1 was again incubated alone, or with histones H3–H4, or H2A–H2B at 150 mM NaCl and the proteins were then fractionated by passage through a Superdex 200 column. Proteins contained within the elution peaks were analyzed by SDS-PAGE. The elution profile of Spn1 was significantly altered when it was incubated with histones H3–H4 prior to fractionation, with its elution peak shifting from 65 to 58 ml (Figure 1C). Analysis of the elution fractions also demonstrated that Spn1 and histones H3–H4 co-eluted (Supplemental Figure S1C). In contrast, when Spn1 was incubated with histones H2A–H2B prior to fractionation, its elution profile was not significantly affected and Spn1 did not elute with histones H2A–H2B (Figure 1D and Supplemental Figure S1D). These results confirm our sucrose gradient sedimentation results, and further indicate that Spn1 is not interacting non-specifically via electrostatic interactions with the basic histones as it clearly has a preference for binding histones H3–H4 over H2A–H2B.
We also assayed Spn1K192N and Spn1141–305 in the sucrose gradient sedimentation assay. The K192N substitution in Spn1 significantly reduces its binding to RNAPII and Spt6 (18) and we wondered if this substitution would also affect the ability of Spn1 to bind histones H3–H4. However, when Spn1K192N was combined with H3–H4, it shifted the H3–H4 sedimentation peak from fraction 10 to fraction 18, indicating that Spn1K192N is competent to bind H3–H4 (Supplemental Figure S2) and that the histone binding site on Spn1 is distinct from the regions required to bind RNAPII and Spt6.
Spn1141–305 contains the highly conserved core domain of Spn1 and can support cellular growth under rich growth conditions (10,11). When Spn1141–305 was assessed for histone H3–H4 binding using sucrose gradient sedimentation it only produced a slight shift (from fraction 10 to fraction 12) in the H3–H4 sedimentation peak (Supplemental Figure S3). This result was not unequivocal so we evaluated the histone binding properties of Spn1141–305 using SEC. Spn1141–305 and histones H3–H4 were incubated at 150 mM NaCl, and then loaded onto a Superdex 200 column. Spn1141–305 and histones H3–H4 eluted from the column in two distinct peaks, with H3–H4 eluting before Spn1141–305 (Supplemental Figure S4), thus indicating that the core domain of Spn1 is not sufficient for histone H3–H4 binding.
Spn1 binds DNA, mononucleosomes and nucleosomal arrays
To investigate whether Spn1 makes direct contacts with chromatin, we tested Spn1 and Spn1141–305 for DNA and nucleosome binding activity. The Spn1K192N protein degrades significantly during purification (Supplemental Figure S5) and therefore, was omitted from the remainder of the in vitro assays presented here. Nucleosomes were assembled on a 147 base-pair DNA fragment bearing the 601-nucleosome positioning sequence (601–147 DNA) (20). Increasing amounts of Spn1 or Spn1141–305 were incubated with free 601–147 DNA or nucleosomes, and the DNA-protein complexes were then assessed by an EMSA. Spn1 bound both DNA and nucleosomes resulting in Spn1 containing complexes that migrated more slowly through the gel (Figure 2). It should be noted that overhanging DNA ends are not present in nucleosomes assembled with 601–147 DNA, thus Spn1 is not simply binding to the ends of the DNA assembled into the nucleosomes. Unlike Spt6, Spn1 does not require Nhp6, a small HMG protein, to bind nucleosomes (7). Spn1141–305 did not bind either DNA or nucleosomes (Figure 2), indicating that the core domain of Spn1 is not sufficient for DNA or nucleosome binding.
Figure 2.

Wild type, full-length Spn1 can bind DNA and nucleosomes. (A) DNA (2.5 μM 601–147) or (B) nucleosomes assembled with histone octamer on 601–147 DNA were incubated with a 2- or 4-fold molar excess of Spn1 (WT) or Spn1141–305 as indicated, for 30 min at room temperature. Free DNA and protein complexes were separated by electrophoresis through a native 5% polyacrylamide gel and the gel stained with ethidium bromide. The length in base pairs of select DNA fragments in the electrophoresis markers (M) is shown to the left of the panels. Lanes with DNA (D) or nucleosomes (N) alone are indicated.
To further characterize the nucleosome binding activity of Spn1, we tested whether Spn1 could bind an array containing 12 nucleosomes, a substrate more similar to chromatin found in vivo. Nucleosomal arrays were reconstituted using native chicken histone octamers and DNA comprised of 12 207 bp repeats of the 601-nucleosome positioning sequence (20). The arrays were reconstituted to homogeneous saturation as determined by sedimentation velocity analytical ultracentrifugation (SV-AUC) (Figure 3). Increasing amounts of full length Spn1 were incubated with the arrays and the resulting complexes were assessed by SV-AUC and EMSA. The homogeneous nucleosomal arrays alone sedimented at 29.4S. With increasing amounts of Spn1, a species that sedimented with a savg of 38.5S was formed (Figure 3A). Consistent with the results of the sedimentation velocity analysis, full length Spn1 retarded the migration of the nucleosomal arrays in the EMSA (Figure 3C).
Figure 3.
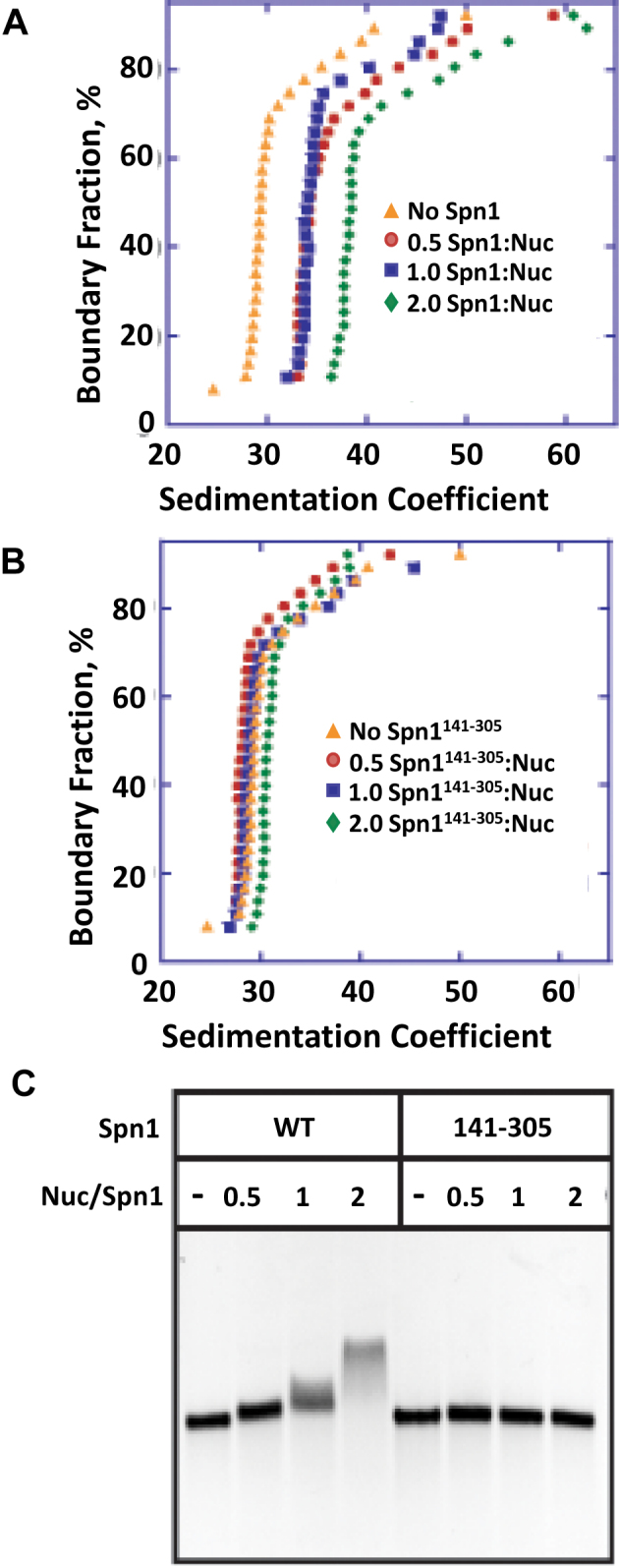
Spn1 binds nucleosomal arrays. The binding of Spn1 or Spn1141–305 to nucleosomal arrays assembled on DNA containing 12 repeats of the 207 base pair, 601 nucleosome positioning sequence was assessed by AUC and EMSA. Sedimentation velocity curves for (A) wild type Spn1 or (B) Spn1141–305 added to the nucleosomal arrays at a Spn1:nucleosome molar ratio of 0.5 (red circles), 1.0 (blue squares) or 2.0 (green diamonds) in the presence of 2.5 mM NaCl. Orange triangles are the arrays alone, (C) EMSA of wild type Spn1 and Spn1141–305 with 207–12 nucleosomal arrays using 1% agarose gels.
While the core domain of Spn1 (Spn1141–305) could not bind mononucleosomes in our previous assay, it remained formally possible that it could bind nucleosomes in the context of an array. Therefore, we included Spn1141–305 in our SV-AUC and EMSA experiments. The addition of increasing amounts of Spn1141–305 did not affect the sedimentation of the nucleosomal arrays (Figure 3B), nor did it result in a shift in the EMSA (Figure 3C). Our SV-AUC and EMSA results are consistent with Spn1 being able to bind nucleosomal arrays and with the nucleosome binding region requiring residues outside the core domain of Spn1.
The binding of Spn1 to nucleosomal arrays does not affect the formation of higher-order structures
The addition of Mg2+ to nucleosomal arrays induces the intrinsic folding pathway that leads to the formation of higher-order, condensed chromatin structures (36,37). In the presence of Mg2+ the Sir3 protein (Sir3p) has been shown to bind nucleosomal arrays and form hyper-condensed higher-order chromatin structures (38). In contrast to Sir3p, the addition of the high mobility group N (HMGN) protein to nucleosomal arrays in the presence of Mg2+ leads to a decompaction of the folded array (39). We evaluated the binding of Spn1 to nucleosomal arrays in the presence of increasing concentrations of Mg2+ (Supplemental Figure S6). Nucleosomal arrays in the presence of 1.0, 1.5 and 1.75 mM Mg2+ sedimented at 32.9S, 35S and 36S, respectively (Supplemental Figure S6A–C). When increasing amounts of Spn1 were added to the arrays, the sedimentation coefficients increased linearly (Supplemental Figure S6D), indicating that Spn1 was binding to the arrays, but was not affecting higher-order chromatin folding. Thus, Spn1 differs from both the Sir3 and HMGN proteins in that Spn1 binding to nucleosomal arrays does not affect salt-dependent chromatin compaction.
Spn1 facilitates H3–H4 deposition onto DNA and has weak nucleosome assembly activity
Given that Spn1 can bind histones H3–H4, DNA, and nucleosomes, we hypothesized that it might also be able to deposit H3–H4 onto DNA and facilitate nucleosome assembly in vitro. The initial step in nucleosome assembly is the deposition of H3–H4 onto DNA, resulting in the formation of an intermediate structure known as the tetrasome (40). In the absence of a histone chaperone, under physiological ionic conditions H3–H4 can interact with DNA to independently form tetrasomes although the efficiency of tetrasome formation is low (41). The histone H3–H4 dimer chaperone, Asf1 has been shown to reduce the aggregation of H3–H4 and to facilitate the assembly of one H3–H4 dimer onto DNA (a disome) in a dose dependent manner (41). To test whether Spn1 can assemble tetrasomes in vitro, we used native gel electrophoresis and a 79 base pair DNA containing the 601 positioning sequence (20), fluorescently labeled H3–H4 and either full length Spn1 or Spn1141–305. We also assayed Asf11–168 and BSA as positive and negative controls, respectively, of H3–H4 deposition onto DNA. Full length Spn1 clearly stimulated tetrasome formation resulting in a 1.8 ± 0.3-fold increase in tetrasome levels, while spn1141–305 did not significantly affect tetrasome formation (Figure 4A and Supplemental Figure S7A). It should be noted, that the H3–H4 deposition assay was completed with relatively low amounts of Spn1 to minimize Spn1 binding to the tetrasome which leads to complexes that do not enter the gel.
Figure 4.

Spn1 facilitates H3–H4 deposition onto DNA and has weak nucleosome assembly activity. (A) Labeled H3–H4 and a 79 bp DNA were incubated with the indicated protein and DNA and DNA–protein complexes resolved by electrophoresis through native 5% polyacrylamide gels (Supplemental Figure S7A). Results from a minimum of three independent experiments were quantified, and the average tetrasome amount with standard deviation is shown. (B) Nucleosome assembly activity of Spn1 or Spn1141–305 was analyzed using a plasmid supercoiling assay as described in the methods. A representative gel from a plasmid supercoiling assay is shown. Lane M: DNA size standards, lane 1: pBR322 DNA, lane 2: relaxed pBR322 DNA, lanes 3–5: relaxed pBR322 DNA with wild type Spn1, Spn1141–305 or Nap1 (as a control), lane 6: relaxed pBR322 DNA with Xenopus octamer, lanes 7–12: relaxed pBR322 DNA with Xenopus octamer, and wild type Spn1, Spn1141–305 or Nap1 as indicated. The position of relaxed and supercoiled (SC) plasmid DNA are indicated at the right above.
To determine whether Spn1 can assemble nucleosomes in vitro, we utilized a plasmid supercoiling assay (27). In this assay, relaxed plasmid DNA is incubated with histones, the protein of interest and then topoisomerase I. If the test protein assembles nucleosomes on the relaxed plasmid, supercoils are introduced into the plasmid and remain after the removal of all protein. The topological state of the plasmid DNA is then evaluated by gel electrophoresis. In the absence of histones none of the test proteins (Spn1, Spn1141–305 or Nap1 as a positive control) affected the topology of the relaxed plasmid, while the addition of histones increased supercoiling slightly (Figure 4B). When Nap1 was added to the relaxed plasmid in the presence of histones, supercoils were introduced into the plasmid indicating that nucleosomes were formed. Full length Spn1 moderately increased supercoiling in the presence of histones, whereas Spn1141–305 did not affect supercoiling. To determine whether nucleosome assembly requires both the N- and C-terminal regions of Spn1, we used the supercoiling assay and included two additional Spn1 truncations (Spn11–305 and Spn1141–410). Interestingly, neither Spn1 truncation significantly increased supercoiling, indicating that both the N- and C-terminal regions of Spn1 are required for nucleosome assembly (Supplemental Figure S7B). Collectively, these results indicate that full length Spn1 facilitates H3–H4 deposition onto DNA and has weak nucleosome assembly activity, and as with its other chromatin based functions, the core domain of Spn1 is not competent for these functions.
Full-length Spn1 has unstructured N- and C-terminal regions
The structure of the core domain of yeast Spn1 has been determined by X-ray crystallography and was shown to consist of eight alpha-helices (7,11). Full-length Spn1 protein is not amenable to crystallographic studies. However, since the N- and C-terminal regions of Spn1 are required for histone, DNA and nucleosome binding, we wondered if there were uncharacterized structural elements in the Spn1 terminal regions. We utilized circular dichroism (CD) spectroscopy and SV-AUC to further investigate the structural features of Spn1. To determine the amount of secondary structure present in each protein, full length Spn1 and Spn1141–305 were expressed in Escherichia coli, purified and analyzed by CD spectroscopy (Figure 5A and Supplemental Table S2). We included Spn1141–305 in the analysis since its structure is so well characterized (7,11). The samples were scanned from 180 nm to 260 nm using a Jasco 720 spectropolarimeter and the resulting CD spectra deconvoluted using CDPro secondary structure prediction algorithms (Supplemental Table S2) (29). CD spectroscopy is most accurate in predicting α-helical structures compared to other types of secondary structure and our results with Spn1141–305 were consistent with this feature of CD. The CD prediction for α-helicity in Spn1141–305 is very similar to the crystallographic values (100–120 versus 107) while the β-sheet (8–16 versus 0) and β-turns (28–34 versus 16) were both higher. The number of unstructured amino acids calculated from the CD spectra is lower than the actual amount in Spn1141–305 (12–16 versus 42).
Figure 5.
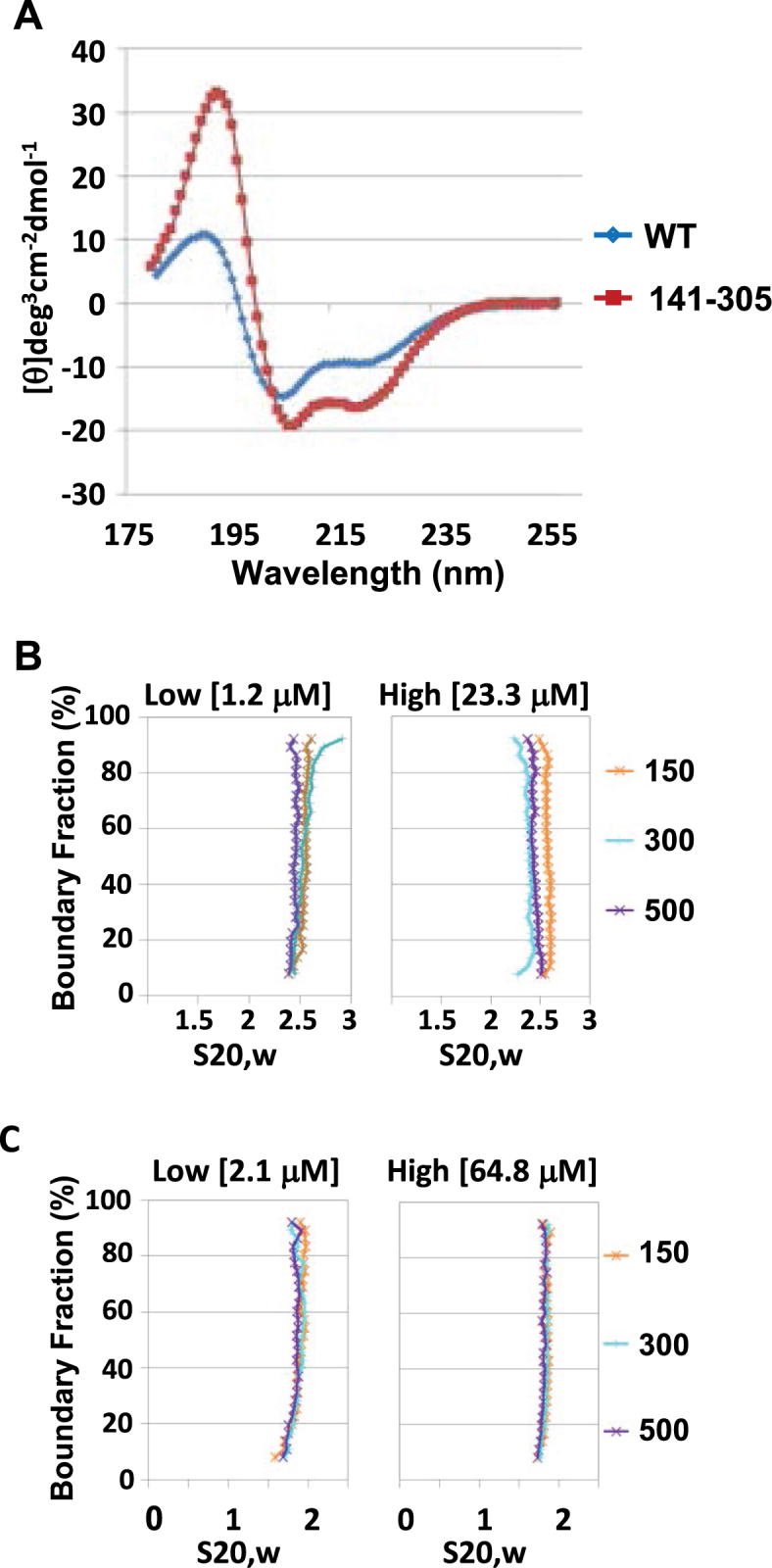
The N- and C-termini of Spn1 are predominantly disordered, and full length Spn1 has an elongated, asymmetric shape. (A) The secondary structure of wild type Spn1 and Spn1141–305 was analyzed using circular dichroism spectroscopy. The far UV spectra for wild type Spn1 and Spn1141–305 is shown. The hydrodynamic properties of Spn1 and Spn1141–305 were analyzed using analytical ultracentrifugation. A sedimentation velocity curve for (B) wild type Spn1 and (C) Spn1141–305 at two protein concentrations, and 150, 300 or 500 mM NaCl in 20 mM Tris–HCl (pH 7.5) is shown.
The CD data for full length Spn1 predicts an approximately equal number of structured and unstructured residues in the N- and C-terminal regions (Supplemental Table S2). Given that Spn1141–305 has 107 α-helical residues, full length Spn1 is expected to have only 28–50 additional residues in an α-helical conformation. The remaining structured residues in Spn1 are predicted to be in β-sheet or β-turns, both of which could be over-estimates based on the results with Spn1141–305. It is probable that the structured residues within the Spn1 termini are distributed in short regions flanked by unstructured residues. The C-terminal region of Spn1 has a small area that is predicted to be structured (42,43) while the entire N-terminus and remainder of the C-terminus are predicted to be disordered (Supplemental Figure S8).
Given that the N- and C-terminal regions of Spn1 are predicted to be predominantly disordered, it is possible that Spn1 can form a range of extended and compact structures. To further evaluate the structure of Spn1 in solution and to verify the CD results, sedimentation velocity experiments were carried out. Wild type Spn1 and Spn1141–305 were assayed over a 20-fold or 30-fold concentration range, respectively and at three different salt concentrations (150, 300 and 500 mM NaCl). Both proteins sediment as well-behaved homogeneous species and no dependencies on salt or protein concentration were detected (Figure 5B and C and Supplemental Table S3). The average sedimentation coefficients for wild type Spn1 and Spn1141–305 are 2.5S and 1.8S, respectively. An extensive 2DSA and Genetic Algorithm analysis was completed using Ultrascan software (version 9.9) for each of the highest protein concentration data sets. Both proteins sediment as a monomer. The frictional ratio is the main parameter calculated from AUC to determine the general shape of a protein in solution, and is calculated by dividing the frictional coefficient of the protein by the frictional coefficient of a sphere with the same mass. A frictional ratio of one indicates that the protein is a perfect sphere while increasingly higher ratios suggest increasing asymmetry in the protein dimensions. The frictional ratio calculated for Spn1141–305 was ∼1.4 while wild type Spn1 was approximately 2 (Supplemental Table S3). This suggests that wild type Spn1 has an elongated, asymmetric shape in contrast to the more compact, globular shape of Spn1141–305. This is consistent with, the N- and C-terminal regions of full-length Spn1 being extensively disordered.
The core domain of Spn1 (Spn1141-305) associates with RNAPII and Spt6 in vivo, but is not pre-bound at the CYC1 promoter
Since Spn1141–305 was defective for histone, DNA, and nucleosome binding we wondered if it was also defective for Spn1 functions in vivo. Spn1 associates with RNAPII, both at the promoter and in the open reading frame (ORF) of genes (17,18). Spn1K192N has reduced RNAPII and Spt6 binding, and has been shown to have drastically reduced occupancy at the CYC1 gene (18). Therefore, we decided to test whether Spn1141–305 could interact with RNAPII. To assess this, we performed a co-immunoprecipitation assay using cell extracts prepared from strains expressing TAP-tagged Spn1 or Spn1141–305. TAP-tagged protein complexes were purified from yeast cells and RNAPII in the complexes detected by immunoblot analysis with monoclonal anti-RNAPII antibodies (Figure 6A). RNAPII specifically co-immunoprecipitated with both full length Spn1 and Spn1141–305 indicating that the core domain of Spn1 is fully competent for associating with RNAPII.
Figure 6.
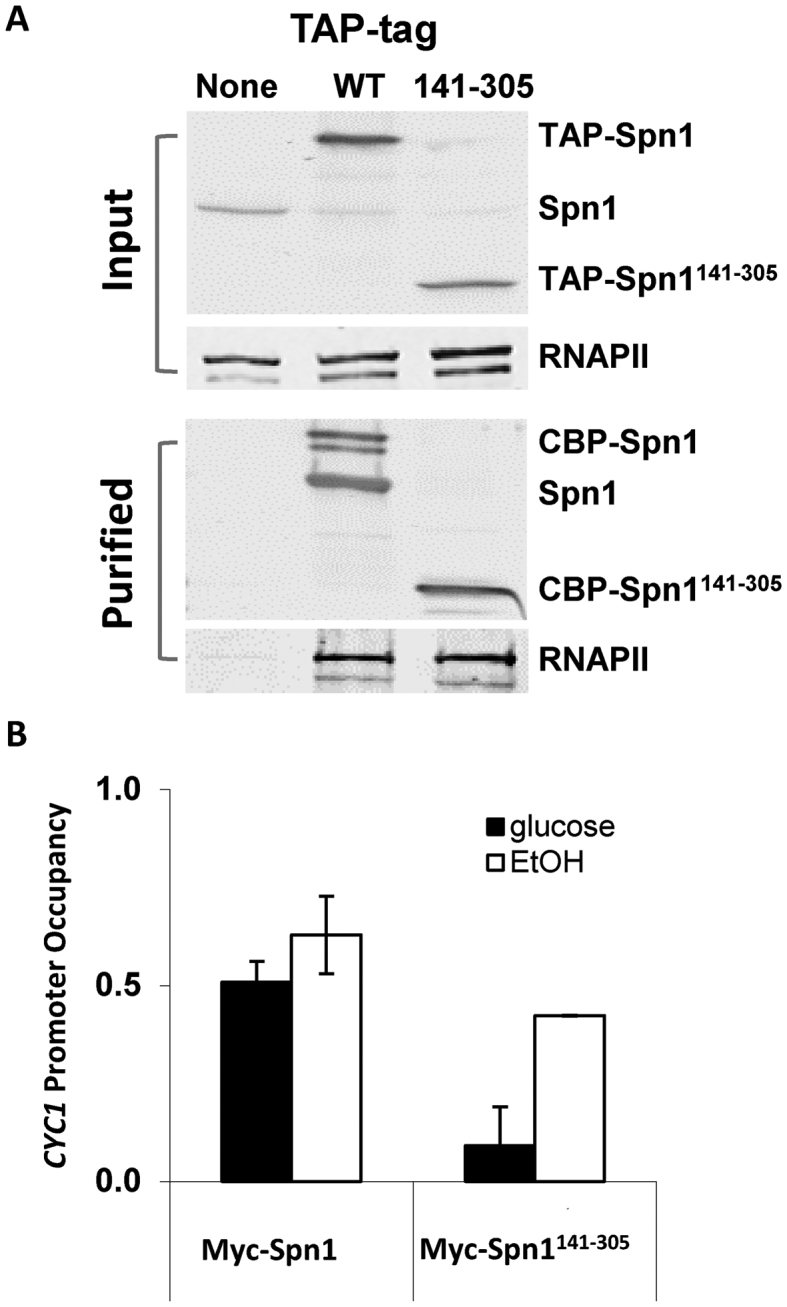
The core domain of Spn1 is competent for interaction with RNAPII, but is not stably recruited to the CYC1 promoter prior to activation of transcription. (A) Cell lysates prepared from the indicated strains were incubated with IgG sepharose beads. After extensive washing, TAP-tagged protein complexes were released from the beads by TEV protease cleavage. Protein extracts loaded onto the IgG sepharose (Input) and the released proteins (purified) were analyzed by immunoblotting with polyclonal anti-Spn1 and monoclonal anti-RNA Polymerase II (Covance 8WG16) antibodies. (B) ChIP analysis of Myc-tagged wild type Spn1 and Spn1141–305 occupancy at the CYC1 promoter under repressive (glucose) and activating (ethanol) growth conditions. Occupancies were normalized to an un-tagged strain and then to the telomere proximal region.
Since the conserved core domain of Spn1 is sufficient for complex formation with Spt6 in vitro (7), we predicted that Spn1141–305 would be functional for Spt6 interactions in vivo. To test this, we completed a co-immunoprecipitation with yeast strains bearing Myc-tagged Spn1 (wild type Spn1, Spn1K192N or Spn1141–305) and HA-tagged Spt6. As expected, Spt6 co-immunoprecipitated with wild type Spn1 and Spn1141–305, but did not precipitate with Spn1K192N (Supplemental Figure S9).
We have previously shown that prior to activation, Spn1 and the general transcription machinery (including TATA-Binding Protein, TFIIH and Ser-5 phosphorylated RNAPII) are bound to the CYC1 promoter (18,33,44). The yeast CYC1 gene encodes iso-1-cytochrome c, a mitochondrial protein that is involved in electron transport (45). CYC1 expression is regulated by carbon source: in the presence of a fermentable carbon source (such as glucose) CYC1 expression is repressed, while in the presence of a non-fermentable carbon source (such as ethanol) it is activated approximately 10-fold (46,47). We asked if Spn1141–305 like wild type Spn1, occupies the CYC1 gene under repressive (glucose) or inducing (ethanol) growth conditions using a chromatin immunoprecipitation assay (ChIP). We found that in glucose, Spn1141–305 occupancy at the CYC1 gene is significantly (2–3-fold) lower than wild type Spn1 occupancy (Figure 6B). In ethanol, Spn1141–305 occupancy increases and reaches a level similar to wild type Spn1. The decreased occupancy of Spn1141–305 at the CYC1 promoter in glucose does not correlate with RNAPII occupancy, which is unaffected by Spn1141–305 (Supplemental Figure S10). Thus, the core domain of Spn1 is not sufficient for stable association with the CYC1 gene prior to the activation of transcription, possibly due to its inability to interact with chromatin in the flanking regions of the CYC1 promoter. When transcription is induced, it is possible that the interaction of Spn1141–305 with Spt6 stabilizes its association with RNAPII, thereby increasing its promoter occupancy.
Since Spn1141–305 does not occupy the CYC1 promoter when cells are grown in glucose and cannot carry out Spn1-chromatin associated functions, we wondered if the chromatin structure at the CYC1 locus differs in the spn1141–305 strain versus the SPN1 strain. To test this possibility, we completed an indirect end-labeling analysis of Micrococcal Nuclease (MNase) digested chromatin purified from spheroplasts of SPN1 and spn1141–305 cells grown in glucose or ethanol. This assay was performed with two biological replicates of each strain and cell culturing, spheroplast isolation and MNase digestion was completed in parallel for the two cell types. The results did not differ for the two replicates. When the cells were grown in glucose, the nucleosome positions and relative occupancies were not significantly different in the spn1141–305 strain versus the SPN1 strain (Supplemental Figure S11), consistent with Spn1 occupancy not affecting chromatin structure at the CYC1 locus when transcription is repressed. The positions of the nucleosome free region (NFR) and the +1, +2 and +3 nucleosomes in our samples, were consistent with those seen in previous genome-wide assays (48,49). However, we found that the region upstream of the promoter is especially sensitive to MNase cleavage, as we did not see a well-positioned, high occupancy nucleosome at the -1 position in the chromatin from either strain (Supplemental Figure S11B). It should be noted that the lack of MNase protection is not due to over-digestion of the chromatin: visualization of the MNase cleaved DNA with Ethidium Bromide shows a ladder of bands consistent with up to six nucleosomes (Supplemental Figure S11A), indicating the MNase sensitivity is specific to the CYC1 locus.
Activation of CYC1 transcription by growth of the strains in ethanol did not change the position of the +1, +2 or +3 nucleosomes in either strain (Supplemental Figure S12B), indicating transcription does not lead to dissociation of nucleosomes within the ORF. Intriguingly, the relative intensity of the cleavage products in the ORF is increased in the spn1141–305 strain compared to the wild type strain (Supplemental Figure S12B). In addition, although the upstream region lacks the regular periodicity expected for nucleosomes, protection within this region is also increased in the spn1141–305 strain. This suggests that Spn1 chromatin functions may be required to reduce nucleosome occupancy within the CYC1 ORF, and potentially within the upstream region, when transcription is occurring.
Spn1141-305 does not cover all the essential Spn1 functions
The core domain of Spn1 (Spn1141–305) is sufficient for growth on a rich medium (YPD). However, we have shown here that this region of Spn1 is not sufficient for binding DNA, histones, or nucleosomes and cannot assemble nucleosomes in vitro. This led us to investigate whether Spn1141–305 is truly covering all of the Spn1 cellular functions. We introduced the spn1141–305 allele into an otherwise wild type strain (BY4741) and assessed the growth of the cells under 10 conditions (Figure 7 and Supplemental Table S4). No obvious growth defects were observed when growth of the strain with Spn1141–305 was compared to the strain with wild type Spn1 or a strain containing Spn1K192N. We previously found that combining the spn1K192N allele with deletion of the genes encoding the transcription elongation factor, TFIIS (DST1) or the Polymerase Associated Factor 1 (PAF1) complex subunit, RTF1 resulted in synthetic growth defects (18). We next introduced the spn1141–305 allele into the dst1Δ and rtf1Δ strains and assessed growth of the resulting strains as described above. The combination of the spn1141–305 allele with deletion of either DST1 or RTF1 resulted in significant growth defects (Figure 7 and Supplemental Table S4). Introduction of the spn1141–305 allele into the rtf1Δ strain was especially deleterious, leading to reduced growth at 30°C on YPD and synthetic lethality when the cells were challenged by non-optimal growth conditions. This indicates that Spn1141–305 is not competent for all Spn1 cellular functions.
Figure 7.
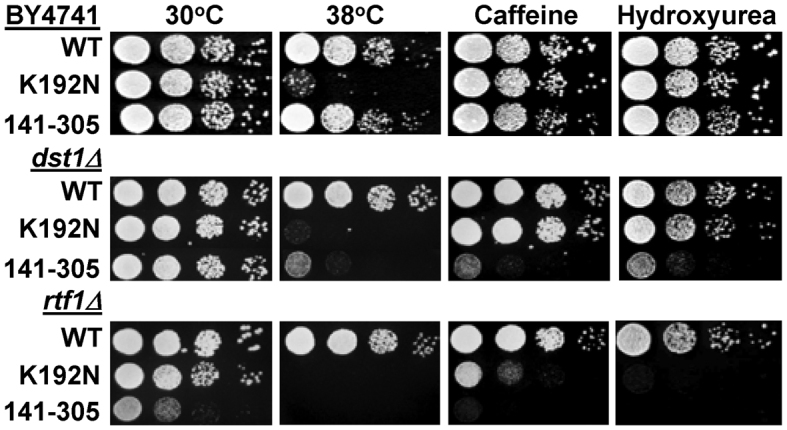
Severe growth defects are seen when the spn1K192N and spn1141–305 alleles are combined with elongation factor deletion strains (dst1Δ, rtf1Δ). Ten-fold serial dilutions of the indicated strains were spotted onto solid YPD, YPD with 10 mM Caffeine, or YPD with 150 mM hydroxyurea and incubated at 30°C unless otherwise noted. Pictures were taken after 2–4 days of growth.
Histone chaperones are known to be involved in multiple cellular processes like transcription, DNA replication, and DNA repair that are affected by the presence of chromatin (reviewed in (50)). Therefore, we reasoned that combining the spn1141–305 allele with histone chaperone deletions might provide insight into the cellular pathways that require Spn1 chromatin associated functions. In this analysis we chose strains in which NAP1, VPS75, HIR1, HIR2, ASF1, and the Chromatin Assembly Factor-1 (CAF-1) subunits CAC1 and CAC3, had been deleted as these histone chaperones are known to have over-lapping functions in transcription, replication and DNA repair. Growth of the strains with Spn1141–305 was again compared to the parental deletion strain (with wild type Spn1) and a strain containing Spn1K192N. The spn1K192N allele had not been previously evaluated in the histone chaperone deletion strains. The combination of the spn1141–305 allele with the histone chaperone deletions produced a range of growth defects (Figure 8 and Supplemental Table S5). The vps75Δ and cac3Δ strains were unaffected, nap1Δ had a mild growth defect when caffeine was present, rtt106Δ, asf1Δ, and cac1Δ had obvious growth defects under several of the growth conditions, while the hir1Δ and hir2Δ strains were profoundly sick when grown on YPD at 30°C, and did not grow at all when challenged by non-optimal growth conditions. The spn1K192N allele did not severely affect the growth of any of the strains, although moderate growth defects were seen with the hir1Δ and hir2Δ strains when the cells were grown on media containing hydroxyurea (Figure 8 and Supplemental Table S5). Collectively, the phenotypes seen with spn1141–305 suggest that the chromatin associated functions of Spn1 could play roles in transcription, DNA replication and/or repair.
Figure 8.

Spn1 interacts genetically with a broad spectrum of histone chaperones. Ten-fold serial dilutions of the indicated strains were spotted onto solid YPD, YPD with 10 mM Caffeine and YPD with 150 mM hydroxyurea and incubated at 30°C unless otherwise noted. Pictures were taken after 2–7 days of growth.
DISCUSSION
The chromatin associated functions of Spn1 overlap with its binding partner, Spt6. Both proteins bind histones H3–H4, DNA and nucleosomes, and can assemble nucleosomes in vitro (3,7,51). However, Spt6 binding to nucleosomes requires Nhp6 and it was suggested that nucleosome binding by Spt6 occurs after the nucleosomes have been destabilized by Nhp6 (7). We have shown here that Spn1 alone is capable of nucleosome binding. The nucleosome and Spn1 binding sites on Spt6 overlap, and Spn1120–410 inhibits nucleosome binding by Spt6, indicating that Spn1 binding regulates the ability of Spt6 to bind nucleosomes (7). Future studies are needed to elucidate whether Spn1 can bridge the binding of Spt6 to nucleosomes or if formation of a Spn1–Spt6 complex mutually inhibits nucleosome binding.
The results of our structural analysis of the N- and C-terminal regions of Spn1 agree with structural models that predict they are intrinsically disordered (42) and have an elongated, asymmetric shape that is consistent with the termini extending away from the core domain. In general, Intrinsically Disordered Regions (IDRs) are thought to increase the binding diversity of a protein by providing sites for binding to multiple proteins and/or DNA (reviewed in (52)). Additionally, amino acids within IDRs are frequently post-translationally modified in association with cellular regulation. The IDRs of Spn1 are required for binding to histones, DNA, and nucleosomes, and 17 residues within the N-terminal and C-terminal IDRs have been shown to be either phosphorylated or ubiquitinated (53).
The presence of IDRs in the N- and C-terminal regions of Spn1 is conserved in higher eukaryotic Spn1 (called Iws1), demonstrating their functional importance in the Spn1/Iws1 family of proteins. The IDRs of human Iws1 have been shown to bind the setd2 histone methyltransferase, the REF1/Aly mRNA export factor and Lens Epithelium Derived Growth Factor/p75 (LEDGF/p75) (9,54,55). Iws1 binding to Spt6 and subsequent recruitment of LEDGF/p75 induces the formation of repressive chromatin at the HIV-LTR, and reduction of Iws1 levels within HeLa cells leads to reactivated HIV expression (55). Post-translational modification of residues within the C-terminal IDR of Iws1 have also been linked to lung cancer in humans (56). Phosphorylation of Iws1 residues, Ser720/Thr721 was shown to regulate alternative splicing of the Fibroblast Growth Factor Receptor-2 (FGFR-2) transcript leading to increased proliferation, migration and invasiveness of lung tumor cells (56). Although the presence of IDRs in Spn1/Iws1 is conserved, the length of these regions is divergent and currently, it is not known if the specific binding functions of the IDRs are conserved. However, the N- and C-terminal IDRs of Spn1/Iws1 are clearly regions that are used to interact with other proteins in carrying out its cellular functions.
We have shown here that Spn1141–305 is sufficient for interacting with RNAPII and Spt6, although it does not appear to be stably recruited to the CYC1 gene prior to the activation of transcription. This suggests that wild type Spn1 interacts with nucleosomes at the CYC1 promoter through its IDRs to stabilize its binding, and since Spn1141–305 does not bind nucleosomes, its binding to the CYC1 promoter is less stable. The absence of both Spn1K192N and Spn1141–305 at the transcriptionally repressed CYC1 promoter indicates that the ability of Spn1 to associate with RNAPII and to interact with nucleosomes (respectively) are required for Spn1 occupancy at CYC1.
The introduction of the spn1141–305 allele into the dst1Δ, rtf1Δ, and histone chaperone deletion strains led to significant synthetic growth defects. Previously, we saw exacerbation of the dst1Δ and rtf1Δ phenotypes with the spn1K192N allele (18), so it was not unexpected that spn1141–305 also affected the growth of these deletion strains. However, the severity of the growth defects seen with spn1141–305 indicates the N- and C-termini of Spn1 are required for critical functions in the absence of TFIIS (Dst1) and Rtf1. TFIIS is a transcription elongation factor that functions in the release of stalled RNAPII (57,58), and human TFIIS has been shown to stimulate transcription through multiple contiguous nucleosomes in vitro (59). Spn1 residues 215–295 and the N-terminal domain of mouse TFIIS exhibit sequence and structural similarity (11,60). It is possible that the K192N substitution destabilizes this conserved structural region in Spn1, leading to the growth defects seen in the dst1Δ strain. However, this structural region is unaffected in Spn1141–305 and therefore, the growth defects seen with this allele must be due to functions that were lost with deletion of the N- and C-termini of Spn1, including the chromatin functions we have elucidated here. Since Spn1141–305 cannot perform Spn1 chromatin functions, it is possible that the phenotypes seen in the dst1Δ strain are due to a further decrease in transcription elongation through nucleosomes.
Rtf1 is a subunit of the PAF1 complex that, like Spn1, has been shown to have multiple functions associated with transcription initiation, elongation and termination, and mRNA processing (reviewed in (61)). The spn1141–305 allele is especially deleterious in the rtf1Δ background, with the double mutants growing poorly under optimal conditions and not at all under stress inducing conditions. This synthetic growth defect is conserved in Arabidopsis where a Spn1/Iws1 mutant could not be combined with deletion of a gene homologous to RTF1, presumably due to embryonic lethality (62). The combination of rtf1Δ with either spnK192N or spn1141–305 produces significant growth defects, suggesting that Spn1 functions that reside throughout the protein (N- and C-termini and the core domain) are necessary when PAF1 complex functions are compromised. Given the number of overlapping functions between the PAF1 complex and Spn1 it is not surprising that combining their mutants reduces cellular viability.
Histone chaperones have redundant functions that are critical for proper transcription, replication and DNA repair (reviewed in (50)). Nap1 was the only H2A–H2B histone chaperone that we tested in our Spn1 genetic assay and we saw a mild growth defect on plates with caffeine, suggesting that Nap1 and Spn1 functions overlap in yeast. To date, Spn1 has not been shown to play a role in DNA replication or repair. However, we found that SPN1 interacts genetically with CAC1 (a CAF-1 subunit gene), ASF1 and RTT106 all of which are known to encode proteins that function in DNA repair and/or replication (50). While Asf1 and Rtt106 also function in transcription (63,64), CAF-1 does not, indicating the growth defects seen with spn1141–305 and cac1Δ are most likely due to negative effects on DNA replication or repair. Finally, spn1141–305 was essentially synthetically lethal when it was combined with deletion of HIR1 and HIR2, which encode subunits of the HIR complex (65,66). The HIR complex in association with Asf1 and Rtt106 has been shown to regulate histone gene expression (67,68), and to function in replication independent histone deposition and the repression of cryptic transcription (66,69). Formosa et al., demonstrated that HIR complex mutants were also synthetically lethal when combined with mutations in the elongation factors FACT (Spt16), DSIF (Spt4/5), Spt6 and the PAF-1 complex (70). Currently, it is not known how the elongation factors and the HIR complex work together in the cell but, it is clear elongation factors, including Spn1, are essential when functions of the HIR complex are compromised.
The loss of Spn1 chromatin functions in the spn1141–305 mutant strain does not affect chromatin structure at the CYC1 locus when cells are grown in glucose and transcription is repressed. However, when CYC1 transcription is activated by growth in ethanol there is an obvious increase in protection from MNase cleavage, both upstream of the CYC1 promoter, and within the ORF. This suggests that Spn1 plays a role in reducing nucleosome occupancy at the CYC1 locus, perhaps to keep the DNA more accessible, especially in the promoter region. Collectively, the results presented here suggest that Spn1 functions in both the assembly and disassembly of nucleosomes. It has been proposed that the Spn1–Spt6 complex monitors the correct assembly of nucleosomes (16). The increase in nucleosome occupancy at the CYC1 locus could be due to defective nucleosome surveillance by the Spn1141–305–Spt6 complex, perhaps due to a loss of Spn1 chromatin functions or a decrease in the regulation of Spt6 functions. Going forward it will be interesting to see how Spn1 and its binding partner Spt6 use their chromatin functions, both together and independently, to regulate chromatin architecture and cellular functions.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Robert Woody and Dr Uma Muthurajan for their expert technical assistance, and Desirée Quiñones Vega and Raira Ank for general lab assistance and cell culturing. We also thank Dr Hataichanok Scherman at CSU Protein Expression and Purification Facility for purified histones.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
American Heart Association [15POST22770011 to A.K., 10PRE4160125 to L.Z.]; International Rett Syndrome Foundation [2822 to A.K.]; National Institutes of Health [GM067777 to K.L.]; National Science Foundation [MCB-1330019 to L.A.S.]; Howard Hughes Medical Institute (to K.L.). Funding for open access charge: National Science Foundation [MCB-1330019].
Conflict of interest statement. None declared.
REFERENCES
- 1. Rando O.J., Winston F.. Chromatin and transcription in yeast. Genetics. 2012; 190:351–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tanny J.C. Chromatin modification by the RNA Polymerase II elongation complex. Transcription. 2014; 5:e988093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bortvin A., Winston F.. Evidence that Spt6p controls chromatin structure by a direct interaction with histones. Science. 1996; 272:1473–1476. [DOI] [PubMed] [Google Scholar]
- 4. Chen S.H., Albuquerque C.P., Liang J., Suhandynata R.T., Zhou H.. A proteome-wide analysis of kinase-substrate network in the DNA damage response. J. Biol. Chem. 2010; 285:12803–12812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krogan N.J.M., Kim M., Ahn S.H., Zhong G., Kobor M.S., Cagney G., Emili A., Shilatifard A., Buratowski S., Greenblatt J.F.. RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Mol. Cell. Biol. 2002; 22:6979–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lindstrom D.L., Squazzo S.L., Muster N., Burckin T.A., Wachter K.C., Emigh C.A., McCleery J.A., Yates J.R.r., Hartzog G.A.. Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Mol. Cell. Biol. 2003; 1368–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McDonald S.M., Close D., Xin H., Formosa T., Hill C.P.. Structure and biological importance of the Spn1–Spt6 interaction, and its regulatory role in nucleosome binding. Mol. Cell. 2010; 40:725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yoh S.M., Cho H., Pickle L., Evans R.M., Jones K.A.. The Spt6 SH2 domain binds Ser2-P RNAPII to direct Iws1-dependent mRNA splicing and export. Genes Dev. 2007; 21:160–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoh S.M., Lucas J.S., Jones K.A.. The Iws1:Spt6:CTD complex controls cotranscriptional mRNA biosynthesis and HYPB/Setd2-mediated histone H3K36 methylation. Genes Dev. 2008; 22:3422–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fischbeck J.A., Kraemer S.M., Stargell L.A.. SPN1, a conserved gene identified by suppression of a postrecruitment-defective yeast TATA-binding protein mutant. Genetics. 2002; 162:1605–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pujari V., Radebaugh C.A., Chodaparambil J.V., Muthurajan U.M., Almeida A.R., Fischbeck J.A., Luger K., Stargell L.A.. The transcription factor Spn1 regulates gene expression via a highly conserved novel structural motif. J. Mol. Biol. 2010; 404:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li L., Ye H., Guo H., Yin Y.. Arabidopsis IWS1 interacts with transcription factor BES1 and is involved in plant steroid hormone brassinosteroid regulated gene expression. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:3918–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Diebold M.L., Koch M., Loeliger E., Cura V., Winston F., Cavarelli J., Romier C.. The structure of an Iws1/Spt6 complex reveals an interaction domain conserved in TFIIS, Elongin A and Med26. EMBO J. 2010; 29:3979–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Adkins M.W., Tyler J.K.. Transcriptional activators are dispensable for transcription in the absence of Spt6-mediated chromatin reassembly of promoter regions. Mol. Cell. 2006; 21:405–416. [DOI] [PubMed] [Google Scholar]
- 15. Kaplan C.D., Laprade L., Winston F.. Transcription elongation factors repress transcription initiation from cryptic sites. Science. 2003; 301:1096–1099. [DOI] [PubMed] [Google Scholar]
- 16. McCullough L., Connell Z., Petersen C., Formosa T.. The abundant histone chaperones Spt6 and FACT collaborate to assemble, inspect, and maintain chromatin structure in Saccharomyces cerevisiae. Genetics. 2015; 201:1031–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mayer A., Lidschreiber M., Siebert M., Leike K., Soding J., Cramer P.. Uniform transitions of the general RNA polymerase II transcription complex. Nat. Struct. Mol. Biol. 2010; 17:1272–1278. [DOI] [PubMed] [Google Scholar]
- 18. Zhang L., Fletcher A.G., Cheung V., Winston F., Stargell L.A.. Spn1 regulates the recruitment of Spt6 and the Swi/Snf complex during transcriptional activation by RNA polymerase II. Mol. Cell. Biol. 2008; 28:1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hainer S.J., Pruneski J.A., Mitchell R.D., Monteverde R.M., Martens J.A.. Intergenic transcription causes repression by directing nucleosome assembly. Genes Dev. 2011; 25:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lowary P.T., Widom J.. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998; 276:19–42. [DOI] [PubMed] [Google Scholar]
- 21. Dyer P.N., Edayathumangalam R.S., White C.L., Bao Y., Chakravarthy S., Muthurajan U.M., Luger K.. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 2004; 375:23–44. [DOI] [PubMed] [Google Scholar]
- 22. Carruthers L.M., Bednar J., Woodcock C.L., Hansen J.C.. Linker histones stabilize the intrinsic salt-dependent folding of nucleosomal arrays: mechanistic ramifications for higher-order chromatin folding. Biochemistry. 1998; 37:14776–14787. [DOI] [PubMed] [Google Scholar]
- 23. Demeler B., van Holde K.E.. Sedimentation velocity analysis of highly heterogeneous systems. Anal. Biochem. 2004; 335:279–288. [DOI] [PubMed] [Google Scholar]
- 24. Park Y.J., Sudhoff K.B., Andrews A.J., Stargell L.A., Luger K.. Histone chaperone specificity in Rtt109 activation. Nat. Struct. Mol. Biol. 2008; 15:957–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winkler D.D., Luger K., Hieb A.R.. Quantifying chromatin-associated interactions: the HI-FI system. Methods Enzymol. 2012; 512:243–274. [DOI] [PubMed] [Google Scholar]
- 26. Mattiroli F., Gu Y., Yadav T., Balsbaugh J.L., Harris M.R., Findlay E.S., Liu Y., Radebaugh C.A., Stargell L.A., Ahn N.G. et al. DNA-mediated association of two histone-bound complexes of yeast Chromatin Assembly Factor-1 (CAF-1) drives tetrasome assembly in the wake of DNA replication. eLife. 2017; 6:e22799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lusser A., Kadonaga J.T.. Strategies for the reconstitution of chromatin. Nat. Methods. 2004; 1:19–26. [DOI] [PubMed] [Google Scholar]
- 28. Andrews A.J., Downing G., Brown K., Park Y.J., Luger K.. A thermodynamic model for Nap1-histone interactions. J. Biol. Chem. 2008; 283:32412–32418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sreerama N., Woody R.W.. Computation and analysis of protein circular dichroism spectra. Methods Enzymol. 2004; 383:318–351. [DOI] [PubMed] [Google Scholar]
- 30. Carruthers L.M., Schirf V.R., Demeler B., Hansen J.C.. Sedimentation velocity analysis of macromolecular assemblies. Methods Enzymol. 2000; 321:66–80. [DOI] [PubMed] [Google Scholar]
- 31. Demeler B., Behlke J., Ristau O.. Molecular parameters from sedimentation velocity experiments: whole boundary fitting using approximate and numerical solutions of Lamm equation. Methods Enzymol. 2000; 321:38–66. [DOI] [PubMed] [Google Scholar]
- 32. Kaiser P., Meierhofer D., Wang X., Huang L.. Tandem affinity purification combined with mass spectrometry to identify components of protein complexes. Methods Mol. Biol. 2008; 439:309–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee S.K., Fletcher A.G., Zhang L., Chen X., Fischbeck J.A., Stargell L.A.. Activation of a poised RNAPII-dependent promoter requires both SAGA and mediator. Genetics. 2010; 184:659–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen X., D’Arcy S., Radebaugh C.A., Krzizike D.D., Giebler H.A., Huang L., Nyborg J.K., Luger K., Stargell L.A.. Histone chaperone Nap1 is a major regulator of histone H2A–H2B dynamics at the inducible GAL locus. Mol. Cell. Biol. 2016; 36:1287–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Collins S.R., Miller K.M., Maas N.L., Roguev A., Fillingham J., Chu C.S., Schuldiner M., Gebbia M., Recht J., Shales M. et al. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature. 2007; 446:806–810. [DOI] [PubMed] [Google Scholar]
- 36. Hansen J.C. Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions. Annu. Rev. Biophys. Biomol. Struct. 2002; 31:361–392. [DOI] [PubMed] [Google Scholar]
- 37. Tremethick D.J. Higher-order structures of chromatin: the elusive 30 nm fiber. Cell. 2007; 128:651–654. [DOI] [PubMed] [Google Scholar]
- 38. McBryant S.J., Krause C., Woodcock C.L., Hansen J.C.. The silent information regulator 3 protein, SIR3p, binds to chromatin fibers and assembles a hypercondensed chromatin architecture in the presence of salt. Mol. Cell. Biol. 2008; 28:3563–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Postnikov Y., Bustin M.. Regulation of chromatin structure and function by HMGN proteins. Biochim. Biophys. Acta. 2010; 1799:62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luger K., Dechassa M.L., Tremethick D.J.. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair?. Nat. Rev. Mol. Cell Biol. 2012; 13:436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Donham D.C. 2nd, Scorgie J.K., Churchill M.E.. The activity of the histone chaperone yeast Asf1 in the assembly and disassembly of histone H3/H4-DNA complexes. Nucleic Acids Res. 2011; 39:5449–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jones D.T., Cozzetto D.. DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics. 2015; 31:857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Buchan D.W., Minneci F., Nugent T.C., Bryson K., Jones D.T.. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013; 41:W349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen J.-L., Attardi L.D., Verrijzer C.P., Yokomori K., Tjian R.. Assembly of recombinant TFIID reveals differential coactivator requirements for distinct transcriptional activators. Cell. 1994; 79:93–105. [DOI] [PubMed] [Google Scholar]
- 45. Sherman F., Stewart J.W., Margoliash E., Parker J., Campbell W.. The structural gene for yeast cytochrome C. Proc. Natl. Acad. Sci. U.S.A. 1966; 55:1498–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guarente L., Lalonde B., Gifford P., Alani E.. Distinctly regulated tandem upstream activation sites mediate catabolite repression of the CYC1 gene of S.cerevisiae. Cell. 1984; 36:503–511. [DOI] [PubMed] [Google Scholar]
- 47. Guarente L., Mason T.. Heme regulates transcription of the CYC1 gene of S.cerevisiae via an upstream activation site. Cell. 1983; 32:1279–1286. [DOI] [PubMed] [Google Scholar]
- 48. Kaplan N., Moore I., Fondufe-Mittendorf Y., Gossett A.J., Tillo D., Field Y., Hughes T.R., Lieb J.D., Widom J., Segal E.. Nucleosome sequence preferences influence in vivo nucleosome organization. Nat. Struct. Mol. Biol. 2010; 17:918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mavrich T.N., Ioshikhes I.P., Venters B.J., Jiang C., Tomsho L.P., Qi J., Schuster S.C., Albert I., Pugh B.F.. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008; 18:1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hammond C.M., Stromme C.B., Huang H., Patel D.J., Groth A.. Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell Biol. 2017; 18:141–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Close D., Johnson S.J., Sdano M.A., McDonald S.M., Robinson H., Formosa T., Hill C.P.. Crystal structures of the S. cerevisiae Spt6 core and C-terminal tandem SH2 domain. J. Mol. Biol. 2011; 408:697–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Babu M.M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 2016; 44:1185–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cherry J.M., Hong E.L., Amundsen C., Balakrishnan R., Binkley G., Chan E.T., Christie K.R., Costanzo M.C., Dwight S.S., Engel S.R. et al. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic acids research. 2012; 40:D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tesina P., Cermakova K., Horejsi M., Prochazkova K., Fabry M., Sharma S., Christ F., Demeulemeester J., Debyser Z., De Rijck J. et al. Multiple cellular proteins interact with LEDGF/p75 through a conserved unstructured consensus motif. Nat. Commun. 2015; 6:7968. [DOI] [PubMed] [Google Scholar]
- 55. Gerard A., Segeral E., Naughtin M., Abdouni A., Charmeteau B., Cheynier R., Rain J.C., Emiliani S.. The integrase cofactor LEDGF/p75 associates with Iws1 and Spt6 for postintegration silencing of HIV-1 gene expression in latently infected cells. Cell Host Microbe. 2015; 17:107–117. [DOI] [PubMed] [Google Scholar]
- 56. Sanidas I., Polytarchou C., Hatziapostolou M., Ezell S.A., Kottakis F., Hu L., Guo A., Xie J., Comb M.J., Iliopoulos D. et al. Phosphoproteomics screen reveals akt isoform-specific signals linking RNA processing to lung cancer. Mol. Cell. 2014; 53:577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Reinberg D., Horikoshi M., Roeder R.G.. Factors involved in specific transcription by mammalian RNA polymerase II: Functional analysis of initiation factors IIA and IID, and identification of a new factor operating at sequences downstream of the initiation site. J. Biol. Chem. 1987; 262:3322–3330. [PubMed] [Google Scholar]
- 58. Reines D., Chamberlin M.J., Kane C.M.. Transcription elongation factor SII (TFIIS) enables RNA polymerase II to elongate through a block to transcription in a human gene in vitro. J. Biol. Chem. 1989; 264:10799–10809. [PubMed] [Google Scholar]
- 59. Guermah M., Kim J., Roeder R.G.. Transcription of in vitro assembled chromatin templates in a highly purified RNA polymerase II system. Methods. 2009; 48:353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ling Y., Smith A.J., Morgan G.T.. A sequence motif conserved in diverse nuclear proteins identifies a protein interaction domain utilised for nuclear targeting by human TFIIS. Nucleic Acids Res. 2006; 34:2219–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tomson B.N., Arndt K.M.. The many roles of the conserved eukaryotic Paf1 complex in regulating transcription, histone modifications, and disease states. Biochim. Biophys. Acta. 2013; 1829:116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Widiez T., El Kafafi el S., Girin T., Berr A., Ruffel S., Krouk G., Vayssieres A., Shen W.H., Coruzzi G.M., Gojon A. et al. High nitrogen insensitive 9 (HNI9)-mediated systemic repression of root NO3- uptake is associated with changes in histone methylation. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:13329–13334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Adkins M.W., Howar S.R., Tyler J.K.. Chromatin disassembly mediated by the histone chaperone Asf1 is essential for transcriptional activation of the yeast PHO5 and PHO8 genes. Mol. Cell. 2004; 14:657–666. [DOI] [PubMed] [Google Scholar]
- 64. Imbeault D., Gamar L., Rufiange A., Paquet E., Nourani A.. The Rtt106 histone chaperone is functionally linked to transcription elongation and is involved in the regulation of spurious transcription from cryptic promoters in yeast. J. Biol. Chem. 2008; 283:27350–27354. [DOI] [PubMed] [Google Scholar]
- 65. Prochasson P., Florens L., Swanson S.K., Washburn M.P., Workman J.L.. The HIR corepressor complex binds to nucleosomes generating a distinct protein/DNA complex resistant to remodeling by SWI/SNF. Genes Dev. 2005; 19:2534–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Green E.M., Antczak A.J., Bailey A.O., Franco A.A., Wu K.J., Yates J.R. 3rd, Kaufman P.D.. Replication-independent histone deposition by the HIR complex and Asf1. Curr. Biol.: CB. 2005; 15:2044–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Eriksson P.R., Ganguli D., Nagarajavel V., Clark D.J.. Regulation of histone gene expression in budding yeast. Genetics. 2012; 191:7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fillingham J., Kainth P., Lambert J.P., van Bakel H., Tsui K., Pena-Castillo L., Nislow C., Figeys D., Hughes T.R., Greenblatt J. et al. Two-color cell array screen reveals interdependent roles for histone chaperones and a chromatin boundary regulator in histone gene repression. Mol. Cell. 2009; 35:340–351. [DOI] [PubMed] [Google Scholar]
- 69. Silva A.C., Xu X., Kim H.S., Fillingham J., Kislinger T., Mennella T.A., Keogh M.C.. The replication-independent histone H3–H4 chaperones HIR, ASF1, and RTT106 co-operate to maintain promoter fidelity. J. Biol. Chem. 2012; 287:1709–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Formosa T., Ruone S., Adams M.D., Olsen A.E., Eriksson P., Yu Y., Rhoades A.R., Kaufman P.D., Stillman D.J.. Defects in SPT16 or POB3 (yFACT) in Saccharomyces cerevisiae cause dependence on the Hir/Hpc pathway: polymerase passage may degrade chromatin structure. Genetics. 2002; 162:1557–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.