


Abstract
CglI is a restriction endonuclease from Corynebacterium glutamicum that forms a complex between: two R-subunits that have site specific-recognition and nuclease domains; and two H-subunits, with Superfamily 2 helicase-like DEAD domains, and uncharacterized Z1 and C-terminal domains. ATP hydrolysis by the H-subunits catalyses dsDNA translocation that is necessary for long-range movement along DNA that activates nuclease activity. Here, we provide biochemical and molecular modelling evidence that shows that Z1 has a fold distantly-related to RecA, and that the DEAD-Z1 domains together form an ATP binding interface and are the prototype of a previously undescribed monomeric helicase-like motor. The DEAD-Z1 motor has unusual Walker A and Motif VI sequences those nonetheless have their expected functions. Additionally, it contains DEAD-Z1-specific features: an H/H motif and a loop (aa 163–aa 172), that both play a role in the coupling of ATP hydrolysis to DNA cleavage. We also solved the crystal structure of the C-terminal domain which has a unique fold, and demonstrate that the Z1-C domains are the principal DNA binding interface of the H-subunit. Finally, we use small angle X-ray scattering to provide a model for how the H-subunit domains are arranged in a dimeric complex.
INTRODUCTION
Helicases are a large class of nucleic acid enzymes with a wide range of biological functions (1). The classical activity for which they are named is the NTP hydrolysis-dependent strand separation of duplex polynucleotides. The single-strand products are important intermediates in processes such as DNA replication and repair, and RNA metabolism. Additionally, helicase enzymes have evolved for nucleic acid processing activities, including dsDNA translocation (e.g. in the chromatin remodelling enzymes (2)), or acting as molecular switches (e.g. in the Type III restriction enzymes (3)). In these enzymes the domains are referred to as ‘helicase-like’, to highlight the lack of unwinding activity (4). Here, we examined the structural and mechanistic features of the H-subunit of the Type I restriction enzyme CglI (5,6), which represents an alternative use of helicase-like activity.
Helicase and helicase-like enzymes are members of the P-loop superfamily of NTPases which are characterised by the Walker A and Walker B catalytic motifs. They can be further identified and classified based on different structural features which help define their function (7–12): (i) helicases are grouped by homologous amino acid sequences into six superfamilies, SF1 – SF6; (ii) the core structural fold of all helicases is a RecA-like domain or the related AAA+ domain, comprising an α–β structure which folds into an antiparallel β-sheet sandwiched by the α-helices. Helicase motifs are on the intervening loops. Additional unique domains can be identified which provide additional activities, such as dsDNA binding; (iii) helicases can be active as either monomers, or higher order assemblies, typically hexameric rings. In all cases, the catalytic NTP binds at the interface between two RecA/AAA+ domains. For the monomeric helicases, a pair of domains are found in one polypeptide and are named N-core (containing the Walker A and B) and C-core. For hexameric helicases, the interface is formed between an adjacent pair of protomers.
Restriction enzymes (REs) such as CglI evolved as prokaryotic immune systems to restrict bacteriophage growth or transformation by other genetic elements, by cleaving the target DNA following sequence specific recognition (13). At least a third of REs have SF2 helicase-like domains and have DNA cleavage activity that is dependent upon ATP hydrolysis (14); these enzymes are classified as Type I, ISP or III (15,16). The Type I RE CglI comprises two subunits, R and H (Figure 1A) (5,6). The 358 aa R-subunit has an N-terminal phospholipase D (PLD) domain and a C-terminal B3-like domain which binds the specific target sequence 5′-GCCGC-3′. The 632 aa H-subunit comprises an N-terminal 229 aa helicase domain (named ‘DEAD’ after the Walker B sequence), an uncharacterised 234 aa Z1-superfamily domain (17), and an uncharacterized 169 aa C-terminal domain with a lack of primary sequence similarity to proteins of known structure. CglI is active as a heterotetrameric R2H2 complex, formed of two dimers R2 and H2, where dimerization occurs via the PLD domains (to generate a dimeric nuclease with a single active site), and via the Z1 domains, respectively (Figure 1B). Following site-specific DNA recognition by the B3 domain(s), extensive nucleotide hydrolysis (∼170 ATP/s) is coupled to stepwise DNA translocation, preferentially in a downstream direction from the target, although upstream translocation is also possible (5). Unusually, translocating motors can transfer onto a new DNA substrate with low efficiency. Upon collision with another enzyme at a target site, DNA cleavage occurs. The most efficient cleavage activity is due to ‘rear-end’, so circular DNA and linear DNA with sites in direct repeat (i.e. head-to-tail) are the best cleavage substrates.
Figure 1.

The restriction endonuclease CglI from Corynebacterium glutamicum. (A) Domain organisation of R- and H.CglI proteins. R.CglI (NCgl1704) has PLD-superfamily nuclease and B3-like DNA binding domains. H.CglI (NCgl1705) has a predicted SF2 helicase/ATPase domain (DEAD) linked to uncharacterized Z1-superfamily and C-terminal domains. Numbers represent predicted amino acid positions of domain boundaries. (B) A possible model of the R2H2.CglI complex (6,79). The R.CglI dimer is presented by the structure of the homologous R.NgoAVII (PDB ID 4RCT) bound to the cognate DNA via the B3 domains (PDB ID 4RD5). Two R.NgoAVII PLD domains from separate subunits (coloured blue and light blue) dimerise to form single active site (nuclease active site residues H104 and K106 are coloured red). B3-like domains (coloured green and light green) are found in plant transcription factors and recognise the 5′-GCCGC-3′ target. DNA is shown as black ribbon. H.CglI contains the DEAD, Z1 and C-terminal domains (separate subunits are white or light grey).
The elevated ATP hydrolysis rate and roadblock displacement activities of CglI are consistent with a dsDNA translocating motor like other Type I and ISP REs (4). The DEAD domain was identified based on the homology of conserved SF2 motifs, including the characteristic Walker A and B (18,19). However, at 229 aa (Figure 1A), the domain is appreciably smaller than the SF2 helicase-like motors of other Type I and ISP enzymes, which are typically >400 aa monomers comprising both N- and C-core RecA-like sub-domains (4,7). This is also the arrangement observed in all other SF2 enzymes (20). The DEAD domain of CglI has a size more consistent with a single RecA fold. Key to understanding the mechanism of CglI is therefore an understanding of the structural arrangement of the unusual DEAD domain.
Based on our simple model for the R2H2 complex (Figure 1B) (6), three structural arrangements for the helicase-like motor can be suggested: (i) The DEAD domain alone has ATP binding and hydrolysis activity, meaning there are two independent ATPase sites per R2H2 complex. This model is disfavoured because all helicases studied to-date, regardless of superfamily, use two RecA folds to generate an NTP binding interface. Moreover, it is the opening-closing motions of the domains during the ATPase cycle that are fundamental to mechanochemical coupling. (ii) The two DEAD domains in the complex dimerise to produce one ATP binding interface. This would allow opening-closing motions of the domains that could be coupled to dsDNA translocation. This model is disfavoured because dimerization between isolated DEAD domains has not been observed (6). Additionally, this arrangement would be unusual for a SF2 enzyme, and interactions between identical domains in SF3-SF6 is via multimerization, not dimerization. (iii) The DEAD domain is only one half of the helicase-like motor, and another domain provides the second RecA-fold. Given the presence of the Walker A and B motifs, the DEAD domain can be defined as the N-core. The most likely candidate for the C-core would be the adjacent 234 aa Z1 domain. This arrangement would be more like other SF2 enzymes where the N- and C-core are contiguous. Therefore, we propose the R2H2 complex would contain two separate DEAD-Z1 helicase-like motors.
To examine whether CglI employs a prototype DEAD-Z1 helicase-like motor and to further expand on our overall understanding of the mechanism of this class of REs, we employed a combination of structural approaches (including X-ray crystallography, small-angle X-ray scattering and molecular modelling) and biochemical analysis (DNA- and ATP-binding, ATP hydrolysis and DNA cleavage) with protein mutagenesis. Our results indicate that DEAD-Z1 do indeed together form a unique ATP- and DNA-binding helicase fold with specific helicase motifs (Loop 163–172, H/H, Motif V and Motif VI), while a crystal structure of the C-terminal domain revealed a novel fold with DNA-binding activity. Despite the independent activities of the domains, we could not reconstitute ATPase or nuclease activity from any combination of the purified domains. We additionally found that when even one of the C-terminal domains in the complex was deleted, DNA cleavage activity was reduced suggesting that both H-subunits (i.e. two motors) cooperate in the process. Finally, using SAXS we are able to determine in more detail the relative arrangement of the H-subunit dimer.
MATERIALS AND METHODS
Expression and purification of CglI proteins
The CglI proteins, their mutants and the isolated domains and domain fusions were expressed and purified as described earlier (6). Se-Met modified H.CglI C-terminal domain was prepared by inhibiting the methionine synthesis pathway and expressing the protein in the presence of d,l-selenomethionine (Sigma) following the published procedure (21). The identity of the purified proteins was confirmed by mass spectrometry. All protein concentrations used in this work are shown in terms of monomer.
Mutagenesis
Mutants of the H.CglI protein and its domains were produced by QuikChange Site-Directed Mutagenesis (22). Sequencing of the entire gene for each mutant confirmed that only the desired mutations had been introduced.
Crystallization and structure determination
The unlabelled and Se-Met labelled C-terminal domains were concentrated to 15 mg/ml in buffer (10 mM Tris–HCl (pH 7.5 at 25°C), 300 mM KCl, 1 mM EDTA, 1 mM DTT). Crystals were grown using the sitting drop vapor diffusion method at 19°C by mixing 0.5 μl of the protein solution with 0.5 μl of the crystallization buffer (0.1 M Tris–HCl (pH 8.5 at 25°C), 7–14% (v/v) Tacsimate, 25–30% (w/v) PEG3350). Crystals belonging to the space group P6122 appeared after few days. The X-ray diffraction datasets for Se-Met modified protein crystals were collected at the EMBL/DESY PetraIII P14 beamline (Germany) at 100 K using reservoir solution supplemented with 20% (v/v) ethylene glycol as cryo-buffer. XDS (23), SCALA and TRUNCATE (24) were used for data processing. Native protein crystals diffracted to lower resolution (2.9 Å) compared to Se-Met crystals (2.4 Å). Therefore, the structure of the Se-Met modified protein was solved and is discussed in this paper. Data collection and refinement statistics are presented in the Supplementary Table S1.
The structure was solved using the 3W-MAD protocol of Auto-Rickshaw: the EMBL-Hamburg automated crystal structure determination platform (25). The input diffraction data were prepared and converted for use in Auto-Rickshaw using programs of the CCP4 suite (24). FA values were calculated using the program SHELXC (26). Based on an initial analysis of the data, the maximum resolution for substructure determination and initial phase calculation was set to 2.40 Å. Two heavy atoms out of the maximum number of two heavy atoms requested were found using the program SHELXD (27). The correct hand for the substructure was determined using the programs ABS (28) and SHELXE (29). Initial phases were calculated after density modification using the program SHELXE (29). 77.53% of the model was built using the program ARP/wARP (30). Manual model rebuilding was performed in COOT (31). The structure was refined with phenix.refine.1.8.3 (32). TLS groups were determined by TLSMD server (http://skuld.bmsc.washington.edu/∼tlsmd/) (33). The crystal contacts were analysed using PISA server (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html) (34). Electrostatic surface potential was calculated using PDB2PQR and APBS (35,36). All molecular scale representations were prepared using the PyMOL Molecular Graphics System (Version 1.2r3pre, Schrödinger, LLC) (37).
Computational sequence analysis and structural modelling
Homologs of H.CglI were collected from UniprotKB (38) by running three iterations of jackhmmer (39) search with the H.CglI region (237–632), covering Z1 and C-terminal domains as the query. Full-length sequences of all the identified homologs (E-value ≤ 10−3) were clustered at 90% sequence identity with CD-HIT (40) and aligned with MAFFT (41). The alignment was further refined by removing sequence fragments and sequences with poor alignment in the N-terminal and/or C-terminal regions.
Detection of H.CglI homologs with known structures was performed using HHpred (42). The alignment obtained as described above (807 sequences) was used as an input to HHpred to search against HMM profiles corresponding to PDB structures.
A structural model of H.CglI region corresponding to both DEAD and Z1 domains was constructed as follows. A number of homology models were generated from high-scoring HHpred alignments using MODELLER (43) with various helicases as structural templates. Estimation of model quality was performed using VoroMQA (44). The representative model was constructed based on the combination of three structural templates: (1) PriA Helicase (45) (PDB ID 4NL4); (2) the HsdR subunit of the EcoR124I Type I RE in complex with ATP (PDB ID 4XJX); and, (3) the DEAD-box protein Dhh1p (46) (PDB ID 1S2M). Long loops and regions not covered by the structural templates were remodelled using I-Tasser (47). The model was refined by fragment-guided molecular dynamics (48). ATP was modelled into H.CglI by simply copying it from the HsdR structure. The approximate position of the bound DNA was inferred from superposition of the H.CglI model with the SsoRad54–DNA complex (49) (PDB ID 1Z63) (Figure 3B).
Figure 3.
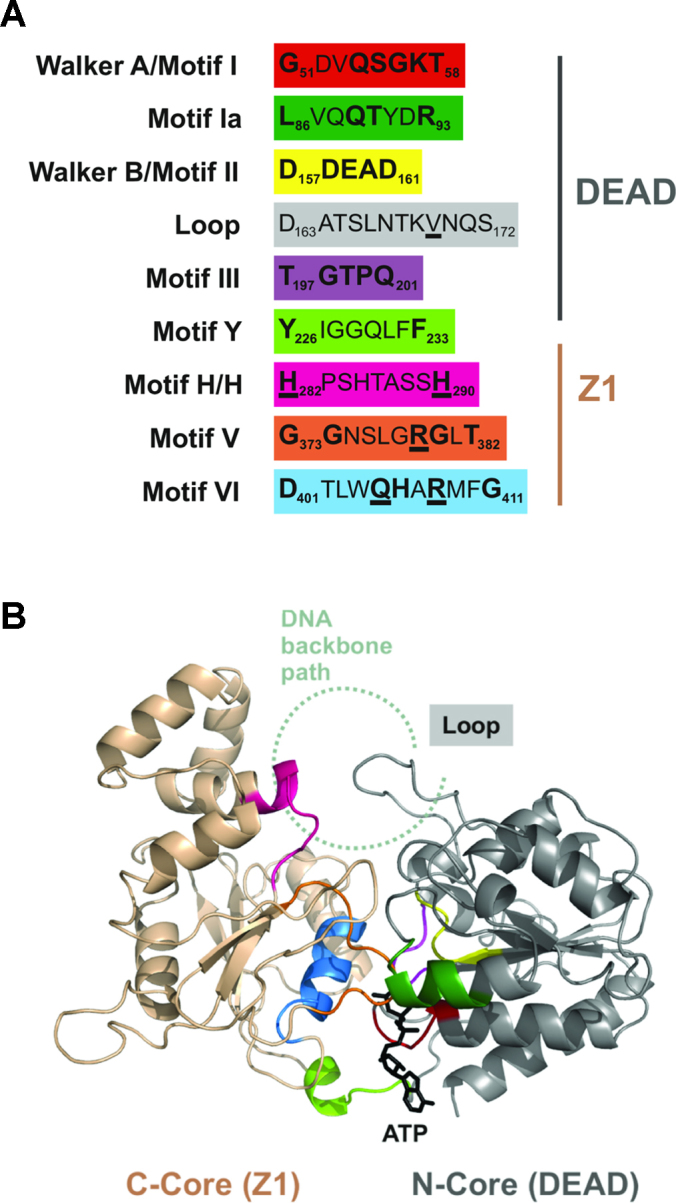
Putative helicase motifs and model for the DEAD-Z1 helicase of H.CglI. (A) H.CglI helicase motifs identified from the alignment of ORFs related to the H-subunit (Supplementary Figure S5). The loop was identified from the model structure shown in panel B. Residues in bold are part of the DEAD-Z1 consensus sequences. Underlining indicates that a mutation was made at that amino acid in this study. (B) Model of the DEAD-Z1 helicase of H.CglI (aa 22 – aa 447). The DEAD domain is coloured in grey, the Z1 domain in gold. The motifs are coloured as in panel A. ATP is shown as a stick model. An approximate location of the path of a DNA backbone was constructed by similarity to the SsoRad54-DNA complex (49), PDB ID 1Z63.
Mant-ATP binding assay
Protein samples at increasing loading concentrations (from 0.1 to 15 μM) were prepared in 160 μl buffer (20 mM Tris–HCl (pH 8.0 at 25°C), 10 mM MgCl2, 100 mM NaCl, 1 μM Mant-ATP). Mant-ATP binding anisotropy values of the same sample were recorded five times using FluoroMax-3 spectrofluorimeter equipped with Glan-Thompson polarizers (JY-Horiba Inc., New Jersey) and a 10 mm quartz cuvette. Fluorescence anisotropy measurements were carried out at room temperature using λex = 356 nm and λem = 440 nm. Dissociation constant values (Kd) for Mant-ATP binding were calculated from 3–5 consecutive measurements by fitting binding data to the hyperbolic Equation (2).
ATPase assay
ATPase reactions were performed at 37°C in reaction buffer (33 mM Tris-acetate (pH 7.0 at 25°C), 10 mM Mg-acetate, 0.1 mg/ml BSA, 1 mM DTT, 150 mM K-acetate) supplemented with 0.62 nM phage λ DNA (contains 181 CglI site) and 2 mM ATP. Reactions were initiated by adding 10 nM H.CglI or its domain(s) and 200 nM R.CglI. Aliquots were removed at fixed time intervals and the reactions were stopped by adding EDTA to a final concentration of 9.5 mM. Phosphate accumulation rate (v, μM × min−1) was measured using Malachite green phosphate assay kit (BioAssay Systems), which is used to measure the ATP hydrolysis rate by detection of liberated free phosphate, following the manufacturer's protocol. The relationship between the absorbance and phosphate concentration was established by using the phosphate solution as a standard. The rates are quoted as the mean of at least three independent experiments.
DNA binding assay
DNA binding by the CglI proteins was analysed by the electrophoretic mobility shift assay (EMSA) using a 25 bp non-cognate oligoduplex (5′-AGATCGAAGACCGTAGCACTGGGCT-3′/3′-TCTAGCTTCTGGCATCGTGACCCGA-5′). The DNA was labelled at the 5′ end on one strand with 33P. DNA (final concentration 100 nM) was incubated with H.CglI proteins or domains (final concentrations from 50 to 5000 nM) for 20 min in 10 μl of the binding buffer containing 30 mM MES-His (pH 6.0 at 25°C), 1 mM DTT, 0.1 mg/ml BSA and 10% (v/v) glycerol at room temperature. Free DNA and protein–DNA complexes were separated by electrophoresis using 8% (w/v) acrylamide gels (29:1 acrylamide/bisacrylamide in 30 mM MES-His, pH 6.0 at 25°C). Samples were separated at room temperature for 1 h at ∼6 V/cm. Radiolabelled DNA was detected and quantified using the Fujifilm FLA-5100 fluorescent image analyzer (Fujifilm, Tokyo, Japan) and the OptiQuant 3.0 software (Packard Instrument) to determine the volume of each band, taking background readings into account. Apparent Kd values for protein-DNA binding were calculated from three independent reactions by fitting the binding data to Equation (1).
DNA cleavage assay
DNA cleavage activity was tested using 10 nM of one-site supercoiled circular p1 plasmid DNA (5). Reactions were performed at 37°C in reaction buffer (see above) supplemented with 4 mM ATP, 20 mM phosphocreatine and 5 U/ml creatine kinase. The reactions were initiated by adding 200 nM H.CglI and 200 nM R.CglI. Reactions were stopped by adding 2× Loading Solution (75 mM EDTA, 0.6% (w/v) orange G, 50% (v/v) glycerol, 0.5% (w/v) SDS, pH 8.0 at 25°C) followed by incubation at 70°C for 15 min. The reaction products were separated by electrophoresis through 1.0% (w/v) agarose gels with 0.5 μg/ml ethidium bromide. The relative amount of dsDNA cleavage was quantified by gel densitometry using BioDocAnalyze image analyzer (Biometra, Göttingen, Germany) and the images were analyzed with OptiQuant 3.0 software (Packard Instrument) to determine the volume of each band, taking background readings into account. The fraction of DNA substrate left in each sample was calculated as volume(DNA substrate left)/(volume(DNA substrate left) + volume(DNA product)) (5). DNA cleavage rates are quoted as the mean of three independent experiments by fitting supercoiled DNA depletion data to a single exponential decay Equations (3) and (4).
DNA cleavage activity of H.CglI domains were tested using 10 nM pBR322 at 37°C for 1 h in the same reaction buffer as above. The reactions were initiated by adding 200 nM H.CglI or its domain(s) and 200 nM R.CglI. The reaction products were analysed as described above.
Small angle X-ray scattering (SAXS) experiments
SAXS data were collected at P12 EMBL beam line at PETRA III storage ring of DESY synchrotron in Hamburg (Germany). The SAXS data collection parameters and processing for H.CglI sample were described in (6). Dummy atoms models were calculated with DAMMIN (version 5.3) (50) and DAMMIF (r6669) (51) applying P1 or P2 symmetry, as the molecular weight of H.CglI in solution corresponds to dimer (6). Both variants, P1 and P2 were very similar. Ten runs of each program were compared and averaged with DAMAVER package (52).
The DEAD-Z1 model and the crystal structure of the C-terminal domain were used for the rigid body modelling against solution scattering data with SASREF program (version 6.0 r6669) (53). As the molecular weight of H.CglI in solution corresponds to dimer (6), model was calculated without applying symmetry and contact conditions were used in order to keep domains interconnected.
Data analysis
Quantitative analysis of protein-DNA binding and DNA cleavage was performed using KyPlot version 2.0 (54). The Kd values for oligonucleotide binding by the CglI proteins were calculated by using a quadratic equation:
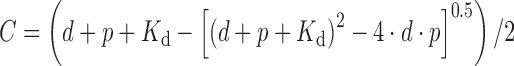 |
(1) |
where C is the formed protein–DNA complex concentration (nM) at each protein concentration p (nM), d is the DNA concentration in binding mixture, Kd is the dissociation constant of protein DNA complex (nM).
The Kd values for Mant-ATP binding by the CglI proteins were calculated by using a hyperbolic equation:
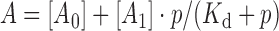 |
(2) |
where A is the observed anisotropy values at each protein concentration p (μM), A0 is anisotropy observed with free Mant-ATP, A1 is the free amplitude, Kd is the dissociation constant of protein–DNA complex (μM).
Rate constants (k1) for DNA cleavage were calculated using one of the following equations:
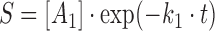 |
(3) |
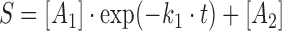 |
(4) |
where S is the amount of substrate left (nM), A1 is the amplitude of the first phase, A2 is the free amplitude, k1 is the rate of the first phase, t is the reaction time in seconds.
RESULTS AND DISCUSSION
Crystallography trials of H.CglI and its domains
In order to understand the helicase-like mechanism of the H-subunit in more detail, we first tried to crystallise the full-length protein (Materials and Methods). A recombinant expression clone of H.CglI was produced and the subunit purified to >90% homogeneity (Supplementary Figure S1A). The CD spectrum suggested a folded protein (Supplementary Figure S1B) and assays demonstrating functional activity are described below. However, despite trying a wide range of conditions, we could not obtain crystals suitable for structure determination. One method to overcome the problem of inefficient crystallization is targeted mutagenesis of surface patches containing residues with large flexible side chains and their replacement with smaller amino acids (alanine). This can reduce surface entropy which in turn could help with formation of the crystal lattice (55). Mutants of the H.CglI were generated according to predictions using the SERp server (56): E22A + K23A (further EK mutant), Q116A + K117A (further QK mutant) and E361A + E362A (further EE mutant). The production and analysis of these mutants is described in the Supplementary Information and Supplementary Figures S1 and S2. Unfortunately, these mutant proteins also gave crystals unsuitable for structure determination. We nonetheless note that the predicted positions of the surface residues that were mutated are in agreement with the DEAD-Z1 model that is described below.
As an alternative approach, we reasoned that the individual domains of the H-subunit, or combinations thereof, might crystallize more efficiently. Recombinant expression clones for the isolated DEAD, Z1, C-terminal domains and the DEAD-Z1 and Z1-C-domain-fusions (6) were used to purify the proteins to >90% homogeneity (Supplementary Figure S1A) (Materials and Methods). The CD spectra were consistent with folded proteins (Supplementary Figure S1B), and assays demonstrating functional activity are described below. The DEAD, Z1, DEAD-Z1 and Z1-C proteins were refractory to crystallization. Mutants of the Z1 domain were generated according to predictions using the SERp server (55): Q295A+E296A (further QE mutant), E361A + E362A (further EE mutant) and K396A (further K mutant). These were also unsuccessful for structure determination. However, crystals of the C-terminal domain were generated, and a structure was solved, which is described in the next section.
Crystal structure of the C-terminal domain
Native and Se-methionine labelled C-terminal domains (residues 464–632) were crystallized by the sitting drop vapour diffusion method and the structure was determined by MAD (Materials and Methods). In the crystal structure 14 residues at the N-terminus and 9 residues at the C-terminus of the domain are disordered. The asymmetric unit of the C-terminal domain crystal contains one protein chain (Figure 2A, Supplementary Figure S3A). Analysis of the protein interfaces in the crystal with the PISA server (34) has not revealed any specific interactions that could result in the formation of stable quaternary structures indicating that the C-terminal domain is a monomer. This agrees with the gel filtration data (6). The C-terminal domain is folded into a 5-stranded mixed β sheet (βC3, βC5, βC6, βC7, βC1) flanked by four α helices, three helices from one side (αC1, αC2, αC3) and one helix (αC4) from the other side (Figure 2A, Supplementary Figure S3A). The domain also contains additional two-stranded antiparallel β sheet (βC2 and βC3) and a helix αC5. Structure comparison using DALI server (57) revealed that the C-terminal domain has a unique fold, which is also consistent with its lack of primary sequence similarity to other proteins (Supplementary Figure S3B). In isolation, the C-terminal domain structure does not provide an immediate answer to the question of its role.
Figure 2.
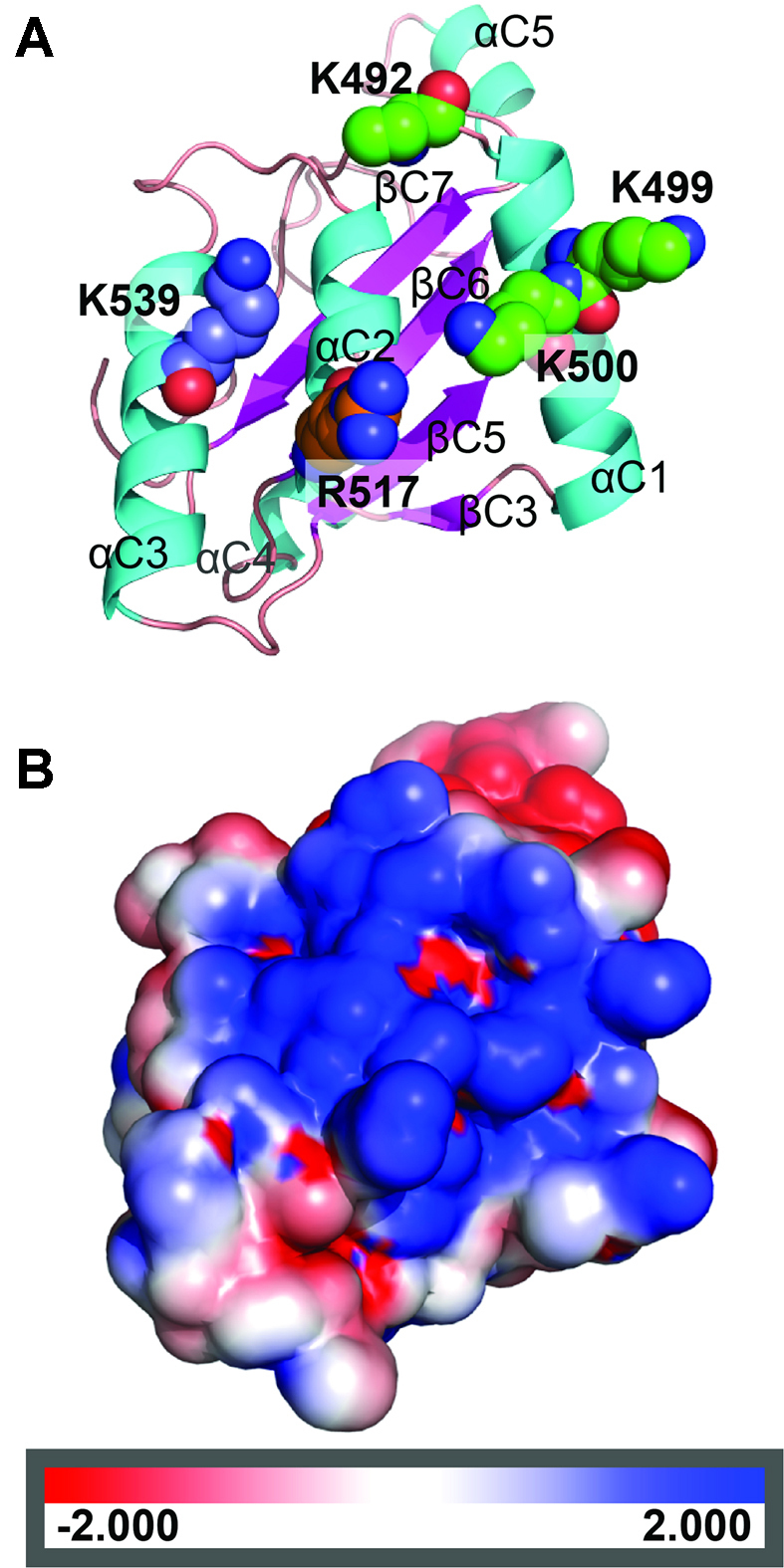
Crystal structure of the H.CglI C-terminal domain. (A) Ribbon representation of the C-terminal domain with possible aa responsible for DNA binding. The positively charged residues subjected to mutagenesis are shown as spacefill. (B) Electrostatic surface potential of the C-terminal domain (the protein orientation is as in (A)).
Analysis of the C-terminal domain surface potential showed a patch of positively-charged residues located mostly on the surface of helices αC1, αC2 and αC3, namely K492, K499, K500, R517, K539 (Figure 2). This feature is characteristic of a DNA-interacting surface. However, there is no data indicating whether this domain is involved in DNA binding by H.CglI. Further biochemical analysis of the C-terminal domain alongside the other H-subunit domains is presented below.
The Z1 domain is characteristic of the second RecA-like domain (C-core) of an SF2 helicase
We reasoned that computational analysis and molecular modelling could be applied to H.CglI to provide information about the domain functions. In addition to the C-terminal domain structure determined above, the H-subunit amino acid sequence has two identifiable domains: (i) a RecA-like domain having Walker A and B motifs characteristic of ATP-dependent DNA/RNA helicases and other helicase-like enzymes (12); and (ii) a so-called Z1 domain (Pfam: PF10593). The Z1 domain having unknown structure and function was initially identified as a C-terminal domain of a SF2 helicase (17). However, at the time Z1 was predicted to have an α+β architecture, which is distinct from the typical α/β architecture of a SF2 C-core (RecA-like) domain. The H.CglI sequence was further analysed computationally, with particular focus on the uncharacterized Z1 domain. Iterative sequence searches dealing with multidomain proteins are prone to the overextension error (58). This type of error often occurs when one of the domains is very popular in nature (in this case DEAD domain), but the other domain (Z1) is uncommon. We therefore used a multi-step approach to provide a confident assignment of the Z1 fold.
Initially, H.CglI homologs were collected from UniprotKB using as a query only the H.CglI region encompassing Z1 and C-terminal domains. This was done in order to avoid inclusion of all the other P-loop ATPases that lack the Z1 domain. Remarkably, nearly all of the detected homologs of the H.CglI Z1 domain had the P-loop ATPase domain featuring Walker A and B motifs preceding the Z1 domain. This observation suggested that the Z1 domain might correspond to the universally present C-core domain of SF2 helicases. To test this hypothesis we used the alignment of H.CglI homologs (see Materials and Methods) as an input to HHpred (42) to search for related proteins in the PDB. The results revealed high confidence matches (HHpred probability >95%) to various DNA/RNA helicases and translocases. As expected, the H.CglI N-terminal region matched the N-core RecA-like fold containing ATP-binding signature (Walker A and B motifs). Notably, the Z1 domain region confidently matched a C-core RecA-like domain indicating that H.CglI has two RecA-like core domains typical of the SF2 helicases, fused with the novel C-terminal domain.
The assignment of Z1 as the RecA-like C-core domain was further scrutinized by using the Z1 domain alone. The Pfam alignment for the Z1 domain sequences (filtered at 75% sequence identity) was downloaded and subsequently used as an input for the HHpred search against PDB. Again, the top matches were all against the C-core RecA-like domains of various helicases/translocases, albeit with slightly lower confidence (the highest HHpred probability around 90%). We therefore suggest that each H-subunit contains a standalone helicase-like motor comprising dual RecA folds.
A structural model of the DEAD-Z1 helicase-like motor of H.CglI
Taken together, the in silico analyses above show that the uncharacterized Z1 domain most likely represents a variant of the RecA-like C-core domain present in other SF2 helicases/translocases. Based on the detected homology with SF2 helicases/translocases, we constructed a structural model for both the N-core (DEAD) and the C-core (Z1) RecA-like domains.
To help understand how consistent the conserved amino acid sequence motifs are located within the putative RecA-like folds, we further sought to re-analyse the primary sequence alignment of the H-subunits to determine amino acid motifs that might be characteristic of this sub-family of helicase-like proteins. The full-length H-subunit alignment used above was analysed for consensus sequences using WebLogo (59). We could clearly identify six characteristic helicase motifs (Figure 3A, Supplementary Figure S5): Motif I/Walker A, Motif Ia, Motif II/Walker B and Motif III within the DEAD domain, and, Motif V and Motif VI in the Z1 domain. The substitution of a glycine for a glutamine marks Motif I as being an uncommon Walker A variant amongst the SF1 and SF2 helicases. Another unique feature is motif VI, where instead of the typical QxxxR signature, a shorter QxxR spacing is found, implying that the side chain of R408 is somewhat differently oriented compared to the ‘arginine-finger’ in a typical motif VI. Nonetheless, the presence of helicase Motifs V and VI in the Z1 domain reemphasizes the potential importance of this domain to helicase-like motor activity.
A Y-motif characterised in other RE helicase-like subunits (60) could be also identified at the interface between the DEAD and Z1 domains. Additionally, we identified a Z1-specific motif, which we called H/H, which to our knowledge has not been observed in other helicases or helicase-like motors so far (Figure 3A). It also coincides with the beginning of an α-helical insertion/subdomain that seems unique to H.CglI and its close homologs. We note that the H/H motif is in a structurally-equivalent position to motif IV of SF2 helicases, which is most commonly involved in interaction with DNA/RNA. Motif IV does not have a common consensus sequence, but the H/H motif appears unique. We note that Motif IV sequences have been difficult to identify in Type I and III restriction enzymes (61). Nonetheless, we would predict a role for the H/H motif in DNA binding during translocation.
Clear motifs in the C-terminal domain could not be confidently discriminated, although solitary conserved residues were observed (e.g. N493, R562, W609, P622 and G632) (Supplementary Figure S5).
Overall, the common helicase motifs in the structural model of H.CglI are in the expected locations in 3-dimensions (Figure 3B). For comparison, the location of the SF2 helicase motifs in Rad54 is shown in Supplementary Figure S6. The N-core of CglI has a typical RecA-like structure with both Walker A (Motif I) and Walker B (Motif II, D158, E159 and D161) present at the ATP binding pocket. One of the specific features of the N-core of H.CglI and its homologs is that the Walker B (DDEAD) motif is followed by an extended structurally-conserved loop (residues 163–172). The first half of this loop shows some sequence conservation (Supplementary Figure S5). Based on the proposed approximate location of the DNA, the more sequence divergent second half of Loop 163–172 has the potential to interact with the translocated DNA. A corresponding loop in an archaeal SWI2/SNF2 protein (SsoRad54 dsDNA translocase), referred to as the ‘switch region’ or Motif IIa, has been shown to be involved in regulation of DNA-binding dependent ATPase activity (49). Corresponding regions in some other helicases, including the Type ISP REs, have also been implicated in the DNA-binding and/or ATPase activity (62–64).
The C-core has some unique features. One of these is a conserved α-helical insertion into the RecA-like fold. This insertion includes the two histidine residues (H282 and H290) of the conserved H/H motif identified above. Although some helicases/translocases do have domain/subdomain insertions in the corresponding region, the helical insertion seems to be unique to Z1 homologues. Furthermore, this insertion might be the reason why Z1 has not been documented previously as the C-core RecA-like domain. Although the modelled structure of the insertion has a low confidence, its relative position suggests that this structural motif might also be involved in binding the translocated DNA.
Mutation of the helicase motifs in the Z1 domain abolish ATPase and nuclease activities
To begin to test our model of DEAD-Z1 as a dual RecA helicase-like motor, we expressed and purified to >90% homogeneity the following H.CglI mutants as full-length H-subunits (Supplementary Figure S1A): helicase motif mutants R379A (Motif V), Q405A (Motif VI) and R408A (Motif VI); H/H motif mutants H282A, H290A and H282A + H290A; and, Loop 163–172 mutants L1 (V169A), L3 (L165A + K168A + V169A) and ΔL (a deletion of the residues 165–172). According to size exclusion gel filtration chromatography, all the H-subunits variants except L3 were dimeric and formed heterotetrameric R2H2 complexes with the wild type R-subunit (Supplementary Table S2). CD spectra of the Loop 163–172 variants showed that L3 was aggregated, so this mutant was not analysed further (Supplementary Figure S1B).
To evaluate characteristic enzyme activities of the wild type and mutant CglI proteins, we measured the following properties (Materials and Methods): (i) ATP-binding, as evaluated by a fluorescence anisotropy assay using Mant-ATP (65); (ii) ATP hydrolysis activity, measured from the phosphate release rates using a malachite green assay (6); (iii) DNA cleavage activity, measured by a gel-based assay using a one-site plasmid DNA which is an efficient substrate for the wild type enzyme; and (iv) DNA binding using an EMSA with a 25 bp non-cognate oligoduplex. Representative ATP binding, ATPase and DNA cleavage data is shown in Figure 4 for the loop and H/H motif mutants, while raw and fitted data for all the mutants are in Supplementary Figures S7–S10 and Tables S3–S6.
Figure 4.

Enzyme properties of Loop and H/H motif mutants. (A) Mant-ATP binding was studied by fluorescence anisotropy measurements. Data points are fitted to a hyperbolic function. (B) Rates of ATP hydrolysis. Reactions contained 0.62 nM phage λ DNA (contains 181 CglI sites), 10 nM H.CglI, 200 nM R.CglI, 2 mM ATP. (C) Rate of cleavage of the first strand (nicking) of one-site supercoiled DNA. Data points are fitted to a single exponential decay function. The fitted values for panels A-C are presented in Supplementary Tables S4–S6. All data is the average of three or more reactions, with the error bars ± SD where shown.
All of the variants were able to bind Mant-ATP with a similar affinity, within experimental error (1.9–5.0 μM), as the wild type H.CglI (3.2 μM, Figure 4 and Supplementary Table S6). The Kd values for ATP binding by other DEAD-box proteins can range between ∼50 nM and >6 mM (66–68). The CglI values are not atypical and SF2 helicase RecG has a similar Kd for Mant-ATP of ∼4 μM (69), while NS3 has a Kd for ATP of ∼2 μM (70). In the case of the R379A and R408A mutants, the amplitude of the anisotropy change was lower than that of the wild type protein, but they could nonetheless still bind to the Mant-ATP (Supplementary Figure S11). However, all of the variants had significantly decreased or unmeasurably-low ATPase activity and DNA cleavage activity close to, or at, background rates (Figure 4 and Supplementary Figures S8–S9 and Tables S4–S5). This suggested that although ATP can bind in all cases, the coupling between ATP turnover and subsequent DNA cleavage has been broken. Similarly, mutations in motif VI of RecD2 only slightly reduced affinity for nucleotide or RNA binding, but decreased ATPase turnover by 10-fold, which is also observed in other DEAD-box helicases (71). Motif VI most likely plays a direct role in ATP catalysis, typical of its role in other helicases and helicase-like enzymes. Structures equivalent to Motif V and Loop 163–172 are implicated in DNA interactions. However, in our EMSAs, little difference was observed between the wild type H.CglI and the loop, helicase or H/H variants in DNA binding affinity (Kd = 241–451 nM, Supplementary Figures S7–S8 and Table S3). The Kd values for polynucleotide-binding among helicases and helicase–like proteins varies greatly, from low nanomolar to low micromolar (66,69–74). Of course there are differences in the nature of the substrate and the buffer conditions which can influence these values, while affinity can vary significantly dependent upon ATP binding (e.g. (75)).
The almost background ATPase rate of the Motif V (R379A) mutant might suggest a more direct role in ATP hydrolysis. The loop and H/H mutants produced a 3- to 8-fold decrease in the ATPase rates and a >100-fold decrease in the subsequent DNA cleavage, which may indicate roles for these motifs in coupling of the ATP hydrolysis activity. The DNA binding data does not support a role for the H/H motif in DNA interactions (Supplementary Figures S7 and S8).
Roles for the H-subunit domains in binding to ATP and DNA
Based on the homology model of the DEAD-Z1 helicase (Figure 3) and the crystal structure of the C-terminal domain (Figure 2), we predicted that different domains or combinations thereof would have different ATP and/or DNA binding properties. Firstly, given that the DEAD and Z1 RecA-like cores should both be required to make an ATPase active site, ATP binding should therefore require both domains, but should not require the C-terminal domain. Secondly, the DEAD, Z1 and C-terminal domains all have features that might be necessary for DNA binding. This may explain why single mutations/deletions were insufficient to prevent DNA binding. To test these ideas, we used the H-subunit domains isolated above and tested the Mant-ATP or non-specific DNA binding activities (Materials and Methods). Representative data is shown in Figure 5A and B and Supplementary Figures S7 and S8, and the fitted Kd values in Supplementary Tables S3 and S6.
Figure 5.

Functional analysis of the domains of H.CglI. (A) Mant-ATP binding by the full-length H.CglI and its isolated domains was studied by fluorescence anisotropy measurements. Data points were fitted to a hyperbolic function. (B) DNA binding by the full-length H.CglI and its domains measured using an EMSA. The reactions contained 1 nM of the 33P-labelled oligoduplex and the proteins indicated at increasing concentrations (nM): 0, 50, 100, 200, 500, 1000, 2000 and 5000 (shown as wide black triangles). Images of separate native polyacrylamide gels are shown, obtained by Fujifilm FLA-5100 fluorescent image analyzer and the OptiQuant 3.0 software. Filled and open triangles correspond to the mobility of the free DNA and a protein–DNA complex, respectively. (C) Cleavage of the 21-site pBR322 plasmid DNA by the mixtures of the R.CglI and H.CglI or its domains, as indicated, for 1 h at 37°C. M – marker (DNA fragments of known length in bp), K – control (pBR322 plasmid DNA), SC – supercoiled circular DNA, OC – open circular DNA (‘nicked’), FLL – full-length linear DNA, CP – cleavage products at multiple sites. (D) Rate of cleavage of the first strand (nicking) of one-site supercoiled DNA by the wt R2H2.CglI and R2HΔH.CglI (lacking a single C-terminal domain within H.CglI) complexes. Data points are fitted to a single exponential function. The fitted values for panels A, B and C are presented in Supplementary Tables S3, S4 and S6. All data points are the average of 3 or more reactions, with the error bars ± SD.
A change in fluorescence anisotropy consistent with binding of Mant-ATP was observed with the full-length H-subunit in isolation and the DEAD-Z1 fusion, but not with the isolated DEAD or Z1 domains (Figure 5A, Supplementary Tables S6). The apparent Kd values are very similar for the full-length H-subunit and the isolated DEAD-Z1 domains (Kd = 3.2 or 3.7 μM, respectively). This provides compelling evidence in support of the model in Figure 3B for the DEAD-Z1 domains being the complete helicase motor.
Titration of the wild type H-subunit against a non-cognate oligoduplex produced a band-shift on a native polyacrylamide gel, consistent with DNA binding (Figure 5B). In addition to a discrete shifted band, a ‘smearing’ of the free DNA was observed which most likely represents more weakly associated complexes that dissociate during the electrophoresis process. DNA binding activity was also observed with the isolated Z1 and C-terminal domains, and with the DEAD-Z1 and Z1-C fusions, but not with the DEAD domain alone (Supplementary Figures S7 and S8). The isolated Z1 domain and Z1-C fusion had an apparent Kd values (Kd = 84 or 46 nM, respectively) that were lower than the full-length protein (Kd = 307 nM) or the DEAD-Z1 fusion (Kd = 844 nM) (Supplementary Table S3), which may indicate that the Z1 domain is autoinhibited by the DEAD domain. The data suggests that Z1-C provides an important DNA contact surface. The lack of binding of DNA by the isolated DEAD domain is perhaps surprising given our prediction of a role in DNA transport and the presence of Loop 163–172. However, it may be that Loop 163–172 only makes transient DNA contacts as part of a mechanochemical cycle that cannot be mimicked by EMSAs in the absence of ATP. An H/H mutant of the Z1 domain had an apparent Kd similar to wild type (Kd = 99 nM) (Supplementary Figures S7 and S8 and Table S3) which is in agreement with the results above. Again, in the absence of ATP we cannot fully rule out a role in DNA interaction during translocation.
Given the availability of the C-terminal domain structure and its DNA binding activity, we also mutated residues on the basic surface patch (Figure 2), in both the isolated domain and the full-length H-subunit: R517E, K539E and 3M (K492E+K499E+K500E). We tested DNA binding, ATPase and nuclease activities (Supplementary Information, Supplementary Figures S7-S10, Supplementary Tables S3-S5). The results support a role for the C-terminal domain basic surface patch in binding to DNA and in the coupling DNA binding to ATP hydrolysis and long-range communication necessary for DNA cleavage (Supplementary Information).
The importance of H-subunit domain connectivity and complete heterotetrameric assembly to the activity of CglI
Since the isolated domains and domain fusions of H.CglI have biochemical activity, we reasoned that it may be possible to reconstitute full RE activity using different combinations of the domains/domain fusions along with the full-length R.CglI. We tested this by mixing different protein combinations at 1:1 stoichiometry, and then following the cleavage of pBR322 (contains 21 CglI target) at a reaction endpoint (37°C for 1 h), conditions under which the wild type proteins cut the DNA multiple times (Figure 5C). Although background levels of DNA cleavage were observed with some combinations, only the full-length H-subunit produced efficient DNA cleavage activity with the R-subunit. A similar conclusion could be drawn from measurements of ATP hydrolysis using combinations of the domains (Supplementary Table S5). This may be the consequence of inter-domain motions during ATP hydrolysis that cause disassembly of the complex unless the domains are connected as a single polypeptide. We note however that the DEAD-Z1 fusion was still capable of ATP hydrolysis, albeit 20-fold reduced compared to the full complex.
This data suggests that the DEAD, Z1 and C-terminal domains are all required for full RE activity. A key mechanistic question is why CglI is active as a tetramer with two H-subunits. Given our model and data, there are two separate helicase motors (Figure 1B). In the cleavage model, an R2H2 complex must translocate along DNA to communicate with a second enzyme at a distant target site. However, it isn’t immediately obvious why two motors would be needed. To begin to approach this problem, we isolated an R2H2 CglI complex without one C-terminal domain (R2HΔH.CglI) (Supplementary Information, Supplementary Figure S4). This complex assembles into the expected heterotetrametric complex (Supplementary Table S2). Surprisingly, this complex had a >5-fold reduction in ATPase activity (Supplementary Table S5) and a corresponding ∼60-fold reduction in the rate of cleavage of a one-site plasmid DNA (Figure 5D, Supplementary Table S4). Despite not being part of the dimerization interface (6), it appears that the C-terminal domains of both H-subunits are necessary for efficient ATPase and DNA cleavage activities.
Small angle X-ray scattering analysis of H.CglI
Based on gel filtration analysis (Supplementary Table S2) and the Mw calculated from SAXS data (76), H.CglI is a dimer in solution with the dimer interface being the RecA-like Z1 domains (6). In the absence of diffracting crystals, we used small angle X-ray scattering to provide further structural information on the overall shape of the H.CglI dimer that could help refine the model in Figure 1B and which could help to resolve whether both motors are cooperating in DNA translocation.
The ab initio estimation of an overall shape of the particle filled by dummy atoms is presented in Figure 6A. According to SAXS data H.CglI dimer in solution has an elongated form.
Figure 6.

The H.CglI dimer modeling using SAXS data. (A) Domain movement (SASREF) model compared with dummy atom model. Dummy atom model of the full-length H.CglI calculated by DAMMIN is shown as transparent spheres. The DEAD and Z1-domains were taken from the DEAD-Z1 model and are coloured green and blue, respectively. The C-terminal domains from the crystal structure (PDB ID 6F1S) are coloured yellow; ATP is shown in red CPK. (B) Fit of the SASREF model (green line) to SAXS data (red points).
We also performed rigid body modelling of the H.CglI dimer using the C-terminal domain crystal structure and the DEAD-Z1 model as separate domains comprising full length H.CglI (Figure 6A). The program used (SASREF 6,0 (53)) allows us to use additional information about contacts between domains. Based on dimerization via the Z1 domains, we placed two copies of the DEAD-Z1 model into the centre and connected a few distal amino acids Cα atoms of both Z1 domains (S253–S253 ∼8 Å and N246–A267 ∼9 Å). The limits are rather loose, reflecting our uncertainty regarding the exact dimerization interface. According to our ab initio shape determination the monomers of H.CglI are also elongated. Based on the proposed orientation of DNA bound by the DEAD-Z1 model we have connected (by very loose restraints), the positively charged surface of the C-terminal domain to residues located in the groove between the DEAD and Z1 domains (N211–D488 ∼8 Å, D186–V494 ∼7 Å, E310–R547, ∼41 Å and D40–Q593 ∼10 Å). These constraints were used in order to keep the presumed DNA-contacting interfaces of DEAD-Z1 model (Figure 3B) and C-terminal domain (Figure 2). As the unmodelled linker between Z1 and C-terminal domains comprises 30 amino acids (residues 448–477), no connectivity between C-terminus of the Z1 domain and N-terminus of the C-terminal domain was used as modelling restraint. Initial position of C-terminal domain was restrained at the distal end of the DEAD domains in the initial orientation of domains based on the ab initio shape obtained from the averaged dummy atom models and assuming dimerization via Z1-domains. The rigid body model obtained could be overlaid with the dummy atoms model (Figure 6A). The comparison of the scattering curve calculated from the rigid body model with the experimental scattering curve is shown in Figure 6B. The comparison shows that the model is smaller than the averaged shape of the particle in solution, as the I(s) values of the model are lower than experimental data. One reason for this could be partial aggregation of the protein sample.
CONCLUSIONS
According to homology modelling and identification of conserved sequence motifs, both the DEAD and Z1 domains of CglI are RecA-like domains that together form an interface where ATP binds and is contacted by key catalytic motifs such as Walker A, Walker B and Motif VI. Biochemical assays using wild type and mutant proteins are consistent with the model (Figure 3B). The Z1 domain is more distantly related RecA-like fold, which may be why it has not been identified as a helicase family domain up to this point (17). We propose that H.CglI is a prototype of a new SF2 helicase sub-family that is both structurally- and functionally-related to, but distinct from, other RE helicase-like motors (Figure 7). The DEAD-Z1 helicase-like motor has several characteristic features which are necessary for ATP and DNA hydrolysis; (i) an unusual Walker A motif sequence compared to other SF1 and SF2 helicases. Some Type ISP-like REs also have an unusual RFGKT Walker A sequence (77), but we note that in the wide P-loop family the first glycine is not conserved well in the Walker A motifs; (ii) an unusual register of the ‘arginine finger’ in the Motif VI compared to other SF1 and SF2 helicases; (iii) a loop following Motif II in the DEAD domain. A loop region that is similarly located in Type ISP REs, but which follows Motif III (Figure 7), may also play a role as a ‘molecular paddle’ during dsDNA translocation (78); and (iv), a previously undescribed H/H motif and structural insertion in the Z1 domain. This motif couples ATP hydrolysis to DNA motion/cleavage but its exact role remains to be confirmed. Although the modelling predicted a role in DNA interaction (equivalent to Motif IV), mutation of the H/H motif did not affect DNA binding activity. Despite the Type I, Type ISP and CglI proteins all undertaking similar dsDNA translocase functions, because of the differences between the proteins at the sequence and structural level, it remains difficult to predict the function of helicase-like proteins without first undertaking biochemical assays.
Figure 7.

Comparison of the helicase-like domains of Types I, ISP and III restriction enzymes. Helicase consensus motifs are shown based on the work here and (61,78,80,81). The Type IIIA consensus sequences are presented. Loop regions and the Type III PIN domain are indicated. ‘h’ represents a hydrophobic residue, ‘x’ represents <85% consensus, ‘…’ represents a variable amino acid spacing. The N- and C-core helicase-like domain structures are shown for (from left to right): H.CglI, Type ISP LlaBIII (PDB ID 4XQK, (78)), Type I HsdR subunit from EcoR124I (PDB ID 2W00, (82)), and Type III Res subunit from EcoP15I (PDB ID 4ZCF, (81)). Helicase motifs are coloured as in the upper panel. The expected position of Motif IV is indicated, although it is less easily identified in restriction enzymes (61). CglI, Type ISP and Type I enzymes are dsDNA translocases that consumes ∼1 ATP/bp (5,77,83). Type III enzymes are molecular switches that consumes 10–20 bp, change conformation, and then move on DNA by thermally-driven DNA sliding (3).
Although the DEAD-Z1 motor has basic ATP and DNA binding activities, and some residual ATPase activity, the complete H-subunit including the C-terminal domain is required for full activity. We were able to solve a crystal structure for the C-terminal domain of H.CglI, showing that it possesses a unique fold with a basic amino acid interface. We could demonstrate that the Z1 and C-terminal domains are responsible for interaction of H.CglI with DNA, but we cannot rule out that other domains or motifs are key to the dsDNA translocation activity. Interestingly, both full-length H-subunits within the R2H2.CglI complex were required for catalytic activity of the complex. We can now confirm that the complex has two complete helicase-like DEAD-Z1 motors and that the H-subunit dimer forms an elongated shape. We suggest a possible structure for the R2H2.CglI complex in Supplementary Figure S12. Whether both motors cooperate in an individual dsDNA translocation event remains to be determined. Future structural work could exploit recent advances in cryo-EM to obtain a structure for a complete R2H2 complex bound to DNA. In the meantime, our improved understanding of the H-subunit domains and motifs will help in more advanced biophysical assays that we are developing to follow the dsDNA translocation activity.
DATA AVAILABILITY
Atomic coordinates and structure factors of the H.CglI C-terminal domain crystal structure has been deposited with the Protein Data Bank under accession number PDB ID 6F1S.
Supplementary Material
ACKNOWLEDGEMENTS
Authors acknowledge Audra Ruksenaite for mass spectrometry, Gleb Burenkov and Alexey Kikhney for assistance with data collection at EMBL P14 and P12 beamline at the PETRA III storage ring, DESY Hamburg. The access to synchrotron has received funding from the European Community's Seventh Framework Programme (FP7/2007–2013) under BioStructX_7869.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Research Council of Lithuania [MIP-56/2015 to M.Z.]; NIH [1RO1GM111507-01 to M.D.S.]. Funding for open access charge: Research Council of Lithuania.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wu C.G., Spies M.. Overview: what are helicases. Adv. Exp. Med. Biol. 2013; 973:1–16. [DOI] [PubMed] [Google Scholar]
- 2. Clapier C.R., Iwasa J., Cairns B.R., Peterson C.L.. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017; 18:407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schwarz F.W., Toth J., van Aelst K., Cui G., Clausing S., Szczelkun M.D., Seidel R.. The helicase-like domains of type III restriction enzymes trigger long-range diffusion along DNA. Science. 2013; 340:353–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Szczelkun M.D. Roles for helicases as ATP-dependent molecular switches. Adv. Exp. Med. Biol. 2013; 767:225–244. [DOI] [PubMed] [Google Scholar]
- 5. Toliusis P., Zaremba M., Silanskas A., Szczelkun M.D., Siksnys V.. CgII cleaves DNA using a mechanism distinct from other ATP-dependent restriction endonucleases. Nucleic Acids Res. 2017; 45:8435–8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zaremba M., Toliusis P., Grigaitis R., Manakova E., Silanskas A., Tamulaitiene G., Szczelkun M.D., Siksnys V.. DNA cleavage by CgII and NgoAVII requires interaction between N- and R-proteins and extensive nucleotide hydrolysis. Nucleic Acids Res. 2014; 42:13887–13896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Singleton M.R., Dillingham M.S., Wigley D.B.. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007; 76:23–50. [DOI] [PubMed] [Google Scholar]
- 8. Erzberger J.P., Berger J.M.. Evolutionary relationships and structural mechanisms of AAA plus proteins. Annu. Rev. Biophys. Biom. 2006; 35:93–114. [DOI] [PubMed] [Google Scholar]
- 9. Tuteja N., Tuteja R.. Unraveling DNA helicases - Motif, structure, mechanism and function. Eur. J. Biochem. 2004; 271:1849–1863. [DOI] [PubMed] [Google Scholar]
- 10. Caruthers J.M., McKay D.B.. Helicase structure and mechanism. Curr. Opin. Struct. Biol. 2002; 12:123–133. [DOI] [PubMed] [Google Scholar]
- 11. Hall M.C., Matson S.W.. Helicase motifs: the engine that powers DNA unwinding. Mol. Microbiol. 1999; 34:867–877. [DOI] [PubMed] [Google Scholar]
- 12. Gorbalenya A.E., Koonin E.V.. Helicases - amino-acid-sequence comparisons and structure-function-relationships. Curr. Opin. Struct. Biol. 1993; 3:419–429. [Google Scholar]
- 13. Samson J.E., Magadan A.H., Sabri M., Moineau S.. Revenge of the phages: defeating bacterial defences. Nat. Rev. Microbiol. 2013; 11:675–687. [DOI] [PubMed] [Google Scholar]
- 14. Roberts R.J., Vincze T., Posfai J., Macelis D.. REBASE-a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015; 43:D298–D299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loenen W.A., Dryden D.T., Raleigh E.A., Wilson G.G.. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2014; 42:20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rao D.N., Dryden D.T.F., Bheemanaik S.. Type III restriction-modification enzymes: a historical perspective. Nucleic Acids Res. 2014; 42:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iyer L.M., Abhiman S., Aravind L.. MutL homologs in restriction-modification systems and the origin of eukaryotic MORC ATPases. Biol. Direct. 2008; 3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Linder P., Lasko P.F., Ashburner M., Leroy P., Nielsen P.J., Nishi K., Schnier J., Slonimski P.P.. Birth of the D-E-A-D box. Nature. 1989; 337:121–122. [DOI] [PubMed] [Google Scholar]
- 19. Gorbalenya A.E., Koonin E.V., Donchenko A.P., Blinov V.M.. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 1989; 17:4713–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fairman-Williams M.E., Guenther U.P., Jankowsky E.. SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol. 2010; 20:313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vanduyne G.D., Standaert R.F., Karplus P.A., Schreiber S.L., Clardy J.. Atomic structures of the human immunophilin Fkbp-12 complexes with Fk506 and Rapamycin. J. Mol. Biol. 1993; 229:105–124. [DOI] [PubMed] [Google Scholar]
- 22. Zheng L., Baumann U., Reymond J.L.. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004; 32:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kabsch W. Xds. Acta Crystallogr D. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. CCP4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994; 50:760–763. [DOI] [PubMed] [Google Scholar]
- 25. Panjikar S., Parthasarathy V., Lamzin V.S., Weiss M.S., Tucker P.A.. Auto-Rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an X-ray diffraction experiment. Acta Crystallogr. D. 2005; 61:449–457. [DOI] [PubMed] [Google Scholar]
- 26. Sheldrick G.M., Schneider T.R.. Direct methods for macromolecules. Nato Sci. Ser. Life. 2001; 325:72–81. [Google Scholar]
- 27. Schneider T.R., Sheldrick G.M.. Substructure solution with SHELXD. Acta Crystallogr. D. 2002; 58:1772–1779. [DOI] [PubMed] [Google Scholar]
- 28. Hao Q. ABS: a program to determine absolute configuration and evaluate anomalous scatterer substructure. J. Appl. Crystallogr. 2004; 37:498–499. [Google Scholar]
- 29. Sheldrick G.M. Macromolecular phasing with SHELXE. Z. Kristallogr. 2002; 217:644–650. [Google Scholar]
- 30. Morris R.J., Zwart P.H., Cohen S., Fernandez F.J., Kakaris M., Kirillova O., Vonrhein C., Perrakis A., Lamzin V.S.. Breaking good resolutions with ARP/wARP. J. Synchrotron Radiat. 2004; 11:56–59. [DOI] [PubMed] [Google Scholar]
- 31. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 32. Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D.. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012; 68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Painter J., Merritt E.A.. TLSMD web server for the generation of multi-group TLS models. J. Appl. Cryst. 2006; 39:109–111. [Google Scholar]
- 34. Krissinel E., Henrick K.. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007; 372:774–797. [DOI] [PubMed] [Google Scholar]
- 35. Dolinsky T.J., Nielsen J.E., McCammon J.A., Baker N.A.. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004; 32:W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A.. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schrödinger, LLC The PyMOL Molecular Graphics System. Version 1.2r3pre.
- 38. Bateman A., Martin M.J., O’Donovan C., Magrane M., Alpi E., Antunes R., Bely B., Bingley M., Bonilla C., Britto R. et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017; 45:D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eddy S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011; 7:e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li W.Z., Godzik A.. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006; 22:1658–1659. [DOI] [PubMed] [Google Scholar]
- 41. Katoh K., Standley D.M.. MAFFT: iterative refinement and additional methods. Methods Mol. Biol. 2014; 1079:131–146. [DOI] [PubMed] [Google Scholar]
- 42. Soding J., Biegert A., Lupas A.N.. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005; 33:W244–W248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sali A., Blundell T.L.. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993; 234:779–815. [DOI] [PubMed] [Google Scholar]
- 44. Olechnovic K., Venclovas C.. VoroMQA: Assessment of protein structure quality using interatomic contact areas. Proteins. 2017; 85:1131–1145. [DOI] [PubMed] [Google Scholar]
- 45. Bhattacharyya B., George N.P., Thurmes T.M., Zhou R.B., Jani N., Wessel S.R., Sandler S.J., Ha T., Keck J.L.. Structural mechanisms of PriA-mediated DNA replication restart. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:1373–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cheng Z.H., Coller J., Parker R., Song H.. Crystal structure and functional analysis of DEAD-box protein Dhh1p. RNAp. 2005; 11:1258–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roy A., Kucukural A., Zhang Y.. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010; 5:725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang J., Liang Y., Zhang Y.. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure. 2011; 19:1784–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Durr H., Korner C., Muller M., Hickmann V., Hopfner K.P.. X-ray structures of the Sulfolobus solfataricus SWI2/SNF2 ATPase core and its complex with DNA. Cell. 2005; 121:363–373. [DOI] [PubMed] [Google Scholar]
- 50. Svergun D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999; 76:2879–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Franke D., Svergun D.I.. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009; 42:342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Volkov V.V., Svergun D.I.. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003; 36:860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Petoukhov M.V., Svergun D.I.. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 2005; 89:1237–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yoshioka K. KyPlot – a user-oriented tool for statistical data analysis and visualization. Comp. Stat. 2002; 17:425–437. [Google Scholar]
- 55. Derewenda Z.S. Rational protein crystallization by mutational surface engineering. Structure. 2004; 12:529–535. [DOI] [PubMed] [Google Scholar]
- 56. Goldschmidt L., Cooper D.R., Derewenda Z.S., Eisenberg D.. Toward rational protein crystallization: a web server for the design of crystallizable protein variants. Protein Sci. 2007; 16:1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Holm L., Rosenstrom P.. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010; 38:W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gonzalez M.W., Pearson W.R.. Homologous over-extension: a challenge for iterative similarity searches. Nucleic Acids Res. 2010; 38:2177–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Murray N.E. Type I restriction systems: Sophisticated molecular machines (a legacy of Bertani and Weigle). Microbiol. Mol. Biol. R. 2000; 64:412–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McClelland S.E., Szczelkun M.D.. Pingoud AM. The type I and III restriction endonucleases: structural elements in molecular motors that process DNA. Restriction Endonucleases. 2004; Berlin, Heidelberg: Springer. [Google Scholar]
- 62. Mohr G., Del Campo M., Turner K.G., Gilman B., Wolf R.Z., Lambowitz A.M.. High-throughput genetic identification of functionally important regions of the yeast DEAD-box protein Mss116p. J. Mol. Biol. 2011; 413:952–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Civril F., Bennett M., Moldt M., Deimling T., Witte G., Schiesser S., Carell T., Hopfner K.P.. The RIG-I ATPase domain structure reveals insights into ATP-dependent antiviral signalling. Embo Rep. 2011; 12:1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chang T.H., Latus L.J., Liu Z., Abbott J.M.. Genetic interactions of conserved regions in the DEAD-box protein Prp28p. Nucleic Acids Res. 1997; 25:5033–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lemaire P.A., Tessmer I., Craig R., Erie D.A., Cole J.L.. Unactivated PKR exists in an open conformation capable of binding nucleotides. Biochemistry-Us. 2006; 45:9074–9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ding H., Guo M., Vidhyasagar V., Talwar T., Wu Y.. The Q motif Is involved in DNA binding but Not ATP binding in ChlR1 helicase. PLoS One. 2015; 10:e0140755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mohlmann S., Mathew R., Neumann P., Schmitt A., Luhrmann R., Ficner R.. Structural and functional analysis of the human spliceosomal DEAD-box helicase Prp28. Acta Crystallogr. D Biol. Crystallogr. 2014; 70:1622–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Talavera M.A., De La Cruz E.M.. Equilibrium and kinetic analysis of nucleotide binding to the DEAD-box RNA helicase DbpA. Biochemistry-Us. 2005; 44:959–970. [DOI] [PubMed] [Google Scholar]
- 69. Toseland C.P., Powell B., Webb M.R.. ATPase cycle and DNA unwinding kinetics of RecG helicase. PLoS One. 2012; 7:e38270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Frick D.N., Banik S., Rypma R.S.. Role of divalent metal cations in ATP hydrolysis catalyzed by the hepatitis C virus NS3 helicase: magnesium provides a bridge for ATP to fuel unwinding. J Mol. Biol. 2007; 365:1017–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kellner J.N., Reinstein J., Meinhart A.. Synergistic effects of ATP and RNA binding to human DEAD-box protein DDX1. Nucleic Acids Res. 2015; 43:2813–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lopez de Victoria A., Moore A.F.T., Gittis A.G., Koculi E.. Kinetics and thermodynamics of DbpA protein's C-terminal domain interaction with RNA. ACS Omega. 2017; 2:8033–8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sisakova E., Stanley L.K., Weiserova M., Szczelkun M.D.. A RecB-family nuclease motif in the Type I restriction endonuclease EcoR124I. Nucleic Acids Res. 2008; 36:3939–3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Richards J.D., Johnson K.A., Liu H., McRobbie A.M., McMahon S., Oke M., Carter L., Naismith J.H., White M.F.. Structure of the DNA repair helicase hel308 reveals DNA binding and autoinhibitory domains. J. Biol. Chem. 2008; 283:5118–5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cao W., Coman M.M., Ding S., Henn A., Middleton E.R., Bradley M.J., Rhoades E., Hackney D.D., Pyle A.M., De La Cruz E.M.. Mechanism of Mss116 ATPase reveals functional diversity of DEAD-Box proteins. J. Mol. Biol. 2011; 409:399–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fischer H., Neto M.D., Napolitano H.B., Polikarpov I., Craievich A.F.. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J. Appl. Crystallogr. 2010; 43:101–109. [Google Scholar]
- 77. Smith R.M., Diffin F.M., Savery N.J., Josephsen J., Szczelkun M.D.. DNA cleavage and methylation specificity of the single polypeptide restriction-modification enzyme LlaGI. Nucleic Acids Res. 2009; 37:7206–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chand M.K., Nirwan N., Diffin F.M., van Aelst K., Kulkarni M., Pernstich C., Szczelkun M.D., Saikrishnan K.. Translocation-coupled DNA cleavage by the Type ISP restriction-modification enzymes. Nat. Chem. Biol. 2015; 11:870–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tamulaitiene G., Silanskas A., Grazulis S., Zaremba M., Siksnys V.. Crystal structure of the R-protein of the multisubunit ATP-dependent restriction endonuclease NgoAVII. Nucleic Acids Res. 2014; 42:14022–14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kulkarni M., Nirwan N., van Aelst K., Szczelkun M.D., Saikrishnan K.. Structural insights into DNA sequence recognition by Type ISP restriction-modification enzymes. Nucleic Acids Res. 2016; 44:4396–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gupta Y.K., Chan S.H., Xu S.Y., Aggarwal A.K.. Structural basis of asymmetric DNA methylation and ATP-triggered long-range diffusion by EcoP15I. Nat. Commun. 2015; 6:7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lapkouski M., Panjikar S., Janscak P., Smatanova I.K., Carey J., Ettrich R., Csefalvay E.. Structure of the motor subunit of type I restriction-modification complex EcoR124I. Nat. Struct. Mol. Biol. 2009; 16:94–95. [DOI] [PubMed] [Google Scholar]
- 83. Seidel R., Bloom J.G., Dekker C., Szczelkun M.D.. Motor step size and ATP coupling efficiency of the dsDNA translocase EcoR124I. Embo J. 2008; 27:1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors of the H.CglI C-terminal domain crystal structure has been deposited with the Protein Data Bank under accession number PDB ID 6F1S.