


Abstract
Transcription factors IRF3, IRF5 and IRF7 (IRF3/5/7) have overlapping, yet distinct, roles in the mammalian response to pathogens. To examine the role that DNA-binding specificity plays in delineating IRF3/5/7-specific gene regulation we used protein-binding microarrays (PBMs) to characterize the DNA binding of IRF3/5/7 homodimers. We identified both common and dimer-specific DNA binding sites, and show that DNA-binding differences can translate into dimer-specific gene regulation. Central to the antiviral response, IRF3/5/7 regulate type I interferon (IFN) genes. We show that IRF3 and IRF7 bind to many interferon-stimulated response element (ISRE)-type sites in the virus-response elements (VREs) of IFN promoters. However, strikingly, IRF5 does not bind the VREs, suggesting evolutionary selection against IRF5 homodimer binding. Mutational analysis reveals a critical specificity-determining residue that inhibits IRF5 binding to the ISRE-variants present in the IFN gene promoters. Integrating PBM and reporter gene data we find that both DNA-binding affinity and affinity-independent mechanisms determine the function of DNA-bound IRF dimers, suggesting that DNA-based allostery plays a role in IRF binding site function. Our results provide new insights into the role and limitations of DNA-binding affinity in delineating IRF3/5/7-specific gene expression.
INTRODUCTION
Pathogen detection by pathogen recognition receptors (PRRs), such as the Toll-like receptors (TLRs) and the RIG-I-like receptors (RLRs), activate a network of transcription factors (TFs) that regulate host defense genes (1). The TFs interferon regulatory factor 3 (IRF3), IRF5 and IRF7 (IRF3/5/7) are central to PRR signaling in response to viruses and intracellular pathogens with distinct, yet overlapping, roles in host defense (1–3). IRF3/5/7 reside predominantly in the cytoplasm and upon PRR-induced phosphorylation they dimerize and translocate to the nucleus to promote gene transcription (4,5). Upon activation, IRF3/5/7 induce both common and factor-specific target genes (1,5–7), but they can also function antagonistically (5,8). For example, IRF3 can repress (IFNA10, IFNA22) or enhance (IFNA1, IFNA7) IRF7-dependent gene activation (5). Despite their central role in the response to pathogens, little is known about the mechanisms by which IRF3/5/7 target overlapping gene programs.
IRFs share a conserved N-terminal DNA-binding domain that recognizes a consensus 5′-AANNGAAA-3′ DNA sequence found upstream of many virus- and IFN-inducible genes (1). Activated IRF3/5/7 function as dimers that recognize the longer composite dimer site 5′-A/GNNGAAANNGAAA-3′ (Figure 1A), referred to as the IFN-stimulated response element (ISRE) (6). Despite a shared ability to bind to consensus binding elements, the inherent DNA-binding differences between IRF3/5/7 may partially account for regulatory differences (6,7,9). In virus-infected BJAB lymphoma cells, expression of exogenous IRF5 or IRF7 induced 568 and 630 target genes, respectively (10); however, only 371 (∼60%) of the genes were induced by both proteins, suggesting that DNA-binding differences led to alternate target genes. Similarly, studies examining the regulation of the type I IFNs by IRF3/5/7 have revealed inherent DNA-binding and regulatory differences for each factor, and attributed regulatory differences to dimer-specific binding to the viral-response elements (VREs) in the type I IFN gene promoters (10–13). The full extent of IRF3/5/7 DNA-binding differences and their impact on dimer-specific gene regulation remains unclear.
Figure 1.

PBM-based analysis of IRF3/5/7 DNA binding. (A) Schematic of IRF dimers and PBM probe design. (B) Phosphomimetic IRF variants used in our PBM experiments. Positions of Ser/Thr to Asp mutations are indicated. (C) PBM z-score distributions for IRF dimer binding to 10 044 synthetic SNV probes. Highlighted are z-scores for literature described IRF binding sites and a high-affinity consensus site bound by all three IRF dimers. (D) PBM-derived DNA-binding logos for IRF3/5/7.
To better understand the scope of IRF3/5/7 DNA-binding differences and their role in defining dimer-specific target genes, we have used protein-binding microarrays (PBMs) to characterize the DNA-binding landscape of IRF3/5/7 dimers. We used constitutively dimeric, phosphomimetic mutants of IRF3 (14,15), IRF5 (7) and IRF7 (16) (Figure 1B) to best characterize the active dimeric form of the IRF proteins. We examined the binding of IRF3/5/7 homodimers to thousands of ISRE-type elements to characterize both common and dimer-specific DNA binding features. To explore the role of binding affinity in the regulation of type I IFN genes, we characterized IRF3/5/7 binding to the VREs of all human and mouse type I IFN genes. Finally, integrating our PBM data with gene expression data, we relate DNA binding affinity to gene regulatory specificity. Our results provide new insights into the role and limitations of affinity as a distinguishing mechanism of IRF3/5/7 gene expression and function.
MATERIALS AND METHODS
IRF3/5/7 expression, purification and mutagenesis
Wild-type human IRF constructs were cloned into the Gateway vector system (LifeTech) for propagation, mutagenesis, and protein expression. Phosphomimetic constructs for IRF5 and IRF7 (IRF5(4D) (7), IRF7(8D) (16)) were made by site-directed mutagenesis using the QuikChange Lightning (Agilent) and NEB Q5 (New England Biolabs) site-directed mutagenesis kits following the manufacturer's instructions. Phosphomimetic construct IRF3(6D) (14,15) was codon optimized for Escherichia coli expression and synthesized as an IDT-gBlock (Integrated DNA Technologies) with Gateway AttB sites then subsequently cloned into the Gateway vector system. GST-IRF5(4D)(pDEST15) and GST-IRF7(8D)(pDEST15) were expressed using the OverExpress C41(DE3) E. coli strain (Lucigen) co-transformed with the pRare tRNA plasmid (Novagen). Transformed bacteria were propagated on Terrific Broth (TB) + 1% glucose + antibiotic plates. Protein expression was carried out in TB + 1% glucose + antibiotic with an initial outgrowth at 37°C up to OD of 0.6 followed by 0.5 mM IPTG induction and expression at room temperature (∼20°C) for 5 h. Addition of glucose to media (to suppress leaky protein expression) and co-transformation with pRare plasmid (to enhance translation) lead to the highest yield of full-length protein. Codon optimized GST-IRF3(6D)-6xHis(pDEST15) protein was expressed using BL21(DE3) E. coli co-transformed with the pLysS plasmid. Transformed bacteria were propagated on TB + antibiotic plates and protein expression was carried out in TB + antibiotic media with an initial outgrowth at 37°C up to an O/D of 0.6 followed by 0.2 mM IPTG induction and expression at room temperature for 5 hours. Bacterial cultures were pelleted and stored at -80°C until lysis and purification. IRF5 and IRF7 were purified with glutathione-S-transferase (GST) affinity chromatography using GSTrapFF columns (GE Healthcare) and an ÄKTApurifier-10 Fast Protein Liquid Chromatography device (GE Healthcare). The binding and elution buffers recommended in the GSTrapFF manual were used and supplemented with 1mM PMSF serine protease inhibitor (binding: PBS, pH 7.3 [140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.3] + 1 mM PMSF; elution: 50 mM Tris–HCl, 10 mM reduced glutathione, pH 8.0 + 1 mM PMSF). Sample was buffer exchanged into binding buffer + 20% glycerol using Amicon 30k MWCO filtration spin units (EMD-Millipore) then snap frozen and stored at –80°C. Protein concentration was estimated using Coomassie Plus Bradford assay (Pierce). IRF3 was tandem affinity purified first using immobilized metal ion affinity chromatography (C-terminal-6xHis tag) followed by GST affinity chromatography (N-term-GST tag). His purification was carried out using HisTrapFF columns (GE Healthcare) and the buffer conditions recommended by the manufacturer (binding buffer: 20 mM sodium phosphate, 500 mM NaCl, 20 mM imidazole, pH 7.4; elution buffer: 20 mM sodium phosphate, 500 mM NaCl, 500 mM imidazole, pH 7.4). Buffers were supplemented with 1 mM PMSF serine protease inhibitor. Sample was buffer exchanged into GST-binding buffer using an Amicon 30k MWCO filtration spin unit. GST-tag purification was carried out as described for IRF5 and IRF7. IRF5(K96S) and IRF7(S101K) DNA binding domain mutations were made with In-vivo Assembly (IVA) site directed mutagenesis (17). IRF-DBD mutant proteins were expressed and purified as described above.
PBM design
Our IRF-specific PBM design included both synthetic and genome-derived IRF binding sites. Synthetic probes: Microarray probes with synthetic sites were based on 108 seed IRF binding sites that were each 20-bp long (for 2-bp half-site spacers), and 54 seed sites that were 21-bp (for 3-bp half-site spacer). Seed IRF sequences were within constant flanking DNA sequence (Figure 1A). For each seed sequence, we included all single-nucleotide variants (SNVs) across the 20-bp or 21-bp long site, for a total collection of 10 044 synthetic IRF sites. For each unique IRF site, six replicate probes were included in each orientation (12 replicates per unique site). The 162 seed sequences were chosen based on IRF binding sites from the literature, available HT-SELEX datasets (18), and preliminary PBM experiments(data not shown). Seed sequences were selected to capture a range of binding affinity, and to include alternate core sequences (i.e. alternates to the canonical 5′-GAAA-3′ core). This seed+SNV design allows us to directly compare IRF-specific DNA binding preferences across the IRF-binding sites in a straightforward manner, and to generate position weight matrices (PWMs) with relatively small number of sequences (see below). Genome-derived probes: Putative IRF binding sites from the promoters of type I IFN genes, and from published cis-regulatory elements for other genes (e.g. CXCL10) were extracted from the genomes as 21-bp genomic fragments and centered on the microarray probes in an identical manner to the synthetic probes. The proximal promoters of the type-1 interferons and other cytokines were scanned using BioPython (v1.68; bio.motifs, bio.SeqUtils.lcc modules) and degenerate IRF consensus motifs with a 2-bp or 3-bp spacer length (Supplementary File 5). Sequences with a low stringency PWM score above zero and a sequence complexity score >0.5 were included in the array. These selection criteria were empirically selected to remove long poly-A runs present in the IFN promoters while still capturing potential IRF binding sites. To reduce redundant genomic probes, sites within 1 bp of each other were removed retaining probes with the PWM hit closest to the end of the PBM probe. To control for potential IRF binding site shifts, trimmed probes were generated by replacing the flanking regions of the IRF-PWM with a low/moderate affinity constant sequence as determined in preliminary array designs (data not shown). Genomic loci and full PBM probe sets are provided (Supplementary Files 1 and 2).
PBM experiments and analysis
PBM experiments were performed using custom-designed microarrays (Agilent Technologies Inc. AMADID 084215, 4 × 180K format). Microarrays were double stranded as previously described (19,20). Wash steps were carried out in coplin jars on an orbital shaker at 125 rpm. Double stranded microarrays were first pre-wetted in PBS containing 0.01% Triton X-100 for five minutes, rinsed in a PBS bath, and then blocked with 2% milk in PBS for 1 h. Following the blocking step, arrays were washed in PBS containing 0.1% Tween-20 for 5 min, then in PBS containing 0.01% Triton X-100 for 2 min and finally briefly rinsed in a PBS bath. Arrays were then incubated with the protein sample(s) for 1 h in a binding reaction containing: 2% milk with 10 mM Tris, pH 7.5; 50 mM NaCl; 2 mM DTT; 0.2 mg/ml BSA; 0.02% Triton X-100; and 0.4 mg/ml salmon testes DNA (Sigma D7656). See Supplementary File 4 for protein samples and concentrations. After protein incubation, microarrays were washed with PBS containing 0.5% Tween-20 for 3 min, then in PBS containing 0.01% Triton X-100 for 2 min followed by a brief PBS rinse. Microarrays were then incubated with 20 μg/ml of Alexa Fluor-488 conjugated Anti-GST antibody (LifeTech, Cat# A-11131) in 2% milk in PBS for 20 min. Excess antibody was removed by washing with PBS containing 0.05% Tween-20 for 3 min, then PBS for 2 min. Microarrays were scanned with a GenePix 4400A scanner and fluorescence was quantified using GenePix Pro 7.2. Exported data were normalized using MicroArray LINEar Regression (19). Microarray probe sequences and fluorescence values from each experiment are provided (Supplementary File 1). IRF dimers exhibit an orientation-specific bias in our PBM experiments; therefore, data from probes in a single orientation (i.e. ‘_o2′ probes in Supplementary File 1) was used in our final analysis. However, all results were observed for probes in both orientations.
PBM experiments for the IRF3/5/7 phosphomimetic dimers were performed at four separate protein concentrations for each IRF dimer (Supplementary File 4). A saturation-binding curve was fit independently to the fluorescence values for each probe sequence:
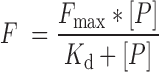 |
(1) |
F is probe fluorescence, Fmax is max fluorescence, [P] is applied protein concentration, Kd is dissociation constant. We previously showed that this approach can accurately estimate relative binding affinities over a wide affinity range (21). Curve fitting was performed in the statistical package R using the optim function (method—‘Brent’, Fmax—highest fluorescence value on PBM at highest protein concentration) with the cost function:
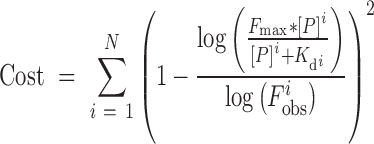 |
(2) |
Kd values were determined for each PBM probe, the median Kd across the six replicate probes was then reported for each unique DNA sequence on the PBM. We refer to these resulting binding constants as KPBM to highlight that these are PBM-derived estimates of relative binding constants. KPBM values were virtually identical (Pearson correlation R = 0.98) when a single value was determined by fitting simultaneously on fluorescence measurements from all replicate probes (i.e. 4 × 6 = 24 fluorescence values). Mutant IRF experiments were performed at a single concentration (see Supplementary File 3). For each DNA sequence, the median fluorescence intensity, over six replicate probe measurements, is used to quantify the binding of the protein to the DNA. We found that KPBM values determined from PBMs done at multiple concentrations (as described above) was approximated well by PBM experiments performed at a concentration ∼100–200 nM (data not shown). For all PBM experiments, z-scores were determined for the log(F) or log(KPBM) values using the mean (μ) and variance (σ2) of the values for the randomly selected background probes: zi = (log(Fi) – μ)/σ. z-scores provide an internally consistent way to quantify the specificity above background for measurements in each experiment.
SNV method for constructing position frequency matrices (PFMs)
PFMs can be constructed using the PBM z-score values of a single seed sequence (i.e. starting sequence), and the 3 × 21 associated SNV sequences. In the low-protein limit (i.e. when [P] << Kd), z-scores are related to log(F) ∼ log(Kd) ∼ E, where E is the binding free energy. Therefore, starting from an individual seed sequence, we compute the relative base preferences for base k at position i as a probability based on the Boltzmann distribution:
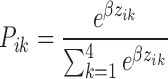 |
(3) |
zik—z-score for this particular seed sequence with SNV k at position i. β—a normalization factor chosen to maximize the correlation between the PFM scores and the PBM-measured log(KPBM) values. We found that β = 1 worked well for all our experiments. Using this approach, we can generate a PWM from a single seed and its complement of SNV probes. To generate a representative PFM for a PBM experiment we determine individual PFMs using the 15 top-scoring seed sequences and average the 15 individual Pik values to determine an average PFM for the experiment. Logos were then generated using the ENOLOGOS webserver (22), with background frequencies set to equally probable.
To identify base positions that are discriminatory for a single dimer, we systematically analyze our PBM data to find single-base changes that abrogate binding of one IRF dimer but not another. Specifically, to identify base positions that discriminate between two dimers, we analyze the binding to all pairs of DNA sequences that differ by a single base (i.e. SNV pairs). SNV pairs are identified where one IRF dimer is bound with high-affinity to both sequences (z-score > 8.0 for both probes, and z-score difference between probes < 3.0) while the other IRF dimer is bound with high-affinity to one probe (z-score > 8.0) but with low affinity to the other probe (z-score < 5.0, and z-score difference between probes > 5.0). Identifying all such SNVs reveals base positions and variants that provide strong discrimination between IRF dimers.
Electro-mobility shift assays (EMSAs)
IRF binding mode EMSAs (Figure 3C): DNA probes were formed by annealing complementary oligos: monomer (5′-GCACCGCTAACCGAAACTGTGC-3′); proximal dimer (5′-GCACCGCTAACCGAAACCGAAACTGTGC-3′) extended dimer (5′-GCACGCTAACCGAAACGCTAACCGAAACTGTGC-3′). Single stranded DNA oligos were annealed at 100 μM concentration in TE buffer + 50 mM NaCl (10 mM Tris, 1 mM EDTA, 50 mM NaCl, pH 8.0). Probes were denatured at 95°C for 2 min in a dry-heating block which was allowed to cool to room temperature. IRF PBM probe EMSAs (Supplementary Figure S1): 60-bp DNA probes were formed by oligo extension: C-2 (5′-GCCTAGCACTAACCGAAACCGAAACCTAAGTGCTAGGTCTTGATTCGCTTGACGCTGCTG-3′); C-3 (5′-GCCTAGCACTAACCGAAACCCGAAACCTAAGTGCTAGTCTTGATTCGCTTGACGCTGCTG-3′); PRD3 (5′-GCCTAGCACATAGGAAAACTGAAAGGGAGGTGCTAGGTCTTGATTCGCTTGACGCTGCTG-3′); Extension primer (5′-CAGCAGCGTCAAGCGAATCAAGAC-3′). Probes were double stranded using BST polymerase (New England Biolabs). Probe and primer were slowly annealed from 95°C to 63°C using a thermocycler with a 0.1°C/s cooling rate in buffer containing 8 uM probe ssDNA oligo, 8uM extension primer, 1× ThermoPol buffer (New England Biolabs), 1.6 mM dNTPs (New England Biolabs). Four units of BST polymerase diluted in 1× ThermoPol buffer were pre-heated to 63°C and quickly added to the annealed mixture. The isothermal double stranding reaction was held at 63°C for 1.5 h, then double stranded probes were purified using a MinElute PCR purification kit according to manufacturer's instructions (Qiagen). DNA probe radiolabeling: Purified dsDNA probes were radiolabeled with [γ-32P]-adenosine triphosphate (PerkinElmer) using T4 Polynucleotide Kinase (New England Biolabs) following manufacturer's protocol. Radiolabeled probes were purified using a QIAquick nucleotide removal kit (Qiagen) according to the manufacturer's instructions. Radiolabeling and purification yield were assumed to be 100% efficient for EMSA probe concentration calculations. EMSAs: Experiments were carried out as described (23). Binding reactions were carried out in 20 ul volumes containing: 1 nM P-32 labeled DNA probe and PBM buffer with nonspecific DNA competitor (10 mM Tris pH 7.5; 50 mM NaCl; 2 mM DTT; 0.08% Triton-X100; 50 ng/ul poly(dI:dC) (LI-COR Biosciences); 0.005 ug/ul salmon sperm DNA (LI-COR Biosciences)). To prevent adsorption to tubes and pipette tips, protein samples were diluted to appropriate concentrations in reaction buffer + 1 mg/ml BSA (New England Biolabs). Reactions were incubated at room temperature for 45 min. Before loading into gels, 1 ul of 50% glycerol and 1 ul Orange Loading Dye were added to each reaction (LI-COR Biosciences). Reactions were resolved in 6% polyacrylamide gels (29:1 crosslinking) with 0.5× TBE running buffer at 10 V/cm in a 4°C water bath until the loading dye front reached the bottom of the gel (∼2–3 h depending on gel length). Before drying under vacuum, EMSA gels were fixed for 30 min using a 20% methanol, 10% acetic acid solution then rinsed in ddH2O for 15 min and transferred to Whatman filter paper. Autoradiography was performed using a BAS storage phosphor screen (GE Healthcare). After overnight exposure, the phosphor screen was scanned using a Typhoon-Trio scanner (GE Healthcare). Typhoon square root space .GEL files were linearized using the ImageJ Linearize GelData plugin. To improve band visibility, the brightness of the linearized TIFF files was decreased by 25 units using Adobe Photoshop CS6.
Figure 3.

IRF3/5/7 binding to ISREs with alternate half-site spacing. (A) Binding of IRF3/5/7 to binding sites found in the PRDI and PRDIII elements of the IFNB promoter (27). Two sites (P1, P2) have a 2-bp spacer between GAAA-type half-sites (2-bp sites); one site (P3) has a 3-bp spacer. Error bars show the standard deviation of PBM-based z-scores to five replicate DNA probe sequences (z-score for each individual sequence was determined by fitting to experiments at different concentrations as described in Materials and Methods). (B) Differential binding (i.e. delta z-scores) of IRF3/5/7 to identical sets of 120 sequence-matched 2- and 3-bp sites that differ by a single base (as shown in schematic). A schematic is shown illustrating how the ‘delta z-score’ for each sequence-matched pairs is calculated. The delta z-score distribution for all 120 sequence pairs is shown for IRF3/5/7. (C) EMSAs are shown for IRF3 binding to three DNA site variants: (monomer)—a single ISRE half-site (i.e. 5′-AANNGAAA-3′); (proximal dimer)—site with a 2-bp spacer between half-sites (2-bp site); (extended dimer)—site with an 8-bp spacer between half-site (8-bp site). (D) Schematic illustrating the proposed binding arrangement of an IRF dimer to the site variants.
Reporter assays
IRF3(6D), IRF5(4D) and IRF7(6D) were cloned into the N-terminal His-tagged protein mammalian expression plasmids (pDest26) (LifeTech). HEK293T cells were cultured in DMEM (Gibco 11965092) + 10% FBS (Gibco 26140079). Cells were plated in tissue culture treated 96-well plates seeded at 12 000 cells per well and allowed to adhere overnight. PEI:DNA complexation reactions were carried out in 500 μl of serum free media and cells were transfected using polyethylenimine (PEI) (Polysciences, Inc.) at a ratio of 2:1 (PEI:DNA). Each 96-well plate well received 10 μl of transfection mixture containing: 12.5 ng of Tk-Luciferase normalization plasmid (pGL4.54); 10 ng of E1α-eGFP carrier DNA; 12.5 ng of reporter plasmid (pNL3.1); and 1.25 ng of His-tagged protein expression plasmid (pDest26) or carrier DNA in background-expression controls. Cells were incubated with transfection reagent overnight and then cell culture media was changed. Cells were lysed and assayed 24 h after transfection using the Nano-Glo Dual Luciferase reporter assay system (Promega). Dual luciferase signal was quantified using a VICTOR-3 plate reader (PerkinElmer). NanoLuc reporter plasmid signal was normalized to the constitutive luciferase signal (i.e. signal from pGL4.54 plasmid) (NanoLuc/Luc). Fold-induction values for each Protein/Reporter combination were calculated relative to the background-expression signal for each reporter plasmid: (protein + reporter)/(control + reporter). Reporter assay conditions had at least three biological replicates and at least three technical replicates per biological replicate. Replicate sample sizes and descriptive statistics are provided in Supplementary File 6.
RESULTS
Characterizing IRF3/5/7 dimer binding with PBMs
PBMs are double-stranded DNA microarrays that enable the in vitro measurement of protein binding to tens of thousands of unique DNA sequences (19,24). To examine the DNA-binding specificity of IRF3/5/7 we used custom-designed PBMs. We designed a PBM that included 10 044 IRF-type binding sites spanning a range of affinities, half-site sequences, and half-site spacing (Figure 1A, Materials and Methods). To query base preferences across the IRF binding site, we designed the PBM to contain 162 distinct IRF binding sites and all possible single-nucleotide variants (SNVs) across a 20-bp sequence centered on the IRF binding site (Figure 1A, Materials and Methods). This SNV-type PBM design allowed us to directly and comprehensively compare the base preferences of the IRF3/5/7 dimers across the IRF binding sites, and to construct position-weight matrix (PWM) descriptions of IRF3/5/7-DNA binding (Figure 1D, Materials and Methods).
PBM and other experiments were performed using phosphomimetic variants of IRF3(6D) (14,15), IRF5(4D) (7) and IRF7(8D) (16) that have been previously shown to form the active, homodimeric form of each protein (Figure 1B, Materials and Methods). Previous studies examining IRF binding used monomeric DNA-binding domains (DBDs) (25), or overexpressed the proteins in HEK293T cells in conditions not known to promote dimerization (18). Hereafter, the phosphomimetic proteins are referred to simply as IRF3, IRF5 and IRF7 (unless otherwise noted). The PBM experiments were performed with purified GST-tagged IRF3/5/7 homodimers at four concentrations, and relative binding constants (KPBM) were determined for each sequence (Materials and Methods). We have previously shown this approach of integrating PBMs at multiple concentrations can reliably define relative protein–DNA binding affinities (21). Log(KPBM) values were transformed into z-scores based on the distribution of scores from 600 random DNA probes (i.e. a background set). We use z-scores to represent our PBM binding data.
To assess the quality of our PBM experiments, we examined the z-scores for known in vivo IRF target sites and our ability to capture known dimer-specific DNA-binding preferences. We find that known IRF binding sites are bound significantly better than the random background sequences (Figure 1C), demonstrating sensitivity in our assay. DNA-binding logos were constructed for the IRF dimers (Figure 1D, Materials and Methods). While clear differences are evident for each IRF dimer, these logos are in agreement with reported ISRE sites (6) and logos generated from a high-throughput HT-SELEX assay (18).
The IRF5 logo from our PBMs resembles a half-site logo with a single core 5′-GAAA-3′ element. This logo matches the HT-SELEX ‘monomer’ logo, as opposed to the ‘extended’ logo that resembles the full-length logos determined for IRF3 and IRF7 that appear to contain two 5′-GAAA-3′ elements (Supplementary Figure S2) (18). For several reasons, we believe that this shorter logo represents a true dimer site but results from an IRF5 preference to engage more strongly to one ISRE half-site. First, if we relax the β parameter in our logo generation procedure (Supplementary Figure S2, Materials and Methods), we see the correct 5′-GAAA’-3′ preferences appearing in the 5-prime half of the logo. This suggests that both half-sites are engaged by IRF5 proteins and that the base preferences are simply weaker for the 5-prime half-site. Second, with our SNV approach to logo generation, we can generate a binding logo for individual sites by altering the DNA base identities and monitoring the change in binding affinity. We find that while the majority of starting seed sequences (13/15) result in the shortened half-site logo (as in Figure 1), logos from a minority of seed sequences (2/15) appear longer, and match the extended-version of the IRF5 logo also identified by HT-SELEX. We find that these extended logos occur when the 5-prime half-site in the seed is a better match to the consensus 5′-GAAA-3′ than for the 3-prime half-site (e.g. 5′-GAAANNGATA-3′). These observations suggest that IRF5 binds as a dimer with an asymmetric half-site preference.
As a further confirmation of the PBM data, we compared PBM z-scores to electro-mobility shift assays (EMSAs) for select high- and low-affinity binding sites (Supplementary Figure S1). We find that EMSAs qualitatively recapitulate the differential binding indicated by our z-scores: IRF3 z-scores 11.4–6.3 correspond to >25-fold change in affinity and 11.4–3.6 correspond to >100-fold change in affinity; IRF5 z-score 16.4–2.2 correspond to >100-fold change in affinity; IRF7 z-scores 16.6–5.1 correspond to > 50-fold change in affinity. These results demonstrate that our PBM data accurately captures the DNA-binding landscape of IRF3/5/7 dimers over a wide range of binding site affinities.
Common and IRF-specific binding sites
To investigate the nature and extent of IRF3/5/7 binding differences we compared the PBM-determined binding profiles for the IRF dimers (Figure 2A). Consistent with binding logo differences, we observed IRF-specific binding preferences indicated by ‘off-diagonal’ data points that represent sequences bound much better by one IRF dimer than another. For comparison, pseudo-replicate IRF experiments showed no such off-diagonal data points (Figure 2B). DNA sequence preferences for the IRF dimers can be queried by examining the distribution of specific sequence subsets. For example, examining the z-score distribution of all sequences that contain IRF sites with the alternate 5′-GATA core elements (i.e. that match the pattern 5′-GATANNGATA-3′) we see clear dimer preferences (Figure 2C). Specifically, IRF7 cannot tolerate these alternate core elements and, therefore, all sequences in the IRF7 experiment have z-scores indistinguishable from background (i.e. z-score near zero). In contrast, both IRF3 and IRF5 can bind sequences with this alternate core sequence. For IRF5, some of these 5′-GATA sequence variants score among the highest in our dataset; however, this is not the case for IRF3 indicating that IRF5 is more tolerant of this alternate core element. Pairwise comparison of our PBM data revealed many differences between the IRF3/5/7 dimers.
Figure 2.

IRF3/5/7 DNA-binding preferences. (A) Pairwise comparison of z-scores for IRF3, IRF5 and IRF7 binding to 10 044 synthetic IRF sites (black dots) and 644 random background sequences (blue dots). (B) Pairwise comparison of PBM z-scores (same sequences as in (A)) for IRF7 performed at two single concentrations (i.e. pseudo-replicate experiments). (C) Shown are the same pairwise comparison data as in (A) with all binding sequences matching the pattern 5′-GATANNGATA-3′ (N is any base) highlighted in red.
In general, base preferences that distinguish the DNA binding of IRF3/5/7 dimers from each other are apparent in their respective DNA-binding logos (Figure 1D). For example, based on the logos, IRF3 appears more tolerant than IRF5 of base variants at most positions; however, this difference is most apparent at position 11 where IRF5 highly prefers a Cyt base. We can directly confirm this selectivity by analyzing our PBM data and identifying single-base changes (i.e. SNVs) that abrogate binding of IRF5 but not IRF3 (Methods and Materials). We identified eight such IRF5-abrogating SNVs in our PBM dataset, and all were C-to-G changes at position 11. For example, 5′-CCGAAACCGAAACC-3′ was bound highly by both IRF3 (z-scores: 10.9) and IRF5 (z-score: 14.8), but the SNV 5′-CCGAAACGGAAACC-3′ was bound well by IRF3 (z-score: 8.2) but not by IRF5 (z-score: 2.9). Base differences at position 11 largely explain the bifurcation seen in the PBM data (Figure 2C) in which IRF5 is bound poorly to a number of DNA sites that are bound with high affinity by IRF3. In a similar manner, we examined the selection against IRF7 binding observed in Figure 2C for the 5′-GATANNGATA-3′ sequences. Comparing the logos of IRF5 and IRF7, we see that IRF7 is primarily more base selective at positions 7 and 8, suggesting that SNVs at these positions may differentially affect IRF5 and IRF7. We identified 50 sequence pairs in which a SNV abrogates IRF7 binding but does not perturb high-affinity IRF5 binding: 41/50 SNVs were A-to-{C, G or T} changes at position 8, which disrupts the IRF7-preferred Ade base, and 2/50 SNVs were A-to-T changes at position 7. For example, IRF5/7 both bound to 5′-CCGAAACCGAAACC-3′ with high affinity (z-scores 16.6 and 16.4, respectively), but the SNV 5′-CCGACACCGAAACC-3′ was bound well by IRF5 (z-score: 13.6) but not by IRF7 (z-score: 4.2). Unexpectedly, we also found 5 A-to-T SNVs at position 14 were selective against IRF7, but not readily expected from the logo comparisons. For example, IRF5/7 both bound to 5′-CCGATACCGAAACC-3′ with high affinity (z-scores: 14.9 and 9.8, respectively), but the SNV 5′-CCGATACCGATACC-3′ abrogated IRF7 binding (z-score: 2.2) while IRF5 was largely unaffected (z-score: 12.0). This IRF5 (and IRF3) tolerance for a Thy at position 14 is indicated by a slightly weaker Ade selectivity in the logo. Pairwise comparison of IRF3/5/7 logos and our SNV binding data reveal features of IRF3/5/7 binding specificity that distinguish the separate dimers and highlight the impact that genomic SNPs may have on relative IRF3/5/7 binding and function.
Despite the binding differences observed for the IRF dimers, we observe common, high affinity binding sites shared by all three proteins. For example, the IRF high affinity probe (Figure 1C) is a top-scoring sequence for each IRF dimer and is consistent with the known canonical ISRE element. This shared specificity landscape for close paralogs, in which both common and dimer-specific binding sequences are observed, has been reported for a number of transcription factor paralog families (reviewed in (26)). The existence of shared and dimer-specific sites provides a potential mechanism for the IRF dimers to regulate both common and dimer-specific genes.
Alternate ISRE half-site spacing
IRF3/5/7 binding has been reported for binding sites with both the canonical 2-bp spacer between each 5′-GAAA-3′ half-site, as well as with a 3-bp spacer, hereafter referred to as 2- and 3-bp sites (27,28). However, DNA-binding logos determined using our PBM dataset (Figure 1), other in vitro methods such as HT-SELEX (18), or learned from ChIP-seq datasets (29) support a dominant 2-bp site. The X-ray crystal structure of IRF3 and IRF7 bound to the IFNB gene promoter demonstrates the binding of alternate IRF3 and IRF7 DBDs that support binding to both a 2-bp site (Figure 3A, PRDIII) and an adjacent 3-bp site (Figure 3B, PRDI). Therefore, we sought to determine whether IRF dimers could bind to both 2-bp and 3-bp sites with similar affinities, and whether specific IRF dimers have individual preferences.
We first examined the binding of the IRF3/5/7 dimers to the variant ISRE elements found in the IFNB promoter for which there is structural evidence for binding to both 2- and 3-bp sites. The X-ray crystal structure supports a simple model of two adjacent IRF3-IRF7 heterodimers bound to the adjacent PRDIII (2-bp) and PRDI (3-bp) sites, P1 and P3, respectively (Figure 3A). However, there is a third possible ISRE element (P2) that spans these canonical elements (Figure 3A). We used PBMs to assay the binding of the IRF3/5/7 dimers to all three possible ISRE sites (Figure 3A). We find that IRF3 and IRF7 bind well to both the proto-typical ISREs PRDIII and PRDI, and IRF5 binds well to PRDI. Notably, all three dimers bind most strongly to the 3-bp PRDI site. While we cannot determine the binding register from our PBM data (i.e., whether canonical residues contact the same positions in the 5′-GAAA-3′-type half-site), X-ray crystal structures (27,28) suggest IRF dimers bind to the PRDI site in a 3-bp binding register. These results demonstrate that in the context of the IFNB promoter the 3-bp PRDI site is the highest affinity IRF binding site. Therefore, a description of IRF-DNA binding that does not include 3-bp sites will fail to capture functional binding sites.
To determine the impact of the spacer length on IRF binding, we examined the binding of IRF3/5/7 to 120 matched pairs of 2-bp/3-bp ISRE sites. Matched pairs differ by a single base in the spacer between half-sites (i.e. GAAANNGAAA → GAAANXNGAAA, Figure 3B). We find that altering the spacing from 2- to 3-bp lowers the binding affinity for all three IRF dimers by a similar amount (mean delta z-score ∼ 5.0) (Figure 3B). These data suggest an explanation for the canonical 2-bp PWMs (i.e., binding models) described in the literature (18,29,30)—2-bp sites are higher affinity. However, despite the lower binding affinity to 3-bp binding site variants, we find that many in our dataset score well above background. For example, IRF7 binds to 559 of 3456 3-bp IRF site variants with z-scores > 4.0. Therefore, many 3-bp sites in the genome are likely of sufficient affinity to be functional (e.g. PRDI in the IFNB promoter).
Previous studies (27,31) have also proposed an alternate binding mode in which IRF dimers are bound to half-sites 8-bp apart (e.g.5′-GAAA(N)8GAAA-3′). The extended binding conformation would allow IRF proteins to reside on the same side of the DNA helix (27) (Figure 3D). We examined the binding of IRF3 to this extended site by electro-mobility shift assay (EMSA) (Figure 3C). We found that IRF3 binding to an 8-bp extended-dimer site was at least 100-fold weaker than to a sequence-matched 2-bp site. These results reaffirm that the preferred binding mode for IRF dimers is to closely-spaced half-sites (i.e. 2-bp and 3-bp sites), and not to an extended dimer site (i.e. 8-bp site).
Differential dimer binding to type I IFN gene VREs
IRF3/5/7 regulate the type I IFN genes (4,6,32) that coordinate immunity to viruses and other intracellular pathogens (4,32). The type I IFN genes consist of IFNB and multiple IFNA genes (Figure 4). IRF3 and IRF7 are the primary type I IFN regulators (1,33,34); however, IRF5 can also regulate select IFNs (6). Induction of the type I IFNs in virus-infected cells is primarily a consequence of IRF regulatory input from the promoter VREs (1,13). Despite their central role in IFN gene regulation, it remains unclear to what extent DNA-binding of IRF3/5/7 differs across the VREs.
Figure 4.

Wild-type and mutant IRF dimer binding to IFNA VREs. (A) Binding profiles of wild-type and mutant IRFs to human IFNA VREs. Binding sites are illustrated as 10-bp blocks in register with the canonical 5-GAAANNGAAA-3′ elements of an IRF binding site. Low-affinity binding sites (z-score < 4.0) are not shown (see Materials and Methods). Z-scores for each panel range from 0 to 13.0. IFNA gene names are shown. Coordinates for VREs elements are provided (Supplementary File 2). (B) Binding profiles to mouse IFNA VREs are shown, all details as in (A). (C) Binding profiles of wild-type IRF3/5/7 to known IRF-binding regulatory elements are shown. Profile details as in (A,B). Coordinates for regulatory element DNA sequences are provided (Supplementary File 2). (D) DNA-binding logos for wild-type and mutant IRF5/7 are shown. Wild-type logos are identical to those in Figure 1 and are shown for contrast. Regions in which the DNA base specificity is altered in the mutant IRFs are highlighted with red shading. DNA logos generated as described in Materials and Methods (i.e. SNV approach). (E) Multiple protein-sequence alignment for human and mouse IRF3/5/7 is shown. Alignment is limited to the portion of protein containing alpha helices 2 and 3 that contains the base-contacting residues that match our criteria to be specificity determining (see main text). Identically conserved residues across all IRFs are indicated with a ‘ * ‘; residues in which IRF3/5/7 are different (but conserved across species) are indicated with a ‘ | ‘. Putative specificity-determining residues are highlighted with shaded bars; our selected specificity-determining residue is highlighted in red.
To address the role of DNA binding in type I IFN gene regulation, we used PBMs to measure the binding of IRF3/5/7 to the promoters of all type I IFN genes from human and mouse. We measured dimer binding to all potential IRF binding sites found in the 250-bp immediately upstream of the IFN genes that encompasses the VREs. Considering only significant binding sites (PBM z-score > 4.0), we found there was little binding to the regions outside of the VRE (data not shown); therefore, we have focused our analysis on the VREs.
For both human and mouse, we observe binding for IRF3 and IRF7 to multiple sites within the VREs (Figure 4A and B). Studies have delineated sub-elements (A/B, C and D) within the IFN VREs that play critical but different roles in IFN regulation (11–13,35) (Figure 4A and B). We find differences in IRF3 and IRF7 binding profiles across the various VREs sub-elements. IRF3 and IRF7 bind with similar, though not identical, patterns to the VRE-C and VRE-D elements. However, they differ strongly in their binding to VRE-A/B elements—IRF7 binds VRE-A/B in most VREs (20/27), whereas IRF3 binds in only three (3/27). It has been proposed, based on studies of mouse Ifna4 and Ifna2 genes (35), that late-phase expression of IFNA genes is controlled by Irf7 binding to VRE-A/B, whereas early-phase expression is controlled by Irf3 binding to VRE-C. We show that for many IFNs (63%, 17/27) it is only IRF7 that binds to VRE-A/B, suggesting that late-phase expression driven by IRF7-VRE-A/B binding may be a common regulatory feature for most IFNs. The sequence logo for IRF sites in the human and mouse VRE-A/B (Supplementary Figure S3) does not indicate a simple explanation for the IRF7 binding preference over IRF3. In contrast, we find that the majority of VRE-C elements are bound by both IRF3 and IRF7, with only the VRE-C of the mouse Ifna4 and Ifnab genes showing IRF3-exclusive binding, suggesting that the regulatory logic of early-phase IFN expression from VRE-Cs may be more complicated.
Unexpectedly, we observed a near complete lack of IRF5 binding to the IFNA VREs. IRF5 does not bind to any human VREs, and only binds three mouse VREs (Figure 4A and B). As a control, we see strong IRF5 binding to regulatory loci for other IRF target genes, such as IFNB (36), IL10 (37), and CXCL10 (1) (Figure 4C). The lack of IRF5 binding to the VREs is broadly consistent with the less prominent role described for IRF5 in the regulation of the IFNA genes (1,6,33). However, given the large number of IRF binding site variants that can be bound strongly by all three IRF dimers (Figure 2A), it is striking that the IRF binding site variants from human and mouse VREs would maintain their inability to bind to IRF5. The sequence logo for IRF sites in the human and mouse VRE regions (Supplementary Figure S3) highlights the sequence variability across individual sites and suggests a possible mechanism for the observed absence of IRF5 binding. There is a nearly complete absence of Cyt bases at positions 11 and 16 flanking the 5′-GAAA-3′ core that are both highly preferred by IRF5 (Figure 1). The evolutionarily conserved absence of IRF5 binding suggests a selective pressure against IRF5 binding to these loci that is based on selective use of binding sites variants bound preferentially by IRF3/7 (addressed more below).
A single amino acid dictates the IRF binding selectivity to IFNA VREs
To examine how IRF5 binding is selected against in the VREs, we examined the multiple protein sequence alignment for IRF3/5/7 to find potential specificity-altering residues. We reasoned that residues critical to distinguishing IRF3/5/7 binding specificity would (i) vary between all three IRFs (but would be conserved between human and mouse orthologs), and (ii) make base-specific contacts with DNA. Based on protein-DNA interactions defined in available IRF3 and IRF7 crystal structures (27,28), we found three residues that fit these criteria (Figure 4E). We examined the IRF sites within the IFNA VREs and determined that selection against IRF5 binding was most likely due to DNA sequence features 3-prime to the canonical 5′-GAAANNGAAA-3′ ISRE sequence; therefore, we chose to examine the residue position in alpha helix α3 shown to contact DNA bases in this region (Figure 4E, red highlight). Given that IRF7 and IRF5 exhibited the largest differences in their binding profiles across the IFNA VREs, we made mutants in which we swapped orthologous residues between IRF5 and IRF7—IRF5(K96S) and IRF7(S101K).
We examined the DNA-binding of the IRF5(K96S) and IRF7(S101K) mutants by PBM experiment. Binding logos generated for the mutants revealed that the amino acid alterations at this position neatly swapped the DNA binding specificity for IRF5 and IRF7 at the 3-prime end of the DNA binding sites (Figure 4D). Mutations at this position had the effect of making IRF5 more tolerant of alternate bases in this region, while making IRF7 less tolerant (e.g., strong selectivity for a cytosine at base position 16). Examining the binding of the mutant IRFs to the IFNA VREs (Figure 4A and B), we found that IRF7(S101K) had drastically reduced binding across the VREs compared to IRF7, while IRF5(K96S) showed increased binding compared to IRF5. These results identify this residue position in helix α3 (Figure 4E) as a key determinant of IRF binding specificity that can alter the base preferences in the 3-prime flanks of the canonical ISRE. Furthermore, base contacts mediated by residues at this position are critical for the selective binding of IRF7 over IRF5 across the IFNA VREs. Therefore, poor binding of IRF5 to VREs is partially explained by the bases flanking the core ISRE elements that are unfavorable for interaction with IRFs containing a lysine residue at this position in the DBD (i.e. IRF5 and IRF7 S101K).
Binding affinity contributes to ISRE function and selectivity
We next sought to determine the extent to which IRF3/5/7 binding differences translate into IRF-specific gene regulation. We examined whether IRF3/5/7-specific binding site variants discovered in our dataset would drive IRF-specific gene activation. Dimer-specific binding site variants (I3, I5, I7) that show preferential binding to IRF3, IRF5 or IRF7, respectively, were chosen from the PBM data (Figure 5A and B). To relate our findings to IFNA gene regulation, we examined the impact of these site variants on gene expression in the context of the 250-bp human IFNA14 promoter. We observed no significant IRF binding to the IFNA14 VRE in our PBM experiments (Figure 4A and B), and others have similarly reported no IRF3/7 binding to this element (12); therefore, it provided a useful promoter context in which to examine the impact of binding site variants on gene expression. Binding site variants were inserted simultaneously into the VRE-C and VRE-D elements of the IFNA14 promoter and we examined the ability of each variant promoter to drive reporter gene expression in the presence of constitutively active IRF dimers: IRF3(6D), IRF5(4D) and IRF7(6D) (Figure 5A, Methods and Materials). For reporter gene experiments, IRF7(6D) was used instead of IRF7(8D) as it is a stronger activating dimer (16) (Supplementary File 3).
Figure 5.

Comparison of DNA binding and gene activation by ISRE variants. (A) Schematic for promoter variants based on IFNA14 VRE. Promoter variants contain simultaneous alterations of C-site and D-site with the binding site shown in the box. Wild-type C-site (WT-C) is shown for context. (B, D) PBM-determined z-scores are shown for IRF3/5/7 to binding site sequence variants listed in (A). Error bars are calculated as in Figure 3. (C, E) Reporter gene activation levels for promoter variants in HEK293T cells over-expressing the phosphomimetic, constitutively active IRF dimers: IRF3(6D), IRF5(4D) and IRF7(6D) (Methods and Materials). Normalized fold-induction (relative to I7–2 promoter levels) is shown for each promoter. Mean and standard error of the mean (SEM) values were calculated over at least 15 replicate measurements (Supplementary File 6).
We found that dimer-specific binding sites can promote dimer-specific gene activation. Promoters with IRF3- and IRF5-specific sites (I3, I5) were selectively activated by IRF3 and IRF5, respectively (Figure 5B and C). However, affinity-independent mechanisms also appear to contribute to IRF binding site function. Promoters with IRF7-specific sites (I7) were strongly activated by IRF7 but, unexpectedly, also by IRF5 (Figure 5B and C). Furthermore, IRF5 drove expression from the I7 promoter at a 4.1-fold higher level than for the I5 promoter, despite the fact that IRF5 binds to I7 with lower affinity than to I5 (z-scores 3.4 and 10.7, respectively). We do not believe that heterodimerization with endogenous IRF proteins contributes to our observation that affinity does not correlate with activity. IRF3 and IRF7 are expressed at low levels in our WT and transfected HEK293T cells (Supplementary Figure S4); however, in our unstimulated cell-culture conditions IRF proteins are not known to be active. Furthermore, we observe very low reporter gene activation with eGFP transfected in place of our active IRF dimers (data not shown), suggesting that the endogenous IRF3/7 are not active and would not contribute to gene expression. These data show that IRF3/5/7-specific gene activation depends on both affinity and affinity-independent mechanisms.
We next tested whether ‘common’ binding sites bound by all three IRF dimers would be functional for each dimer. We found that IRF3/5/7 all drove reporter gene expression from the C-2 promoter bound with high affinity by all three IRFs (Figure 5B and C). However, expression from the C-2 promoter was not the highest for any of the IRFs, despite C-2 being the highest-affinity binding site for all three IRF dimers (Figure 5B). For example, IRF7 expression was 6.1-fold lower from the C-2 promoter than from the I7 promoter despite having a much higher binding affinity (z-score 17.0 versus 9.5). This diminished activity for the C-2-promoters suggests that for IRF3/5/7 dimers high-affinity binding may depress gene activation (discussed more below).
Affinity-independent mechanisms of ISRE function
To understand affinity-independent mechanisms of ISRE function, we first investigated whether ISRE half-site spacing contributed to their activity. We compared the activity of binding site variants that differed only in their half-site spacer length (i.e. 2-bp versus 3-bp sites). Specifically, we compared the activity of a 2-bp (I7–2) and 3-bp (I7) version of our IRF7-specific site, and a 2-bp (C-2) and 3-bp (C-3) version of a high-affinity site common to IRF3/5/7 (Figure 5A). These matched pairs of binding-site variants differed only by a single base in the spacer sequence (Figure 5D).
Spacer variants for the common high-affinity sites (C-2 and C-3) actually led to similar expression levels for all three IRFs (Figure 5E), despite the fact that all IRFs bind with lower affinity to the C-3 variant (Figure 5D). Similarly, spacer variants for the IRF7-specific site (I7–2, I7) also led to similar expression levels for IRF7 despite lower binding affinity to the 3-bp variant. This surprising congruence in activity for IRF-binding-site spacer variants suggests that either (i) 3-bp sites are more functionally active and can make-up for a lower binding affinity; or (ii) that DNA-sequence features, which are virtually identical between spacer pairs, are critical to the activity level of IRF sites.
Additional promoter comparisons clarify that DNA-sequence features beyond spacer length influence the activity of IRF binding sites. First, binding sites with similar IRF binding affinity can exhibit different activity: C-3 and I7 have similar IRF7 binding affinity (z-scores 11.2 and 9.5), and the same spacer lengths, but I7 induces 6.5-fold higher gene activation (Figure 5D and E). Second, we noted that the wild-type IFNA14 promoter, intended as a control in our assay, is activated by IRF7 despite low binding affinity to the WT-C and WT-D sites on PBM (z-scores < 4.0) (Figure 5D and E). These data support a model in which IRF-site DNA sequence features, beyond half-site spacing, contribute to the regulatory activity of an IRF site in an affinity-independent manner.
To determine the DNA sequence features of IRF binding sites that may affect their activity, we compared the DNA sequences of the variant sites tested in reporter assays (Figure 5A). The I7–2 and C-2 sites promote different levels of gene activation (from 2.6-fold for IRF3 to 6.1-fold for IRF7) despite having identical core ISRE sequences—C-2 and I7–2 share an identical 5′-CCGAAACCGAAA-3′ core. Furthermore, the most transcriptionally active of these sites, I7–2, is lower affinity than C-2. This differential activity and DNA sequence similarity indicates that DNA bases flanking the 12-bp core element can modulate the transcriptional activity of an IRF site even at the expense of binding affinity.
DISCUSSION
IRF3/5/7 are central regulators of the host-defense program to pathogens (1–3). Here, we addressed the ability of inherent IRF3/5/7 DNA-binding differences to define dimer-specific gene regulation. We characterized the DNA-binding preferences of IRF3/5/7 homodimers, and identified both common and dimer-specific DNA-binding preferences. We demonstrate that dimer-specific binding sites can promote dimer-specific reporter gene expression, showing that sufficient DNA-binding differences exist for IRF3/5/7 to induce unique target gene sets. We also found that affinity-independent mechanisms contribute to IRF3/5/7 binding site activity and, therefore, may also contribute to dimer-specific gene regulation (discussed more below). Currently, there are no genome-wide chromatin immunoprecipiation (ChIP) studies for IRF7 and IRF5 that would allow a comparison of in vivo IRF3/5/7 binding to our PBM data. Specifically, there are no ChIP studies for IRF7, and the single IRF5 study (38) found no ISRE motif enriched in the IRF5-bound ChIP peaks, suggesting that under the conditions assayed IRF5 might not function as a canonical DNA-bound dimer. Genome-wide binding studies for activated IRF3/5/7 dimers would help to clarify whether the inherent DNA-binding differences described here, and in other studies (18), define the distinct global binding patterns and gene regulatory programs for IRF3/5/7.
IRF3/5/7 are central regulators of type I IFN expression, and the host-defense response to viruses and intracellular pathogens (4,6,32). To understand the role of DNA binding in IFN regulation, we mapped the binding of IRF3/5/7 dimers to all human and mouse IFN gene VREs. We found that IRF3 and IRF7 bind across many of the VRE sub-elements, but that the VRE-A/B sub-element is bound almost exclusively by IRF7. This suggests that the previously described role for IRF7-VRE-A/B binding to control late-phase IFN expression (35) may be common to many IFN genes. Examining the landscape of IRF3/5/7 binding to the individual ISRE sites within the VREs, we find that for the 61 sites bound by at least one IRF dimer 93% (57/61) bound to IRF7, 60% (37/61) bound to IRF3, and only 5% (3/61) bound to IRF5. These data support the current understanding that IRF3 and IRF7 are the primary regulators of the type I IFNs (1,6).
The most striking results from our analysis of the type I IFN VREs was absence of IRF5 homodimer binding to all human and most mouse VREs. Our PBM binding data (Figure 2) demonstrate that there are many IRF site variants that can be bound by all 3 IRF dimers; therefore, the conspicuous absence of IRF5 binding to VREs from two evolutionarily distant species suggests that there has been selection against IRF5 homodimer binding to these loci. We demonstrate that a single amino acid difference between IRF7 and IRF5 was critical to their differential binding profiles across the VREs. Furthermore, this amino acid altered the binding preference for DNA bases immediately 3-prime to the canonical ISRE site (Figure 4D). Therefore, IRF5 homodimer binding to the VREs is inhibited by the evolutionary retention of specific ISRE variants with 3′-flanking bases that are unfavorable for IRF5.
IRF5 can regulate specific IFNA genes in a cell- and stimulus-specific manner (6,9,39,40). Furthermore, IRF5 forms heterodimers with IRF3, and IRF3 can enhance the recruitment of IRF5 to IFNA promoters in virus-infected cells (39,40). This suggests that IRF3:IRF5 heterodimers may be the dimer species critical for IRF5-dependent regulation of the IFNA genes, as opposed to IRF5 homodimers, which we find do not bind well to IFNA VREs. We propose that a heterodimer of IRF3:IRF5 could avoid the unfavorable IRF5 binding that we observe if the 3′-end of the binding site was occupied by IRF3. In other words, heterodimerizing with IRF3 would allow IRF5 to bind the VREs by avoiding the non-optimal half-site sequence. Future studies with IRF heterodimers should further clarify these selection rules for IRF proteins for the IFNA VREs.
Finally, analyzing the role of DNA-binding affinity in IRF3/5/7-dependent gene regulation revealed clear affinity-independent mechanisms. We found that DNA sequence features of IRF binding sites could enhance their activity even at the expense of binding affinity. Comparison of sequence variants revealed that these differences did not need to occur in the core ISRE motif, but could be in the flanking bases. For example, the C-2 and I7 sites have identical 5′-CCGAAACCGAAA-3′ core sequences yet very different IRF7 binding affinity (z-scores 17.0 and 9.5, respectively) and IRF7-dependent gene expression activity (6.1-fold higher for I7) (Figure 5). The mechanism of this uncoupling of affinity and activity remains unclear. However, a plausible mechanism is DNA-based allostery, in which IRF dimers adopt alternate protein conformations based on the DNA sequence of the binding sites and these structural differences affect gene activation. DNA-based allostery has been described for other factors, such as glucocorticoid receptors (41,42) and NF-κB (43), where in certain situations affinity and activity do not correlate. Future studies should clarify these details and provide a clearer picture of how affinity-dependent and affinity-independent mechanisms regulate IRF activity. The PBM dataset of IRF binding sites sequences and affinities generated here will provide an invaluable framework for dissecting the roles of affinity and activity.
ACCESSION NUMBER
GEO accession number: GSE109117.
Supplementary Material
ACKNOWLEDGEMENTS
Wild-type human IRF5 isoform D plasmids were gifts from the labs of Dr Betsy Barnes and Dr Nancy Reich. The wild type human IRF7 isoform A and human IRF3(5D) isoform 1 plasmids were a gift from the lab of Dr Rongtuan Lin. We thank Dr Thomas Gilmore, Dr Juan Fuxman-Bass, David Bray and Ashley Penvose for comments on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
Boston University; National Institutes of Health (NIH) [R01AI116820]. Funding for open access charge: NIH [R01AI116820].
Conflict of interest statement. None declared.
REFERENCES
- 1. Honda K., Taniguchi T.. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006; 6:644–658. [DOI] [PubMed] [Google Scholar]
- 2. Lazear H.M., Lancaster A., Wilkins C., Suthar M.S., Huang A., Vick S.C., Clepper L., Thackray L., Brassil M.M., Virgin H.W. et al. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. 2013; 9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stetson D.B., Medzhitov R.. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006; 24:93–103. [DOI] [PubMed] [Google Scholar]
- 4. Honda K., Takaoka A., Taniguchi T.. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006; 25:349–360. [DOI] [PubMed] [Google Scholar]
- 5. Barnes B., Lubyova B., Pitha P.M.. On the role of IRF in host defense. J. Interferon Cytokine Res. 2002; 22:59–71. [DOI] [PubMed] [Google Scholar]
- 6. Tamura T., Yanai H., Savitsky D., Taniguchi T.. The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 2008; 26:535–584. [DOI] [PubMed] [Google Scholar]
- 7. Cheng T.-F., Brzostek S., Ando O., Van Scoy S., Kumar K.P., Reich N.C.. Differential activation of IFN regulatory factor (IRF)-3 and IRF-5 transcription factors during viral infection. J. Immunol. 2006; 176:7462–7470. [DOI] [PubMed] [Google Scholar]
- 8. Negishi H., Miki S., Sarashina H., Taguchi-Atarashi N., Nakajima A., Matsuki K., Endo N., Yanai H., Nishio J., Honda K. et al. Essential contribution of IRF3 to intestinal homeostasis and microbiota-mediated Tslp gene induction. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:21016–21021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yanai H., Chen H.-M., Inuzuka T., Kondo S., Mak T.W., Takaoka A., Honda K., Taniguchi T.. Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:3402–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barnes B.J., Richards J., Mancl M., Hanash S., Beretta L., Pitha P.M.. Global and distinct targets of IRF-5 and IRF-7 during innate response to viral infection. J. Biol. Chem. 2004; 279:45194–45207. [DOI] [PubMed] [Google Scholar]
- 11. Civas A., Génin P., Morin P., Lin R., Hiscott J.. Promoter organization of the interferon-A genes differentially affects virus-induced expression and responsiveness to TBK1 and IKKepsilon. J. Biol. Chem. 2006; 281:4856–4866. [DOI] [PubMed] [Google Scholar]
- 12. Yeow W.S., Au W.C., Juang Y.T., Fields C.D., Dent C.L., Gewert D.R., Pitha P.M.. Reconstitution of virus-mediated expression of interferon alpha genes in human fibroblast cells by ectopic interferon regulatory factor-7. J. Biol. Chem. 2000; 275:6313–6320. [DOI] [PubMed] [Google Scholar]
- 13. Génin P., Lin R., Hiscott J., Civas A.. Differential regulation of human interferon A gene expression by interferon regulatory factors 3 and 7. Mol. Cell. Biol. 2009; 29:3435–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen W., Srinath H., Lam S.S., Schiffer C.A., Royer W.E. Jr, Lin K.. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. J. Mol. Biol. 2008; 379:251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin R., Mamane Y., Hiscott J.. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol. Cell. Biol. 1999; 19:2465–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Caillaud A., Hovanessian A.G., Levy D.E., Marié I.J.. Regulatory serine residues mediate phosphorylation-dependent and phosphorylation-independent activation of interferon regulatory factor 7. J. Biol. Chem. 2005; 280:17671–17677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. García-Nafría J., Watson J.F., Greger I.H.. IVA cloning: a single-tube universal cloning system exploiting bacterial in vivo assembly. Sci. Rep. 2016; 6:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jolma A., Yan J., Whitington T., Toivonen J., Nitta K.R., Rastas P., Morgunova E., Enge M., Taipale M., Wei G. et al. DNA-binding specificities of human transcription factors. Cell. 2013; 152:327–339. [DOI] [PubMed] [Google Scholar]
- 19. Berger M.F., Philippakis A.A., Qureshi A.M., He F.S., Estep P.W., Bulyk M.L.. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 2006; 24:1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berger M.F., Bulyk M.L.. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat. Protoc. 2009; 4:393–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Siggers T., Duyzend M.H., Reddy J., Khan S., Bulyk M.L.. Non-DNA-binding cofactors enhance DNA-binding specificity of a transcriptional regulatory complex. Mol. Syst. Biol. 2011; 7:555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Workman C.T., Yin Y., Corcoran D.L., Ideker T., Stormo G.D., Benos P.V.. enoLOGOS: a versatile web tool for energy normalized sequence logos. Nucleic. Acids. Res. 2005; 33:W389–W392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hellman L.M., Fried M.G.. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2007; 2:1849–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Siggers T., Chang A.B., Teixeira A., Wong D., Williams K.J., Ahmed B., Ragoussis J., Udalova I.A., Smale S.T., Bulyk M.L.. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-κB family DNA binding. Nat. Immunol. 2011; 13:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Badis G., Berger M.F., Philippakis A.A., Talukder S., Gehrke A.R., Jaeger S.A., Chan E.T., Metzler G., Vedenko A., Chen X. et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009; 324:1720–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Andrilenas K.K., Penvose A., Siggers T.. Using protein-binding microarrays to study transcription factor specificity: homologs, isoforms and complexes. Brief. Funct. Genomics. 2015; 14:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Panne D., Maniatis T., Harrison S.C.. An atomic model of the interferon-beta enhanceosome. Cell. 2007; 129:1111–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Escalante C.R., Nistal-Villán E., Shen L., García-Sastre A., Aggarwal A.K.. Structure of IRF-3 bound to the PRDIII-I regulatory element of the human interferon-beta enhancer. Mol. Cell. 2007; 26:703–716. [DOI] [PubMed] [Google Scholar]
- 29. Freaney J.E., Kim R., Mandhana R., Horvath C.M.. Extensive cooperation of immune master regulators IRF3 and NFkB in RNA Pol II recruitment and pause release in human innate antiviral transcription. CELREP. 2013; 4:959–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mathelier A., Fornes O., Arenillas D.J., Chen C.-Y., Denay G., Lee J., Shi W., Shyr C., Tan G., Worsley-Hunt R. et al. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2016; 44:D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dragan A.I., Hargreaves V.V., Makeyeva E.N., Privalov P.L.. Mechanisms of activation of interferon regulator factor 3: the role of C-terminal domain phosphorylation in IRF-3 dimerization and DNA binding. Nucleic Acids Res. 2007; 35:3525–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stetson D.B., Medzhitov R.. Type I interferons in host defense. Immunity. 2006; 25:373–381. [DOI] [PubMed] [Google Scholar]
- 33. McNab F., Mayer-Barber K., Sher A., Wack A., O’Garra A.. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015; 15:87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Honda K., Yanai H., Negishi H., Asagiri M., Sato M., Mizutani T., Shimada N., Ohba Y., Takaoka A., Yoshida N. et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005; 434:772–777. [DOI] [PubMed] [Google Scholar]
- 35. Civas A., Island M.-L., Génin P., Morin P., Navarro S.. Regulation of virus-induced interferon-A genes. Biochimie. 2002; 84:643–654. [DOI] [PubMed] [Google Scholar]
- 36. Barnes B.J., Kellum M.J., Field A.E., Pitha P.M.. Multiple regulatory domains of IRF-5 control activation, cellular localization, and induction of chemokines that mediate recruitment of T lymphocytes. Mol. Cell. Biol. 2002; 22:5721–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krausgruber T., Blazek K., Smallie T., Alzabin S., Lockstone H., Sahgal N., Hussell T., Feldmann M., Udalova I.A.. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011; 12:231–238. [DOI] [PubMed] [Google Scholar]
- 38. Saliba D.G., Heger A., Eames H.L., Oikonomopoulos S., Teixeira A., Blazek K., Androulidaki A., Wong D., Goh F.G., Weiss M. et al. IRF5:RelA interaction targets inflammatory genes in macrophages. CELREP. 2014; 8:1308–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Barnes B.J., Moore P.A., Pitha P.M.. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon alpha genes. J. Biol. Chem. 2001; 276:23382–23390. [DOI] [PubMed] [Google Scholar]
- 40. Barnes B.J., Field A.E., Pitha-Rowe P.M.. Virus-induced heterodimer formation between IRF-5 and IRF-7 modulates assembly of the IFNA enhanceosome in vivo and transcriptional activity of IFNA genes. J. Biol. Chem. 2003; 278:16630–16641. [DOI] [PubMed] [Google Scholar]
- 41. Meijsing S.H., Pufall M.A., So A.Y., Bates D.L., Chen L., Yamamoto K.R.. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009; 324:407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Watson L.C., Kuchenbecker K.M., Schiller B.J., Gross J.D., Pufall M.A., Yamamoto K.R.. The glucocorticoid receptor dimer interface allosterically transmits sequence-specific DNA signals. Nat. Struct. Mol. Biol. 2013; 20:876–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang V.Y.-F., Huang W., Asagiri M., Spann N., Hoffmann A., Glass C., Ghosh G.. The transcriptional specificity of NF-kB dimers is coded within the kB DNA response elements. CELREP. 2012; 2:824–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.