


Abstract
Short read massive parallel sequencing has emerged as a standard diagnostic tool in the medical setting. However, short read technologies have inherent limitations such as GC bias, difficulties mapping to repetitive elements, trouble discriminating paralogous sequences, and difficulties in phasing alleles. Long read single molecule sequencers resolve these obstacles. Moreover, they offer higher consensus accuracies and can detect epigenetic modifications from native DNA. The first commercially available long read single molecule platform was the RS system based on PacBio's single molecule real-time (SMRT) sequencing technology, which has since evolved into their RSII and Sequel systems. Here we capsulize how SMRT sequencing is revolutionizing constitutional, reproductive, cancer, microbial and viral genetic testing.
INTRODUCTION
Modern medical genomics research and diagnostics relies heavily on DNA sequencing. Sequencing technologies are used in a wide range of applications during the entire human lifespan, from prenatal diagnostics, to newborn screening, to diagnosing rare diseases, hereditary forms of cancer, pharmacogenetics testing and predisposition testing for a plethora of diseases. It can even include testing for future generations in terms of carrier screening and pre-implantation genetic diagnoses (1,2).
The history of sequencing technologies can be broken up into three phases: first-, second- and third-generation sequencing (3). Though earlier first-generation technologies provided ground breaking discoveries, the big revolution in sequencing began with the invention of the ‘chain-termination’ or dideoxy technique, or what is today called Sanger sequencing (3,4). Improvements in chemistry and switching from gels to capillary based electrophoresis led to the current Sanger machines that provide low-throughput, high quality reads of up to ∼1 kb. Sanger sequencing is still often referred to as the gold standard and is commonly used for diagnosing Mendelian disorders (5) and targeted validation of higher-throughput sequencing results.
The first decade of the 21st century brought forth the development of multiple new methods of DNA sequencing (6). As opposed to first-generation platforms, these new second-generation technologies have considerably shorter reads (up to a few hundred bps), but at massively higher throughput (up to billions of reads per run). Common short-read platforms based on fluorescence include Illumina's bridge amplification and sequencing by synthesis technologies (e.g. HiSeq and MiSeq), Roche 454 pyrosequencers, and Applied Biosystem's sequencing by oligonucleotide ligation and detection (SOLiD) platforms. Additional short-read platforms include the Ion Torrent sequencers that detect nucleotides by the difference in pH as a result of hydrogen ions emitted during polymerisation, as opposed to light signals. Though these short-read platforms have permitted scientists to quickly hunt for causative mutations in a panel of disease genes, the exome, or even the entire human genome in both research and clinical settings (7), they all share common pitfalls and drawbacks. The short read lengths hinder assigning reads to complex parts of the genome (8), phasing of variants (9), resolving repeat regions (10) and introduce gaps and ambiguous regions in de novo assemblies (11,12). The amplification steps during library preparation and/or the actual sequencing reaction also introduce chimeric reads (13), variation in repeat size, and an underrepresentation of GC-rich/poor regions. Taken together, these drawbacks hinder the utility of diagnostic variant detection.
Third-generation is in general characterized as single molecule sequencing and is fundamentally different from clonal based second generation sequencing methods. Helicos provided the first commercial application of single molecule sequencing based on fluorescence detection and sequencing by synthesis. Though lacking amplification biases, such as underrepresentation of GC-rich/poor regions, this early single molecule sequencing still produced short (often 35 bp) read lengths (14). Two newer technologies, single molecule real-time (SMRT) sequencing by Pacific Biosciences (PacBio) (15) and nanopore sequencing by Oxford Nanopore Technologies (16), offer the advantages of single molecule sequencing, including exceptionally long read lengths (>20 kb). These platforms permit sequencing/assembly through repetitive elements, direct variant phasing, and even direct detection of epigenetic modifications (17,18). Sequencing also only lasts several hours, which is very appealing in a diagnostic setting. Though simple and low-cost nanopore based technologies (reviewed in (18–20)) are catching on and likely represent future platforms, SMRT sequencing is currently more matured and therefore diagnostically applicable at this time. Here we review how SMRT sequencing is being implemented in human genetic diagnostics.
SMRT SEQUENCING TECHNOLOGY AND TERMINOLOGY
Before SMRT sequencing, a library needs to be prepared from double stranded DNA input material (Figure 1A). Typically this often requires five or more micrograms of DNA which can limit some applications. The library preparation consists of simply ligating hairpin adapters onto DNA molecules, thereby circularizing them into a construct termed a SMRTbell (Figure 1B) (21). Next, a primer and a polymerase are annealed to the adapter whereupon the library is loaded on a SMRT Cell containing 150 000 nanoscale observation chambers (Zero Mode Waveguides (ZMWs)) for the RSII system and up to a million on the newer Sequel platform. The polymerase bound SMRTbells are then loaded into the ZMWs (Figure 1C). Ideally as many ZMWs should be loaded with exactly one SMRTbell as possible to maximise throughput and read lengths. For a good run, this is around one third to one half of the ZMWs per SMRT cell. Hence a SMRT cell typically produces ∼55 000 reads for the RSII system and 365 000 reads for the Sequel system (Table 1). The actual sequencing reaction occurs within each ZMW, whose small diameter only permits the smallest available volume for light detection (22). The polymerase within each ZMW incorporates fluorescently labeled nucleotides, emitting a fluorescent signal that is recorded by a camera in real-time (Figure 1C). These signals are converted to long sequences termed continuous long reads (CLR) (22), linear reads, or polymerase reads. For a short insert library, the circular structure of the molecule results in the insert sequence being covered multiple times by the CLR. Each pass of an original strand is termed a subread. In addition, all subreads from the same molecule can be combined into one highly accurate consensus sequence termed a circular consensus sequence (CCS) or reads-of-insert (ROI) (Figure 1F–H, left panel). These two terms are often used interchangeably, but by definition the difference is CCS requires two full sequencing passes of the insert whereas ROI can be defined starting from even a partial pass.
Figure 1.
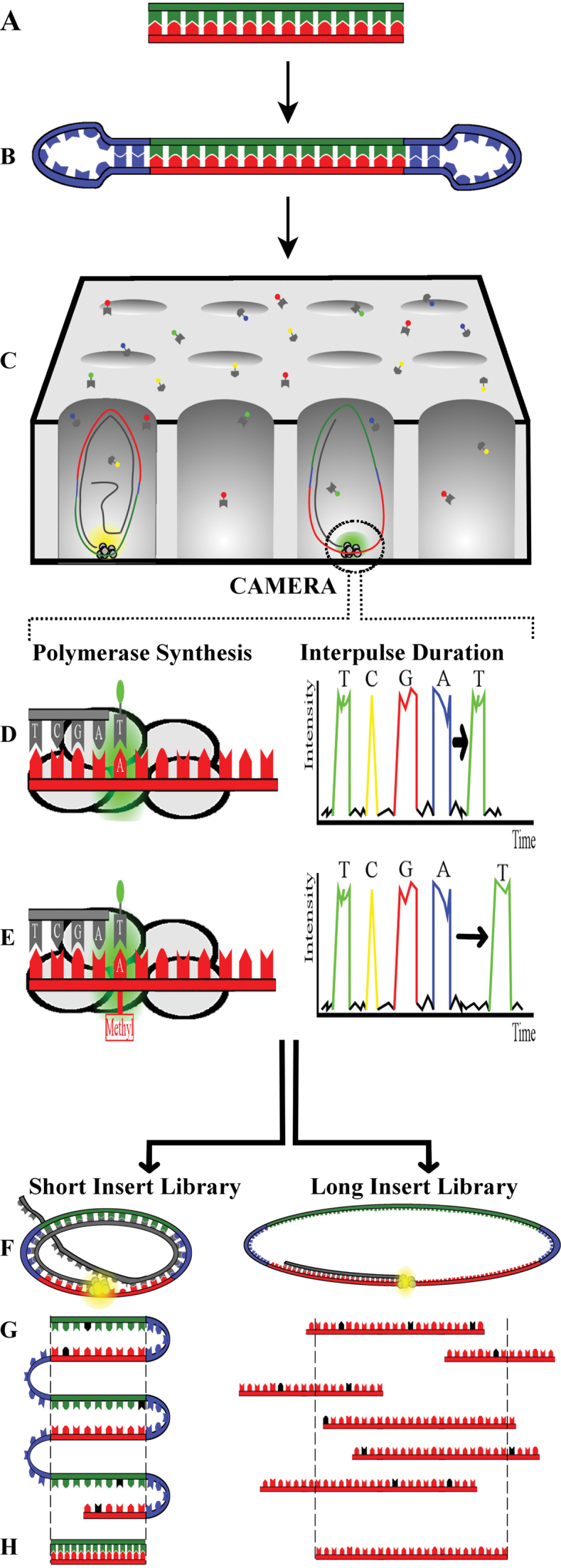
Overview of SMRT Sequencing Technology. Sequencing starts with preparing a library from double stranded DNA (A) to which hairpin adapters are ligated (B). This library is thereafter loaded onto a SMRT Cell made up of nanoscale observation chambers (Zero Mode Waveguides (ZMWs)). The DNA molecules in the library will be pulled to the bottom of the ZMW where the polymerase will incorporate fluorescently labelled nucleotides (C). Note that not all ZMWs will contain a DNA molecule because the library is loaded by diffusion. The fluorescence emitted by the nucleotides is recorded by a camera in real-time. Hence, not only the fluorescence color can be registered, but also the time between nucleotide incorporation which is called the interpulse duration (IPD) (D, right panel). When a sequencing polymerase encounters nucleotides on the DNA strand containing an (epigenetic) modification, like for example a 6-methyl adenosine modification (E, left panel), then the IPD will be delayed (E, right panel) compared to non-methylated DNA (D, right panel). Due to the circular structure of the library, a short insert will be covered multiple times by the continuous long read (CLR). Each pass of the original DNA molecule is termed a subread, which can be combined into one highly accurate consensus sequence termed a circular consensus sequence (CCS) or reads-of-insert (ROI) (F–H, left panel). Though SMRT sequencing always uses a circular template, long insert libraries typically only have a single pass and hence generate a linear sequence with single pass error rates (black nucleotides) (FG, right panel). Afterwards, overlapping single passes can be combined into one consensus sequence of high quality (H, right panel). Overall, CCS reads have the advantage of being very accurate while single passes stand out for their long read lengths (>20 kb).
Table 1. Comparison of PacBio sequencing platforms to two current industry standards.
| Platform | Read length | Number reads | Error rate | Run rime |
|---|---|---|---|---|
| PacBio RSII (per SMRT cell) | Average 10–16 kb | ∼55 000 | 13–15% | 0.5–6 hours |
| PacBio Sequel (per SMRT cell) | Average 10–14 kb | ∼365 000 | 13–15% | 0.5–10 hours |
| Illumina HiSeq 4000 | 2 × 150 bp | 5 billion | ∼0.1% | <1–3.5 days |
| Illumina MiSeq | 2 × 300 bp | 25 million | ∼0.1% | 4–55 hours |
Numbers from personal experience and company website (www.pacb.com and www.illumina.com) queries on 14 November 2017.
Due to the real-time detection of the nucleotide incorporation rate, the pace of the polymerase progressing through the DNA strand is registered during sequencing (23). The time between nucleotide incorporations is termed the interpulse duration (IPD) and varies with epigenetic changes on the DNA (Figure 1D and E). Since a polymerase is not holding a single nucleotide during sequencing, but approximately twelve nucleotides, an epigenetic change on one nucleotide can actually affect the incorporation rate of surrounding nucleotides. This results in a ‘fingerprint,’ (24) some of which have been characterized, such as for 6-mA, 4-mC and (Tet-converted) 5-mC.
In addition to fewer but longer reads (Table 1), PacBio data differs from short read sequencing technologies in several aspects. Reads are not a set read length, but a distribution of read lengths depending on how long each individual polymerase is active. Since there is no need for amplification during the library preparation, nor during the sequencing process, biases such as GC-skewing are near absent. In contrary to second-generation platforms, raw PacBio reads also differ in error types (more indels than mismatches) and have a much higher abundance (∼13–15%, Table 1), though they are spread randomly across the reads (25,26). This randomness enables highly accurate consensuses (>99%) to be build up rapidly by sequencing multiple times the same molecule (CCS reads) (15) or by combining different CLRs derived from the same locus (Figure 1G and H). Also, diffusion loading creates a preference towards shorter molecules which might negatively impact sequencing runs. This loading bias can be mitigated by using magbead loading which keeps molecules <1 kb from binding to the bottom of ZMWs, size selection to remove short molecules, and/or by adding polyethylene glycol during loading to enhance packing of large DNA molecules. It is possible that a complete length independent loading can be achieved in the (near) future by applying an electrical field to force charged molecules into ZMW’s (27).
To address these inherently different reads, bioinformatic analyses require adapting current tools and/or developing new methods, such as for alignment (26,28–32) and assembly (33–39). Many PacBio specific tools and pipelines (including those for demultiplexing, creating CCS reads, long amplicon analyses, de novo assemblies (34) and epigenetic analyses) are available in PacBio's SMRT analysis suite (openly available, www.pacb.com/support/software-downloads/) via the command line or their SMRT Portal and SMRT Link graphical user interfaces.
CONSTITUTIONAL
Tandem repeat disorders
Tandem repeats cause more than 40 neurological, neurodegenerative or neuromuscular diseases when mutated (40). Unfortunately, sequencing those DNA elements is difficult with short-read platforms because the reads are too short to span most tandem repeats. The first tandem repeat studied by SMRT sequencing was the FMR1 CGG repeat (41). Healthy individuals carry around 30 CGG units which is mostly interrupted by one or two AGG units. An expansion of the repeat to more than 200 units causes the Fragile X Syndrome (FXS), which is one of the most frequent causes of inherited intellectual disability and autism. Loomis et al. (41) showed they could sequence through a long full mutation allele of 750 units which equals 2 kb of 100% GC and repetitive content. Interestingly, expansions to full mutations only occur upon maternal transmission whereby the risk directly correlates with increasing repeat size and fewer AGG interruptions (42). SMRT sequencing can be used to determine the repeat size and the detection of the number of interrupting AGG units (43). A main advantage of this approach is the unambiguous separation of the two CGG repeats on the different X chromosomes of females thereby outperforming all other (PCR) approaches. Afterward, the information generated by SMRT sequencing is used clinically for improved genetic counselling of woman weighing the risk of having a child with FXS (43–45). Another example of tackling a tandem repeat by SMRT sequencing is the ATTCT repeat embedded in intron 9 of the Spinocerebellar ataxia type 10 gene (SCA10) (10). For the first time the full length of an expanded ATTCT repeat was completely sequenced using SMRT technology. The repeat was reconstructed by assembly and both known and novel interruptions were detected (10). The presence of those interruptions influence the phenotype of SCA10 patients and hence knowing the exact repeat structure allows for better genotype-phenotype correlations. It will be interesting to use SMRT sequencing in the near future for other tandem repeats with interruptions like Myotonic Dystrophy (46) and Friedreich's Ataxia (47) to increase our knowledge on tandem repeat configuration, its influence on stability of the repeat, and phenotype of an individual.
Where all of the above applications use PCR, novel amplification free enrichment methods are currently being developed. Methods using amplification are very error-prone, especially when amplifying (tandem) repeats (41), and remove all epigenetic marks (48). Thus using amplification impedes a complete genetic and epigenetic characterization of tandem repeats. Currently two methods are under development. The first method presented by Pham et al. (48) is based on type IIS restriction enzyme digestions, customized hairpin adapters especially designed to anneal at the targeted digest overhangs, and a ‘capture-hook’ method. A second and more recent method (bioRxivhttps://doi.org/10.1101/203919) is based on restriction enzyme digestion followed by cleavage of SMRT bells containing the target of interest using the CRISPR/Cas9 system. By ligating a specific capture adapter at the CRISPR/Cas9 DNA cleavage sites, the SMRT bell molecules of interest can then be selectively pulled down by magnetic beads targeting the capture adapter. The high throughput of SMRT sequencing enables different targets (e.g. FMR1 CGG repeat, C9ORF72 GGGGCC repeat, HTT CAG repeat, Sca10 ATTCT repeat, etc.) from one DNA sample to be simultaneously enriched and sequenced in a single run (bioRxivhttps://doi.org/10.1101/203919).
Both methods have been used to target the FMR1 CGG repeat and showed for the first time the true biological CGG repeat variation in human cell lines (48) (bioRxivhttps://doi.org/10.1101/203919). Besides avoiding amplification biases, these methods permit native DNA capture and hence direct detection of epigenetics. In the future, this technique can possibly be used diagnostically to screen for full mutations and assess the methylation status of the FMR1 CGG repeat, both of which influence the phenotype of FXS (49–51). Traditionally this would be determined by Southern blots, a labour intensive and inaccurate method. Thus replacing Southern Blots with faster and more direct SMRT sequencing will greatly enhance FMR1 and additional repeat disorder diagnostics (49–52). PacBio's enrichment technique has also been used to study patients with expanded Sca10 ATTCT repeats (53). Here, SMRT sequencing revealed a complete absence of interruptions which could be linked to the parkinsonism phenotype of the patient.
Polymorphic regions
Genotyping the human leukocyte antigen (HLA) region, or the human major histocompatibility complex (MHC), is crucial for diagnosing autoimmune disorders and selection of donors in organ and stem cell transplantation. Genes in the region can be highly polymorphic, HLA-B being the most variable with >2000 alleles already annotated in 2012 (54). The high variability in sequence make this region exceptionally difficult to map with short reads (54). HLA can be divided into three molecule classes and regions, termed class I, II and III, though the first two are primarily studied. Amplicons of ∼400–900 bp have been used with 454 sequencing to target specific exons of class I genes (55,56). However, considering these genes are ∼3kb in length, entire alleles, as opposed to exons, can be sequenced in a single PacBio read. Class II genes can exceed 10kb making them more difficult, but still possible. Full length class I HLA alleles have been targeted in humans with hybrid PacBio-Illumina approaches (57) and PacBio only approaches (58,59). Many large HLA typing labs, such as the Anthony Nolan Research Institute (58,59), are utilizing or developing SMRT sequencing pipelines of their own or using commercial kits, such as those offered by GenDx (Utrecht, The Netherlands), to now target class I, as wells as many class II genes. This is rapidly expanding the number of known HLA alleles (57) and is becoming a gold standard for organ transplant genotyping and blood stem cell transplantation.
Similarly complex regions can also be analyzed with these approaches. The killer cell immunoglobulin-like receptor (KIR) region, whose genes encode proteins with domains that recognize HLA proteins, was recently analyzed with SMRT sequencing and for the first time multiple haplotypes were phased without imputation (60).
Pseudogene discrimination
The high sequence similarity between pseudogenes and their homologous functional genes makes distinguishing variation between the two extremely difficult when using short read technologies. In general, long reads spanning the actual gene regions can be used to anchor to unique regions and/or phase variants to discriminate between the pseudogene and the actual gene. For diagnostics it is common to target a specific locus or set of loci of interest as a cost effective way to overcome the limited throughput of current generation SMRT sequencing platforms. The easiest option to enrich for specific loci is amplifying the targets by doing a (multiplex) long-range PCR (up to 10 kb). To differentiate samples, barcodes can be added directly during PCR via primers (61,62), by a nested PCR approach (57,61,63,64), or by ligating hairpin adapters containing barcodes during library preparation (Pacific Biosciences Product Note: www.pacb.com/wp-content/uploads/2015/09/ProductNote-Barcoded-Adapters-Barcoded-Universal-Primers.pdf). Therefore, for multiplexed long-amplicon tests only a single library preparation is needed after pooling the barcoded amplicons, as opposed to fragmentation and multiple barcoded library preparations for short-read platforms. This therefore enables fast, cheap library preparations that can be sequenced in just a few hours, permitting the next step in complex gene loci diagnoses.
One application is using barcoded 6–8 kb amplicons, and potential nested amplicons, to target the drug metabolism gene CYP2D6 (61,63). This gene has homologous pseudogenes and copy number variants which impair reliable genotyping with short-read platforms (61,63). After SMRT sequencing, reads can then be aligned and variants called using alignment based or ‘Long Amplicon Analysis’ (LAA, included in SMRT analysis) based pipelines. LAA is particularly powerful in that it enables reference free analyses and phasing of the two alleles (61). The pipeline first demultiplexes reads (if needed), then looks for overlap, performs clustering (i.e. determines different amplicons), phases the clustered reads (i.e. determines different alleles), and determines consensus sequences with Quiver (34). LAA may require optimization, such as the minimal number of reads used for clustering. Too many can result in false alleles and long run times, whereas too little may result in allelic dropouts. Once assembled, alleles can be compared to each other or to a reference genome for annotation. Overall, SMRT sequencing permits expanding from targeting specific CYP2D6 variants/exons, to identification of phased variants across the entire loci, including up/downstream and all introns, that will enhance identification of metabolizer phenotypes in tested individuals and enhance personalized medicine (61). Similar long-range PCR with PacBio applications have been used to genotype and discriminate other genes from pseudogenes (Table 2), including PKD1 for diagnosing autosomal-dominant polycystic kidney disease (64) and IKBKG for diagnosing primary immunodeficiency diseases in patients suffering from life-threatening invasive pyogenic bacterial infections (65).
Table 2. Applications of human SMRT sequencing and clinical utility.
| Target | Disease | Ref. |
|---|---|---|
| Tandem repeat sequencing | ||
| FMR1 | Fragile X Syndrome | (43)a |
| HTT | Huntington's Disease | a |
| C9orf72 | Amyotrophic Lateral Sclerosis (ALS) | a |
| SCA10 | Spinocerebellar ataxia type 10, Parkinson's disease | (10,53)a |
| Highly polymorphic regions | ||
| HLA | Autoimmune disorders & transplantation | (57–59) |
| KIR | Autoimmune diseases & transplantation | (60) |
| Pseudogene discrimination | ||
| CYP2D6 | Drug metabolism | (61,63) |
| PKD1 | Autosomal-dominant polycystic kidney disease | (64) |
| IKBKG | Primary immunodeficiency diseases | (65) |
| Cancer | ||
| BCR-ABL1 | Chronic Myeloid Leukemia (CML) | (69) |
| TP53 | Myelodysplastic Syndromes (MDS) and Acute Myeloblastic Leukemia (AML) | (70) |
| Reproductive genomics | ||
| TCOF1 | Treacher Collins syndrome | (67) |
| PTPN11 | Noonan syndrome | (67) |
a bioRxiv https://doi.org/10.1101/203919.
REPRODUCTIVE GENOMICS
Reproductive genomic medicine and associated counseling, including pre-implantation genetic diagnosis (PGD), relies heavily on the ability to haplotype or phase alleles in embryos, patients, and parents. Long reads enable direct phasing of amplicons from targeted loci which can be used to determine parent-of-origin alleles in embryos or patients (66,67). In a family having one child with Treacher Collins syndrome, SMRT amplicons sequencing was used to confirm the paternal transmission of a TCOF1 variant that affects splicing of the gene and potentially causes the disease (67). For apparent de novo mutations that are a result of germ line mosaicism, determining the frequency of damaging alleles is informative in predicting recurrence in future offspring. For a couple with multiple miscarriages and suspected Noonan syndrome in the fetuses, SMRT amplicon sequencing identified a disease causing PTPN11 variant in 37% of the father's sperm (67). Digital Droplet PCR showed no signs of the variant in the father's blood, but confirmed the 40% frequency in the fathers sperm (67). This therefore enabled an estimate of recurrent risk for subsequent pregnancies. Whole-genome single-cell haplotyping based on arrays is already being used in practice for embryo selection before implantation, though phasing still requires additional family members (68). We envision a profound impact on future PGD applications by incorporating long-read whole-genome sequencing for direct phasing to eliminate the need for analyzing additional family members.
CANCER
During treatment of cancer patients, it is crucial to monitor low frequency mutations that can lead to a proliferative advantage of malignant cells. Chronic myeloid leukemia (CML) is a blood cancer that is caused by a translocation between chromosomes 9 and 22, giving rise to the BCR-ABL1 fusion protein. CML patients are normally treated with tyrosine kinase inhibitors (TKIs) to suppress BCR-ABL1, but the therapy can induce point mutations leading to drug resistance. It is therefore important to screen the BCR-ABL1 gene in CML patients responding poorly to TKI treatment and study the mutational landscape. In a study by Cavelier et al. (69), a ∼1.5 kb amplicon was constructed from BCR-ABL1 cDNA. SMRT sequencing allowed for detection of TKI resistance mutations down to a level of 1%, a significantly lower detection threshold as compared to the 15–20% reached by Sanger sequencing. Moreover, it was possible to phase co-existing mutations thereby giving new information about the clonal distribution of resistance mutations in BCR-ABL1, and also to identify a number of distinct splice isoforms. Apart from BCR-ABL1, a number of other cancer genes are suitable targets for clinical SMRT sequencing (Table 2). In a study of loss-of-function mutations in the tumor suppressor TP53, SMRT sequencing revealed that tumors from acute myeloblastic leukemia (AML) and myelodysplatic syndrome (MDS) patients harbor multiple TP53 mutations distributed in different alleles (70). In the future, detailed information about the subclonal heterogeneity of TP53 could be used to guide the treatment of these patients. Minor variants can also be detected in other types of somatic variation, unrelated to cancer. Gudmunsson et al. (71) used SMRT sequencing to obtain phasing information of somatic mosaicism mutations in GJB2 that led to the repair of skin lesions in a patient with keratitis-ichthyosis-deafness syndrome.
Whole genome and transcriptome sequencing (addressed in later sections) is at the moment only affordable for research, but in the near future will become a diagnostic option. Already whole genome and transcriptome SMRT sequencing has been applied to breast cancer cell models identifying novel gene fusion events with the known oncogene Her2 (Case Study: www.pacb.com/wp-content/uploads/Case-Study-Scientists-deconstruct-cancer-complexity-through-genome-and-transcriptome-analysis.pdf). Whole transcriptome sequencing of prostate cell models has also identified novel RLN1 and RLN2 gene fusions in prostate cancer (72). Importantly, SMRT sequencing can give a more precise view of the cancer gene structure, as was demonstrated in a study by Kohli et al. where a cryptic exon was detected in AR-V9 that was previously thought to be present only in AR-V7 (73). AR-V7 has been studied as a potential biomarker for drug resistance in prostate cancer, based on knockdown experiments that have in fact targeted both isoforms. Thus, AR-V9 may actually be a predictive biomarker for resistance.
Global changes in epigenetics is also a hallmark in cancer. Single molecule real-time bisulfite sequencing (SMRT-BS) enables quantitative and highly multiplexed detection of methylation in 1.5–2 kb amplicons (74,75). This is an improvement of the previous technologies that could only target typical bisulfite PCR sizes (∼300–500 bp) and potentially enables ∼91% of CpG islands in the human genome to be evaluated (75). To date this has been applied to multiple cancer cell lines, including those from an acute myeloid leukemia, chronic myeloid leukemia, anaplastic large cell lymphoma, plasma cell leukemia, Burkitt lymphoma, B-cell lymphoma and multiple myelomas (75). Expanding to genome wide diagnostics, when whole genome SMRT sequencing is performed on non-amplified material it is theoretically possible to determine epigenetic status across all nucleotides based on IPD ratios. Therefore, we envision in the near future cancer genomes, transcriptomes and epigenomes will commonly be characterized at previously unparalleled resolution.
VIRAL AND MICROBIAL MEDICAL SEQUENCING
In infectious disease, SMRT sequencing has been used to analyse influenza viruses (76), hepatitis B viruses (HBV) (77), hepatitis C viruses (HCV) (77,78) and human immunodeficiency viruses (HIV) (79,80) (Table 3). HCV and HIV are RNA molecules of a length of approximately 9 kb, while HBV is a circular DNA virus of size 3 kb. These viruses are suitable subjects for SMRT sequencing, since the entire virus genome can easily be contained in a single read. For example, Bull et al. (77) developed an assay where the resulting reads covered nearly the entire sequence for all six major HCV genotypes. In addition to determining the genome sequence of the infecting viruses, it is also possible to monitor mutations that are developing as a result of drug treatment. For HCV, resistance associated variants (RAVs) in the NS5A gene occurring at a frequency of <0.5% were successfully identified in samples from patients undergoing treatment by direct acting antiviral drugs (DAAs) (78). By full-length sequencing of the HIV-1 provirus, a 9700 bp molecule that encodes nine major proteins via alternative splicing, Ocwieja et al. (80) detected at least 109 different spliced RNAs, including two of which encode new proteins. The fact that this relatively small study could generate a lot of novel information about HIV-1, a molecule that has already been studied in great detail, demonstrates the advantage of full-length RNA sequencing to study the distribution of splicing isoforms in specific genes. Results from these types of experiments could possibly open up novel therapeutic opportunities in infectious disease.
Table 3. Medically relevant microbial SMRT sequencing.
| Target/disease | Ref. |
|---|---|
| Hepatitis B/C virus | (77,78) |
| HIV | (79,80) |
| Influenza viruses | (76) |
| Tuberculosis bacteria | (85) |
| E. coli / Hemolytic–Uremic Syndrome | (86) |
| Salmonella enterica subsp. enterica serovar/gastroenteritis | (87) |
| Leishmania | (88) |
| Leptospira interrogans/leptospirosis | (90) |
| Helicobacter pylori strains/gastrointestinal diseases | (91) |
For bacteria, a single SMRT Cell often provides enough data to de novo assemble Escherichia coli size genomes into single contigs. HGAP is the most widely used assembler and works by taking a selection of longest reads and error correcting them with all reads, followed by Celera assembly (81,82), and finalized by polishing with all reads aligned to the final assembly (34). These long reads and new algorithms enable PacBio assemblies to be more complete and accurate compared to second-generation sequencing methods (83,84). Clinically relevant bacterial assemblies include a strain of the Tuberculosis bacteria Mycobacterium tuberculosis (85), the E. coli strain that caused a Hemolytic–Uremic Syndrome outbreak in Germany in 2011 (86), and strains of Salmonella enterica subsp. enterica serovar that cause gastroenteritis in humans (87) (Table 3). Pacbio sequencing and HGAP have also been used to assemble pathogenic single-cell eukaryote genomes that are more complex than a single chromosome, such as for a new Leishmania reference genome (88), a protozoan parasite that kills >30 000 people each year.
Though long reads permit superb microbial assemblies, what truly differentiates SMRT sequencing from second-generation machines is the ability to directly determine the epigenetics of these organisms. DNA methylation is overall ubiquitous in bacterial genomes (89), which simplifies SMRT analysis of epigenetic characteristics in these organisms. Analyses can be performed using IPD ratios of cases versus controls or vs an in silico control compared to known epigenetics signatures for 6-mA, 4-mC and (Tet converted) 5-mC (available in SMRT analysis). This has been used to discriminate virulent from avirulent Leptospira interrogans, a cause of leptospirosis in humans (90). The genome sequences have no major differences between strains, but higher levels of methylation are found in the avirulent strain (90). Methylation analysis has also been used to identify virulence factor genotype-dependent motifs in eight different H. pylori strains, a bacteria that can lead to gastrointestinal diseases (91). The simplicity to sequence, assemble, and call nucleotide, structural and epigenetic variation for a complete genome from a single SMRT Cell makes SMRT sequencing a truly revolutionizing technology in microbiology.
FUTURE: WHOLE TRANSCRIPTOME AND GENOME SEQUENCING
Traditionally RNA is converted to cDNA and then fragmented for short read sequencing (RNA-seq). Assembling the host of exons detected from RNA-seq into individual transcripts is extremely difficult and error prone. SMRT sequencing eliminates the need for fragmentation, instead sequencing cDNAs from the 5′ end of transcripts to the poly-A tail, termed Iso-Seq. This is an ideal method for complete cDNA sequencing (92). Iso-Seq has been used to sequence full transcriptomes from the blood of a normal Chinese adult male (93), a pool of 20 RNAs from different normal human tissues and organs (92), a trio of lymphoblastoid transcriptomes (94), and analyse prostate and breast cancer cell models (73) (Case Study: www.pacb.com/wp-content/uploads/Case-Study-Scientists-deconstruct-cancer-complexity-through-genome-and-transcriptome-analysis.pdf). As opposed to complex short-read alignment and re-assemblies, these papers demonstrate long-reads can easily detect splicing isoforms in human genes. Besides detecting a vast number of known isoforms, this method has also identified novel splicing forms and genes that have not previously been detected by short-read sequencing (93). Similar to genomic variant phasing, for gene loci with transcribed single nucleotide variants, these can be used to determine precisely which allele isoforms are expressed from (94). Though Iso-Seq is exceptional for transcript structure determination, the lower throughput when compared to second-generation platforms currently limits its usage for expression analysis. However, as costs drop and throughput increases, unbiased PacBio expression and isoform detection will become routine in the near future.
Whole genome sequencing (WGS) has become a widely used method to study variation in the human genome, and several 100’s of thousands of human genomes have been sequenced with short-reads during the last few years. However, the nature of these reads permit only relatively small assemblies and alignments provide only limited information on variation outside of SNPs and small insertions/deletions. SMRT sequencing is greatly expanding the utility of WGS, permitting a factor greater in assembly completeness (93,95) (BioRxiv: https://doi.org/10.1101/067447), even nearing reference genome contig sizes and including diploid aware assemblies by applying algorithms like FALCON-unzip (37). These PacBio WGS’s also demonstrate a vast repertoire of variation missed by short read WGSs. Low coverage (4–8×) sequencing recently was used to characterize structural variation in chromothrypsis-like chromosomes (96) and identify a pathogenic heterozygous 2184 bp deletion in a patient who presented with Carney complex that could not be identified by short-read sequencing (97). Higher coverage sequencing (∼60×) of two haploid genomes has also been used to identify a vast array of structural variations (461 553 from 2 bp to 28 kb in length), including >89% being missed in the analysis of data from the 1000 Genomes Project (98). From this study, Huddleston et al. (98) estimate a 5× increase in discovering indels >7 bp and additional SVs <1 kb which in total bps represents a majority of the difference between genomes. Additional remarkable findings from individual human de novo assemblies is that there seems to exist several megabases of novel sequence, i.e. sequences that are absent from the current (GRCh38) version of the human reference. For example, Shi et al. (93) reported 12.8 Mb of novel sequence in their de novo assembled individual genome, which would correspond to over 0.4% of the entire human genome of size ∼3 Gb. At this point, it is not known whether this novel sequence is common between all human individuals (and thereby missing from GRCh38) or if it mainly represents sequence variation found only in some specific individuals or population groups. Overall, these WGS studies demonstrate long-read sequencing can identify a substantial number of variation missed by short read platforms, including those relevant to clinical diagnoses.
CONCLUSIONS
The myth that SMRT sequencing is too error prone to be diagnostically useful is being expunged and replaced by evidence that it offers advantages over short-read sequencers. SMRT sequencing is opening up new diagnostic avenues, such as the ability to determine tandem repeat lengths, interruptions, and even epigenetics in a single test at base pair resolution. Long read sequencing is already considered the gold standard for some applications, such as for HLA genotyping for tissue transplants. While large scale implementation appears to be hampered by the cost and community expertise, this is likely to change rapidly. In addition to systematic price reductions and a growing customer base, new single molecule technologies such as nanopore based systems are likely to propel the field. Just as second-generation platforms stepped beyond Sanger sequencing and enabled a revolution in genomics medicine, third-generation single molecule sequencing platforms will likely be the next genetic diagnostic revolution.
ACKNOWLEDGEMENTS
We wish to thank Vicky Van Sandt (Belgian Red Cross-Flanders) for constructive comments on HLA typing.
FUNDING
KU Leuven [SymBioSys PFV/10/016, GOA/12/015 to J.R.V.]; Hercules foundation [ZW11–14]; Agency for Innovation by Science and Technology (IWT) (PhD grant) [SB/131787]. Funding for open access charge: KU Leuven [SymBioSys PFV/10/016, GOA/12/015 to J.R.V.]; Hercules foundation [ZW11–14]; Agency for Innovation by Science and Technology (IWT) (PhD grant) [SB/131787].
Conflict of interest statement. None declared.
REFERENCES
- 1. Katsanis S.H., Katsanis N.. Molecular genetic testing and the future of clinical genomics. Nat. Rev. Genet. 2013; 14:415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vermeesch J.R., Voet T., Devriendt K.. Prenatal and pre-implantation genetic diagnosis. Nat. Rev. Genet. 2016; 17:643–656. [DOI] [PubMed] [Google Scholar]
- 3. Heather J.M., Chain B.. The sequence of sequencers: The history of sequencing DNA. Genomics. 2016; 107:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanger F., Nicklen S., Coulson R.. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U.S.A. 1977; 74:5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krier J.B., Kalia S.S., Green R.C.. Genomic sequencing in clinical practice: applications, challenges, and opportunities. Dialogues Clin. Neurosci. 2016; 18:299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levy S.E., Myers R.M.. Advancements in next-generation sequencing. Annu. Rev. Genomics Hum. Genet. 2016; 17:95–115. [DOI] [PubMed] [Google Scholar]
- 7. Koboldt D.C., Steinberg K.M., Larson D.E., Wilson R.K., Mardis E.R.. The next-generation sequencing revolution and its impact on genomics. Cell. 2013; 155:27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011; 27:2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Browning S.R., Browning B.L.. Haplotype phasing: existing methods and new developments. Nat. Rev. Genet. 2011; 12:703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McFarland K.N., Liu J., Landrian I., Godiska R., Shanker S., Yu F., Farmerie W.G., Ashizawa T.. SMRT Sequencing of Long Tandem Nucleotide Repeats in SCA10 Reveals Unique Insight of Repeat Expansion Structure. PLoS One. 2015; 10:e0135906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schatz M.C., Delcher A.L., Salzberg S.L.. Assembly of large genomes using second-generation sequencing. Genome Res. 2010; 20:1165–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alkan C., Sajjadian S., Eichler E.E.. Limitations of next-generation genome sequence assembly. Nat. Methods. 2011; 8:61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guan P., Sung W.K.. Structural variation detection using next-generation sequencing data: a comparative technical review. Methods. 2016; 102:36–49. [DOI] [PubMed] [Google Scholar]
- 14. Harris T.D., Buzby P.R., Babcock H., Beer E., Bowers J., Braslavsky I., Causey M., Colonell J., Dimeo J., Efcavitch J.W. et al. . Single-molecule DNA sequencing of a viral genome. Science. 2008; 320:106–109. [DOI] [PubMed] [Google Scholar]
- 15. Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., Peluso P., Rank D., Baybayan P., Bettman B. et al. . Real-time DNA sequencing from single polymerase molecules. Science. 2009; 323:133–138. [DOI] [PubMed] [Google Scholar]
- 16. Clarke J., Wu H.C., Jayasinghe L., Patel A., Reid S., Bayley H.. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009; 4:265–270. [DOI] [PubMed] [Google Scholar]
- 17. Flusberg B.A., Webster D.R., Lee J.H., Travers K.J., Olivares E.C., Clark T.A., Korlach J., Turner S.W.. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods. 2010; 7:461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jain M., Olsen H.E., Paten B., Akeson M.. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol. 2016; 17:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deamer D., Akeson M., Branton D.. Three decades of nanopore sequencing. Nat. Biotechnol. 2016; 34:518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu H., Giordano F., Ning Z.. Oxford Nanopore MinION Sequencing and Genome Assembly. Genomics Proteomics Bioinformatics. 2016; 14:265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Travers K.J., Chin C.S., Rank D.R., Eid J.S., Turner S.W.. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res. 2010; 38:e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rhoads A., Au K.F.. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics. 2015; 13:278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., Peluso P., Rank D., Baybayan P., Bettman B. et al. . Real-time DNA sequencing from single polymerase molecules. Science. 2009; 323:133–138. [DOI] [PubMed] [Google Scholar]
- 24. Schadt E.E., Banerjee O., Fang G., Feng Z., Wong W.H., Zhang X., Kislyuk A., Clark T.A., Luong K., Keren-Paz A. et al. . Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases. Genome Res. 2013; 23:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chaisson M.J., Wilson R.K., Eichler E.E.. Genetic variation and the de novo assembly of human genomes. Nat. Rev. Genet. 2015; 16:627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carneiro M.O., Russ C., Ross M.G., Gabriel S.B., Nusbaum C., Depristo M.A.. Pacific biosciences sequencing technology for genotyping and variation discovery in human data. BMC Genomics. 2012; 13:375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Larkin J., Henley R.Y., Jadhav V., Korlach J., Wanunu M.. Length-independent DNA packing into nanopore zero-mode waveguides for low-input DNA sequencing. Nat. Nanotechnol. 2017; 12:1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li H., Durbin R.. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010; 26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chaisson M.J., Tesler G.. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics. 2012; 13:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krizanovic K., Echchiki A., Roux J., Sikic M.. Evaluation of tools for long read RNA-seq splice-aware alignment. Bioinformatics. 2017; doi:10.1093/bioinformatics/btx668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu T.D., Reeder J., Lawrence M., Becker G., Brauer M.J.. GMAP and GSNAP for genomic sequence alignment: enhancements to speed, accuracy, and functionality. Methods Mol. Biol. 2016; 1418:283–334. [DOI] [PubMed] [Google Scholar]
- 32. Liu B., Guan D., Teng M., Wang Y.. rHAT: fast alignment of noisy long reads with regional hashing. Bioinformatics. 2016; 32:1625–1631. [DOI] [PubMed] [Google Scholar]
- 33. Koren S., Schatz M.C., Walenz B.P., Martin J., Howard J.T., Ganapathy G., Wang Z., Rasko D.A., McCombie W.R., Jarvis E.D. et al. . Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012; 30:693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chin C.S., Alexander D.H., Marks P., Klammer A.A., Drake J., Heiner C., Clum A., Copeland A., Huddleston J., Eichler E.E. et al. . Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods. 2013; 10:563–569. [DOI] [PubMed] [Google Scholar]
- 35. Vaser R., Sovic I., Nagarajan N., Sikic M.. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017; 27:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kamath G.M., Shomorony I., Xia F., Courtade T.A., Tse D.N.. HINGE: long-read assembly achieves optimal repeat resolution. Genome Res. 2017; 27:747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chin C.S., Peluso P., Sedlazeck F.J., Nattestad M., Concepcion G.T., Clum A., Dunn C., O’Malley R., Figueroa-Balderas R., Morales-Cruz A. et al. . Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods. 2016; 13:1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xiao C.L., Chen Y., Xie S.Q., Chen K.N., Wang Y., Han Y., Luo F., Xie Z.. MECAT: fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. Nat. Methods. 2017; 14:1072–1074. [DOI] [PubMed] [Google Scholar]
- 39. Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M.. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017; 27:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schmidt M.H., Pearson C.E.. Disease-associated repeat instability and mismatch repair. DNA Repair (Amst.). 2016; 38:117–126. [DOI] [PubMed] [Google Scholar]
- 41. Loomis E.W., Eid J.S., Peluso P., Yin J., Hickey L., Rank D., McCalmon S., Hagerman R.J., Tassone F., Hagerman P.J.. Sequencing the unsequenceable: expanded CGG-repeat alleles of the fragile X gene. Genome Res. 2013; 23:121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yrigollen C.M., Martorell L., Durbin-Johnson B., Naudo M., Genoves J., Murgia A., Polli R., Zhou L., Barbouth D., Rupchock A. et al. . AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J. Neurodev. Disord. 2014; 6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ardui S., Race V., Zablotskaya A., Hestand M.S., Van Esch H., Devriendt K., Matthijs G., Vermeesch J.R.. Detecting AGG interruptions in male and female FMR1 premutation carriers by single-molecule sequencing. Hum. Mutat. 2017; 38:324–331. [DOI] [PubMed] [Google Scholar]
- 44. Chen L., Hadd A., Sah S., Filipovic-Sadic S., Krosting J., Sekinger E., Pan R., Hagerman P.J., Stenzel T.T., Tassone F. et al. . An information-rich CGG repeat primed PCR that detects the full range of fragile X expanded alleles and minimizes the need for southern blot analysis. J. Mol. Diagn. 2010; 12:589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hayward B.E., Usdin K.. Improved assays for AGG interruptions in fragile X premutation carriers. J. Mol. Diagn. 2017; 19:828–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Musova Z., Mazanec R., Krepelova A., Ehler E., Vales J., Jaklova R., Prochazka T., Koukal P., Marikova T., Kraus J. et al. . Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am. J. Med. Genet. A. 2009; 149:1365–1374. [DOI] [PubMed] [Google Scholar]
- 47. Holloway T.P., Rowley S.M., Delatycki M.B., Sarsero J.P.. Detection of interruptions in the GAA trinucleotide repeat expansion in the FXN gene of Friedreich ataxia. Biotechniques. 2011; 50:182–186. [DOI] [PubMed] [Google Scholar]
- 48. Pham T.T., Yin J., Eid J.S., Adams E., Lam R., Turner S.W., Loomis E.W., Wang J.Y., Hagerman P.J., Hanes J.W.. Single-locus enrichment without amplification for sequencing and direct detection of epigenetic modifications. Mol. Genet. Genomics. 2016; 291:1491–1504. [DOI] [PubMed] [Google Scholar]
- 49. Pretto D., Yrigollen C.M., Tang H.T., Williamson J., Espinal G., Iwahashi C.K., Durbin-Johnson B., Hagerman R.J., Hagerman P.J., Tassone F.. Clinical and molecular implications of mosaicism in FMR1 full mutations. Front. Genet. 2014; 5:318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pretto D.I., Eid J.S., Yrigollen C.M., Tang H.T., Loomis E.W., Raske C., Durbin-Johnson B., Hagerman P.J., Tassone F.. Differential increases of specific FMR1 mRNA isoforms in premutation carriers. J. Med. Genet. 2015; 52:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Usdin K., Hayward B.E., Kumari D., Lokanga R.A., Sciascia N., Zhao X.N.. Repeat-mediated genetic and epigenetic changes at the FMR1 locus in the Fragile X-related disorders. Front. Genet. 2014; 5:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dion V., Wilson J.H.. Instability and chromatin structure of expanded trinucleotide repeats. Trends Genet. 2009; 25:288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schule B., McFarland K.N., Lee K., Tsai Y.C., Nguyen K.D., Sun C., Liu M., Byrne C., Gopi R., Huang N. et al. . Parkinson's disease associated with pure ATXN10 repeat expansion. NPJ Parkinsons Dis. 2017; 3:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Trowsdale J., Knight J.C.. Major histocompatibility complex genomics and human disease. Annu. Rev. Genomics Hum. Genet. 2013; 14:301–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gabriel C., Danzer M., Hackl C., Kopal G., Hufnagl P., Hofer K., Polin H., Stabentheiner S., Proll J.. Rapid high-throughput human leukocyte antigen typing by massively parallel pyrosequencing for high-resolution allele identification. Hum. Immunol. 2009; 70:960–964. [DOI] [PubMed] [Google Scholar]
- 56. Erlich R.L., Jia X., Anderson S., Banks E., Gao X., Carrington M., Gupta N., DePristo M.A., Henn M.R., Lennon N.J. et al. . Next-generation sequencing for HLA typing of class I loci. BMC Genomics. 2011; 12:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Albrecht V., Zweiniger C., Surendranath V., Lang K., Schofl G., Dahl A., Winkler S., Lange V., Bohme I., Schmidt A.H.. Dual redundant sequencing strategy: Full-length gene characterisation of 1056 novel and confirmatory HLA alleles. HLA. 2017; 90:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mayor N.P., Robinson J., McWhinnie A.J., Ranade S., Eng K., Midwinter W., Bultitude W.P., Chin C.S., Bowman B., Marks P. et al. . HLA typing for the next generation. PLoS One. 2015; 10:e0127153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Turner T.R., Hayhurst J.D., Hayward D.R., Bultitude W.P., Barker D.J., Robinson J., Madrigal J.A., Mayor N.P., Marsh S.G.E.. Single molecule real-time (SMRT(R)) DNA sequencing of HLA genes at ultra-high resolution from 126 International HLA and Immunogenetics Workshop cell lines. Hla. 2017; doi:10.1111/tan.13184. [DOI] [PubMed] [Google Scholar]
- 60. Roe D., Vierra-Green C., Pyo C.W., Eng K., Hall R., Kuang R., Spellman S., Ranade S., Geraghty D.E., Maiers M.. Revealing complete complex KIR haplotypes phased by long-read sequencing technology. Genes Immun. 2017; 18:127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Buermans H.P., Vossen R.H., Anvar S.Y., Allard W.G., Guchelaar H.J., White S.J., den Dunnen J.T., Swen J.J., van der Straaten T.. Flexible and scalable full-length CYP2D6 long amplicon PacBio sequencing. Hum. Mutat. 2017; 38:310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hestand M.S., Van Houdt J., Cristofoli F., Vermeesch J.R.. Polymerase specific error rates and profiles identified by single molecule sequencing. Mutat. Res. 2016; 784–785:39–45. [DOI] [PubMed] [Google Scholar]
- 63. Qiao W., Yang Y., Sebra R., Mendiratta G., Gaedigk A., Desnick R.J., Scott S.A.. Long-read single molecule real-time full gene sequencing of cytochrome P450-2D6. Hum. Mutat. 2016; 37:315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Borras D.M., Vossen R., Liem M., Buermans H.P.J., Dauwerse H., van Heusden D., Gansevoort R.T., den Dunnen J.T., Janssen B., Peters D.J.M. et al. . Detecting PKD1 variants in polycystic kidney disease patients by single-molecule long-read sequencing. Hum. Mutat. 2017; 38:870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Frans G., Meert W., Van der Werff Ten Bosch J., Meyts I., Bossuyt X., Vermeesch J.R., Hestand M.S.. Conventional and single-molecule targeted sequencing method for specific variant detection in IKBKG whilst bypassing the IKBKGP1 pseudogene. J. Mol. Diagn. 2017; doi:10.1016/j.jmoldx.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 66. Mensah M.A., Hestand M.S., Larmuseau M.H., Isrie M., Vanderheyden N., Declercq M., Souche E.L., Van Houdt J., Stoeva R., Van Esch H. et al. . Pseudoautosomal region 1 length polymorphism in the human population. PLoS Genet. 2014; 10:e1004578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wilbe M., Gudmundsson S., Johansson J., Ameur A., Stattin E.L., Anneren G., Malmgren H., Frykholm C., Bondeson M.L.. A novel approach using long-read sequencing and ddPCR to investigate gonadal mosaicism and estimate recurrence risk in two families with developmental disorders. Prenat. Diagn. 2017; 37:1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dimitriadou E., Melotte C., Debrock S., Esteki M.Z., Dierickx K., Voet T., Devriendt K., de Ravel T., Legius E., Peeraer K. et al. . Principles guiding embryo selection following genome-wide haplotyping of preimplantation embryos. Hum. Reprod. 2017; 32:687–697. [DOI] [PubMed] [Google Scholar]
- 69. Cavelier L., Ameur A., Haggqvist S., Hoijer I., Cahill N., Olsson-Stromberg U., Hermanson M.. Clonal distribution of BCR-ABL1 mutations and splice isoforms by single-molecule long-read RNA sequencing. BMC Cancer. 2015; 15:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lode L., Ameur A., Coste T., Menard A., Richebourg S., Gaillard J.B., Le Bris Y., Bene M.C., Lavabre-Bertrand T., Soussi T.. Single-molecule DNA sequencing of acute myeloid leukemia and myelodysplastic syndromes with multiple TP53 alterations. Haematologica. 2017; 103:e13–e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gudmundsson S., Wilbe M., Ekvall S., Ameur A., Cahill N., Alexandrov L.B., Virtanen M., Hellstrom Pigg M., Vahlquist A., Torma H. et al. . Revertant mosaicism repairs skin lesions in a patient with keratitis-ichthyosis-deafness syndrome by second-site mutations in connexin 26. Hum. Mol. Genet. 2017; 26:1070–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tevz G., McGrath S., Demeter R., Magrini V., Jeet V., Rockstroh A., McPherson S., Lai J., Bartonicek N., An J. et al. . Identification of a novel fusion transcript between human relaxin-1 (RLN1) and human relaxin-2 (RLN2) in prostate cancer. Mol. Cell Endocrinol. 2016; 420:159–168. [DOI] [PubMed] [Google Scholar]
- 73. Kohli M., Ho Y., Hillman D.W., Van Etten J.L., Henzler C., Yang R., Sperger J.M., Li Y., Tseng E., Hon T. et al. . Androgen receptor variant AR-V9 Is coexpressed with AR-V7 in prostate cancer metastases and predicts abiraterone resistance. Clin. Cancer Res. 2017; 23:4704–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yang Y., Scott S.A.. DNA methylation profiling using long-read single molecule real-time bisulfite sequencing (SMRT-BS). Methods Mol. Biol. 2017; 1654:125–134. [DOI] [PubMed] [Google Scholar]
- 75. Yang Y., Sebra R., Pullman B.S., Qiao W., Peter I., Desnick R.J., Geyer C.R., DeCoteau J.F., Scott S.A.. Quantitative and multiplexed DNA methylation analysis using long-read single-molecule real-time bisulfite sequencing (SMRT-BS). BMC Genomics. 2015; 16:350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nakano K., Shiroma A., Shimoji M., Tamotsu H., Ashimine N., Ohki S., Shinzato M., Minami M., Nakanishi T., Teruya K. et al. . Advantages of genome sequencing by long-read sequencer using SMRT technology in medical area. Hum. Cell. 2017; 30:149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bull R.A., Eltahla A.A., Rodrigo C., Koekkoek S.M., Walker M., Pirozyan M.R., Betz-Stablein B., Toepfer A., Laird M., Oh S. et al. . A method for near full-length amplification and sequencing for six hepatitis C virus genotypes. BMC Genomics. 2016; 17:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bergfors A., Leenheer D., Bergqvist A., Ameur A., Lennerstrand J.. Analysis of hepatitis C NS5A resistance associated polymorphisms using ultra deep single molecule real time (SMRT) sequencing. Antiviral Res. 2016; 126:81–89. [DOI] [PubMed] [Google Scholar]
- 79. Dilernia D.A., Chien J.T., Monaco D.C., Brown M.P., Ende Z., Deymier M.J., Yue L., Paxinos E.E., Allen S., Tirado-Ramos A. et al. . Multiplexed highly-accurate DNA sequencing of closely-related HIV-1 variants using continuous long reads from single molecule, real-time sequencing. Nucleic Acids Res. 2015; 43:e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ocwieja K.E., Sherrill-Mix S., Mukherjee R., Custers-Allen R., David P., Brown M., Wang S., Link D.R., Olson J., Travers K. et al. . Dynamic regulation of HIV-1 mRNA populations analyzed by single-molecule enrichment and long-read sequencing. Nucleic Acids Res. 2012; 40:10345–10355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Myers E.W., Sutton G.G., Delcher A.L., Dew I.M., Fasulo D.P., Flanigan M.J., Kravitz S.A., Mobarry C.M., Reinert K.H., Remington K.A. et al. . A whole-genome assembly of Drosophila. Science. 2000; 287:2196–2204. [DOI] [PubMed] [Google Scholar]
- 82. Miller J.R., Delcher A.L., Koren S., Venter E., Walenz B.P., Brownley A., Johnson J., Li K., Mobarry C., Sutton G.. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008; 24:2818–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Miyamoto M., Motooka D., Gotoh K., Imai T., Yoshitake K., Goto N., Iida T., Yasunaga T., Horii T., Arakawa K. et al. . Performance comparison of second- and third-generation sequencers using a bacterial genome with two chromosomes. BMC Genomics. 2014; 15:699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Powers J.G., Weigman V.J., Shu J., Pufky J.M., Cox D., Hurban P.. Efficient and accurate whole genome assembly and methylome profiling of E. coli. BMC Genomics. 2013; 14:675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Miyoshi-Akiyama T., Satou K., Kato M., Shiroma A., Matsumura K., Tamotsu H., Iwai H., Teruya K., Funatogawa K., Hirano T. et al. . Complete annotated genome sequence of Mycobacterium tuberculosis (Zopf) Lehmann and Neumann (ATCC35812) (Kurono). Tuberculosis (Edinb.). 2015; 95:37–39. [DOI] [PubMed] [Google Scholar]
- 86. Rasko D.A., Webster D.R., Sahl J.W., Bashir A., Boisen N., Scheutz F., Paxinos E.E., Sebra R., Chin C.S., Iliopoulos D. et al. . Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N. Engl. J. Med. 2011; 365:709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yao K., Muruvanda T., Roberts R.J., Payne J., Allard M.W., Hoffmann M.. Complete Genome and Methylome Sequences of Salmonella enterica subsp. enterica Serovar Panama (ATCC 7378) and Salmonella enterica subsp. enterica Serovar Sloterdijk (ATCC 15791. Genome Announc. 2016; 4:e00133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Dumetz F., Imamura H., Sanders M., Seblova V., Myskova J., Pescher P., Vanaerschot M., Meehan C.J., Cuypers B., De Muylder G. et al. . Modulation of Aneuploidy in Leishmania donovani during Adaptation to Different In Vitro and In Vivo Environments and Its Impact on Gene Expression. MBio. 2017; 8:e00599-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Blow M.J., Clark T.A., Daum C.G., Deutschbauer A.M., Fomenkov A., Fries R., Froula J., Kang D.D., Malmstrom R.R., Morgan R.D. et al. . The epigenomic landscape of prokaryotes. PLoS Genet. 2016; 12:e1005854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Satou K., Shimoji M., Tamotsu H., Juan A., Ashimine N., Shinzato M., Toma C., Nohara T., Shiroma A., Nakano K. et al. . Complete genome sequences of low-passage virulent and high-passage avirulent variants of pathogenic Leptospira interrogans Serovar Manilae Strain UP-MMC-NIID, originally isolated from a patient with severe Leptospirosis, determined using PacBio single-molecule real-time technology. Genome Announc. 2015; 3:e00882-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Satou K., Shiroma A., Teruya K., Shimoji M., Nakano K., Juan A., Tamotsu H., Terabayashi Y., Aoyama M., Teruya M. et al. . Complete genome sequences of eight Helicobacter pylori strains with different virulence factor genotypes and methylation profiles, isolated from patients with diverse gastrointestinal diseases on Okinawa Island, Japan, determined using PacBio single-molecule real-time technology. Genome Announc. 2014; 2:e00286-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sharon D., Tilgner H., Grubert F., Snyder M.. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013; 31:1009–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shi L., Guo Y., Dong C., Huddleston J., Yang H., Han X., Fu A., Li Q., Li N., Gong S. et al. . Long-read sequencing and de novo assembly of a Chinese genome. Nat. Commun. 2016; 7:12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tilgner H., Grubert F., Sharon D., Snyder M.P.. Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:9869–9874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Seo J.S., Rhie A., Kim J., Lee S., Sohn M.H., Kim C.U., Hastie A., Cao H., Yun J.Y., Kim J. et al. . De novo assembly and phasing of a Korean human genome. Nature. 2016; 538:243–247. [DOI] [PubMed] [Google Scholar]
- 96. Masset H., Hestand M.S., Van Esch H., Kleinfinger P., Plaisancie J., Afenjar A., Molignier R., Schluth-Bolard C., Sanlaville D., Vermeesch J.R.. A distinct class of chromoanagenesis events characterized by focal copy number gains. Hum. Mutat. 2016; 37:661–668. [DOI] [PubMed] [Google Scholar]
- 97. Merker J.D., Wenger A.M., Sneddon T., Grove M., Zappala Z., Fresard L., Waggott D., Utiramerur S., Hou Y., Smith K.S. et al. . Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet Med. 2018; 20:159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Huddleston J., Chaisson M.J.P., Steinberg K.M., Warren W., Hoekzema K., Gordon D., Graves-Lindsay T.A., Munson K.M., Kronenberg Z.N., Vives L. et al. . Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 2017; 27:677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]