


Abstract
Pentatricopeptide repeat (PPR) proteins are helical-repeat proteins that offer a promising scaffold for the engineering of proteins to bind specified RNAs. PPR tracts bind RNA in a modular 1-repeat, 1-nucleotide fashion. An amino acid code specifying the bound nucleotide has been elucidated. However, this code does not fully explain the sequence specificity of native PPR proteins. Furthermore, it does not address nuances such as the contribution toward binding affinity of various repeat-nucleotide pairs or the impact of mismatches between a repeat and aligning nucleotide. We used an in vitro bind-n-seq approach to describe the population of sequences bound by four artificial PPR proteins built from consensus scaffolds. The specificity of these proteins can be accounted for by canonical code-based nucleotide recognition. The results show, however, that interactions near the 3′-end of binding sites make less contribution to binding affinity than do those near the 5′-end, that proteins with 11 and 14 repeats exhibit similar affinity for their intended targets but 14-repeats are more permissive for mismatches, and that purine-binding repeats are less tolerant of transversion mismatches than are pyrimidine-binding motifs. These findings have implications for mechanisms that establish PPR–RNA interactions and for optimizing PPR design to minimize off-target interactions.
INTRODUCTION
Evolution has produced a diversity of protein architectures that are capable of binding RNA in a sequence-specific fashion (reviewed in 1). Among these, the PUF and pentatricopeptide repeat (PPR) protein families offer particularly promising scaffolds for the engineering of proteins that bind specified RNA sequences in a single-stranded context. PUF and PPR proteins belong to the alpha solenoid superfamily, and are characterized by regularly spaced helical repeating units that contact the Watson–Crick face of consecutive RNA nucleotides (reviewed in 1–3). Each repeat in PUF and PPR proteins discriminates among nucleotides based on the identities of amino acids at several positions, and these comprise amino acid ‘codes’ for nucleotide recognition (4,5). TALE proteins have a similar architecture and an analogous amino acid code, but bind to specific DNA sequences in a double-stranded context (6,7). These features have been exploited to engineer proteins that bind specific RNA or DNA sequences, but the optimization of binding affinity and specificity to minimize off target action is an ongoing effort. For example, different repeats in PUF, PPR and TALE proteins can vary in their degree of nucleotide specificity and their contribution to binding affinity (8–11). The ability to predict how binding affinity and specificity are distributed along a repeat tract and how repeat tract length impacts this distribution is crucial for harnessing the full potential of these tools.
PPR proteins function primarily in the expression of mitochondrial or chloroplast genes. They mediate various aspects of organelle gene expression that emerged post endosymbiosis, such as RNA stabilization, group II intron splicing, RNA editing and translational activation (3). The PPR family is notable for its evolutionary plasticity. For example, the size of the PPR family varies dramatically among organisms, with fewer than ten members in metazoa and more than 400 members in plants (12,13). Furthermore, PPR proteins can have as few as two repeats or as many as 30, and the length of their repeating units can vary: canonical ‘P’ motifs have 35 amino acids (14), but a subfamily in plants, the PLS PPRs, consist of alternating P, long (L) and short (S) motifs (12,14,15). PPR motifs exhibit considerable sequence variation, but consensus amino acids can be identified at many positions (14,15). Each PPR motif adopts a helix-turn-helix fold, and consecutive repeats stack to form a right handed super helix (16,17). PPR tracts bind RNA in a parallel orientation, with the N-terminal repeats binding to the 5′ end of the binding site (5,17). The nucleotide bound by many PPR motifs is specified by the identities of amino acids at two positions, which form a combinatorial nucleotide recognition code (5,18–20). This ‘PPR code’ has been used to reprogram native PPR proteins (5,10,21,22) and to engineer artificial PPR proteins with customized sequence specificity (20,23,24).
A challenge in the design of customizable nucleic acid binding proteins is the optimization of binding affinity and specificity to maximize occupancy of the intended target while minimizing off-target binding. Although the PPR code largely accounts for sequence preferences exhibited when recombinant proteins have been queried with a limited number of RNA ligands, a comprehensive analysis of the sequence specificity of the native protein PPR10 revealed a more complex picture: four of the 17 nucleotides in PPR10’s minimal binding site are specified in a manner that is not explained by the PPR code (10). Are these idiosyncratic features of PPR10, or do artificial PPR tracts built from consensus PPR motifs also have the capacity for alternative RNA binding modes? Furthermore, the PPR motifs in PPR10 vary widely in their contribution toward binding affinity (10). Does this result from sequence differences among PPR10’s PPR motifs, or is this variation intrinsic to particular PPR code-nucleotide combinations or to the context of the repeat in the protein? To address these and related questions, we used a bind-n-seq approach to comprehensively probe the RNA sequence specificity of four artificial PPR proteins built from consensus PPR motifs, comparing two different repeat tract lengths, two types of PPR consensus design, and two arrangements of the same set of PPR motifs. The results show that canonical code-based nucleotide recognition accounts for the repertoire of sequences bound by these proteins, and provide no evidence for alternative binding modes such as that exhibited by PPR10. However, the contribution of PPR-nucleotide pairings to binding affinity differs between purine and pyrimidine-binding motifs, and is strongly influenced by the position along the PPR–RNA interface: repeats near the C-terminus exhibited little sequence specificity and mismatches near the center of the binding site had very different effects in the context of proteins with 11 or 14 repeats. Notably, extending PPR tract length from eleven to fourteen motifs had little effect on binding affinity but increased tolerance to mismatches. These patterns provide a basis for the rational design of synthetic PPR proteins with minimized off target effects.
MATERIALS AND METHODS
Protein design, expression and purification
The SCD11A, SCD11B and SCD14 proteins were designed according to the scheme described in ref (24). In brief, N- and C-terminal segments of PPR10 (amino acids 37–208 and 737–786, respectively) flank consensus PPR motifs designed to bind the sequences shown in Figure 1A. MCD14 is identical to SCD14 except that the consensus PPR motifs were tailored to the targeted nucleotide based on correlations observed in several native PPR–RNA pairs that had strong experimental support. The G-binding consensus was created from PPR10 repeat 3, CRP1 repeat 9, MRL1 repeats 6 and 8, PGR3 repeats 6 and 7, PPR53 repeats 2, 3 and 6, and RPF3 repeats 5, 8, 12 and 5. The A-binding consensus was created from PPR10 repeat 5, CRP1 repeat 8, MRL1 repeats 7 and 10, PGR3 repeat 5, RPF1 repeat 10, RPF2 repeats 2, 3 and 4, RPF3 repeats 6 and 9, and RPF5 repeats 2 and 8. The C-binding consensus was created from PPR10 repeats 7 and 8, PGR3 repeat 4, MRL1 repeat 9, RPF1 repeat 10, RPF2 repeats 10, 11, 12 and 14, RPF3 repeat 10, and RPF5 repeat 4. The U-binding consensus was created from CRP1 repeat 7, PPR10 repeat 6, RPF1 repeats 3, 4, 5, 7 and 9, RPF2 repeats 5 and 13, RPF3 repeats 1, 2, 4, 7 and 11, RPF5 repeats 3, 7, 9 and 12, PPR10 repeat 9, and PPR53 repeats 1 and 5. Positions 23 and 26 were changed to serines as described by Gully et al. (25).
Figure 1.

Design of bind-n-seq experiments. (A) Design of single-consensus design (SCD) proteins. These proteins used the consensus PPR scaffold and PPR10-derived capping helices described by (24). The sequences of the consensus motifs for binding each nucleotide are shown at top, with amino acids numbered according to the scheme in (5). The first PPR motif is derived from PPR10 and binds G in PPR10’s native RNA ligands (10,30). The remaining PPR motifs have the consensus sequences shown above. PPR motifs 1 through 11 are identical in SCD11A and SCD14. The sequences to which these proteins were designed to bind are shown below each protein. (B) Design of MCD14. MCD14 is identical to SCD14 except that the consensus PPR motifs were tailored to the targeted nucleotide based on correlations observed in native PPR–RNA pairs with strong experimental support. Amino acids that differ from the SCD consensus are underlined. (C) Affinity of synthetic PPR proteins for their intended RNA sequence based on gel mobility shift assays. Gel images are shown in Supplementary Figure S1B. Binding curves generated with different preparations of SCD11A and SCD14 in 4-h binding reactions confirmed that they bind their intended RNA ligand with similar affinity (Supplementary Figure S1C). (D) Oligoribonucleotides used for the bind-n-seq assays. The target site is the expected binding site of MCD14 and SCD14, which were the only proteins assayed with partially randomized oligoribonucleotides. Positions with randomized nucleotides are indicated with N.
The proteins were expressed as fusions to maltose-binding protein (MBP) using the pMAL-TEV vector and E. coli Rosetta 2 cells (Novagen), purified using amylose affinity chromatography, cleaved with TEV protease to remove MBP, and further purified by size exclusion chromatography, as described for PPR10 (26). The purified proteins were dialyzed into 50 mM Tris–HCl, pH 7.5, 200 mM NaCl, 50% glycerol and 5 mM β-mercaptoethanol and stored at –20°C.
Bind-n-seq assays
Bind-n-seq assays were performed as described previously (10) with minor modifications. Synthetic 16-mer oligoribonucleotides (IDT) were designed as shown in Figure 1D, with a 5′-phosphate and using hand-mixed nucleotide pools at the randomized positions. To perform binding reactions, 52 μl of a 2.5× RNA pool (11.25 μM suspended in 10 mM Tris–HCl pH 7.5, 1 mM EDTA) was denatured at 95°C for 3 min and then snap cooled on ice. This was combined with an equal volume of 2.5× BNS buffer (100 mM Tris–HCl pH 7.5, 250 mM NaCl, 10 mM DTT, 1 U/ μl RNAsin [Promega], 0.25 mg/ml BSA, 1.25 mg/ml heparin) and 26 μl of protein at a concentration equivalent to 5-fold the desired final concentration. The final binding reactions contained 4.5 μM RNA, either 50, 100 or 200 nM PPR protein as indicated, 50 mM Tris–HCl pH 7.5, 140 mM NaCl, 10% glycerol, 4 mM DTT, 0.4 U/μl RNAsin, 0.1 mg/ml BSA, 0.5 mg/ml heparin. The binding reactions were incubated at 25°C for 4 h; this incubation time was chosen based on pilot binding reactions involving the specific RNA ligand in trace amounts, which we found took several hours to reach equilibrium. The reactions were resolved in a 5% polyacrylamide gel in 1× THE (34 mM Tris base, 66 mM HEPES, 0.1 mM EDTA) at 4°C for 30 min at 15 W. To mark the position of protein–RNA complexes in the gel, a separate binding reaction involving radiolabeled RNA pool (400 000 cpm per reaction) and 1 μM protein was electrophoresed in an adjacent lane. The gel was exposed briefly to a phosphor screen to identify the position of RNA–protein complexes, and the corresponding region from the non-radioactive reactions was excised, eluted in 400 μl TESS (10 mM Tris–HCl pH 8, 1 mM EDTA, 100 mM NaCl, 0.1% SDS) at 4°C overnight, and purified by phenol-chloroform extraction followed by ethanol precipitation. Sequencing libraries were generated using the NEXTflex Small RNA-Seq Kit v3 (BIOO Scientific).
Computational analysis of RNA bind-n-seq data
Sequence data from experiments involving partially randomized oligonucleotide pools were analyzed essentially as described previously for PPR10 bind-n-seq experiments (10). In brief, the enrichment of each k-mer at each nucleotide position in the bound fractions was calculated as the frequency of the k-mer at that position in the bound fraction divided by its frequency at the same position in the input RNA library. K-mers of 7, 8 or 9 nt were analyzed; results are presented only for 7-mers because these were most effective at revealing enriched motifs. Sequences harboring 7-mers enriched above the designated threshold (> 5 standard deviations above the mean) were used to generate sequence logos using weblogo 3.4 (27), after weighting the sequences harboring those 7-mers according to their enrichment value. Sequences that were not detected in the input library were assigned an enrichment value of 1; however, removal of these sequences from the set used to generate the logos did not change the appearance of the logos.
In a separate analysis of the data from the partially randomized pools, specificity scores were calculated from sequences harboring highly enriched 7-mers (>5 SD above the mean) using the method described in reference (28). If the frequency of a nucleotide at a specific position in the bound fraction was greater than that in the input fraction, the specificity score was calculated as [nucleotide frequency(bound)-nucleotide frequency(input)]/[1-nucleotide frequency (input)]. If the bound nucleotide frequency was less than the input nucleotide frequency, the specificity score was calculated as [nucleotide frequency(bound)-nucleotide frequency(input)]/[nucleotide frequency(input)].
Data from assays that employed fully randomized oligonucleotides were analyzed in two ways. First, position-independent k-mer enrichment was calculated as the frequency of the k-mer at all positions in the bound fraction divided by its frequency in the input RNA library. K-mers of 7, 8, or 9 nt were analyzed; results are presented only for 9-mers because these were most effective at revealing enriched motifs. 9-mers enriched above the designated threshold (> 10 standard deviations above the mean) were weighted according to their enrichment value and aligned using MUSCLE with a high gap open penalty so as to prevent gaps. The weighted MUSCLE-aligned 9-mers were used to generate sequence logos using weblogo 3.4. In a separate analysis, the enrichment of sequences matching a degenerate target site was compared to that of sequences with single transversions at each position. Because pyrimidine-binding motifs discriminate poorly between U and C (5,10), the degenerate ‘wild-type’ site allowed either U or C at each position harboring a pyrimidine. In addition, any nucleotide was permitted at the first position of the target site (which aligns with the ‘G’ binding repeat from PPR10). The degenerate sites were queried in all registers capable of accommodating the full sequence. For example, the 14-nt binding site can be found in three different registers in the 16-nt randomized RNA, and the occurrence of the query sequence in all three registers was summed. Positions flanking the degenerate sites were allowed to be any nucleotide. Enrichment values were calculated as the frequency of sequences that matched the degenerate target site in the bound fraction divided by its frequency in the input fraction.
Gel mobility shift assays
Gel mobility shift assays were performed as previously described (29), with minor modifications. In brief, synthetic RNA oligonucleotides (IDT) were 5′-end labeled with T4 polynucleotide kinase and [γ-32P]-ATP. The binding reactions (25 μl) contained 15 pM RNA, 40 mM Tris–HCl pH 7.5, 140 mM NaCl, 10% glycerol, 4 mM DTT, 10 U RNAsin, 0.1 mg/ml BSA, 0.5 mg/ml heparin and protein at the indicated concentrations. Binding reactions were incubated for 30 min (Figure 1C) or 4 hours (all other assays) at 25°C and resolved on 5% polyacrylamide gels in 1× THE at 4°C for ∼30 min at 15 W. Results were imaged with a phosphorimager and quantified with Image Studio Lite. Curves were fit to the data using Prism software.
RESULTS
Design of artificial PPR proteins
We designed four PPR proteins that vary in the sequence of the consensus PPR scaffold, the intended binding site and repeat tract length. Three proteins were modeled on the consensus PPR design of Shen and coworkers (24); these contained 10 or 13 artificial PPR motifs with the same consensus scaffold sequence, flanked by N- and C-terminal segments of PPR10 to support protein solubility (Figure 1A). The PPR10-derived sequences add one PPR motif to the N-terminus, which binds G in PPR10’s native RNA ligands (10,30). We refer to these proteins as ‘single consensus design’ proteins SCD14, SCD11A and SCD11B. The specificity-determining amino acids (positions 6 and 1’, according to the nomenclature in (5)) of SCD14 and SCD11A were chosen to bind the sequence GUCUAGAUCUAGUU or the first 11 nucleotides of this sequence, respectively. SCD11B was designed to bind the same nucleotides as SCD11A but in shuffled order (Figure 1A). A fourth protein, MCD14, was designed to bind the same sequence as SCD14 but used a ‘multi-consensus design’ (MCD) involving a different consensus PPR scaffold for each targeted nucleotide (see Figure 1B). The MCD and SCD motifs used the same 6/1’ amino acid combinations to specify U, G and C. However, A was specified by S6N1’ and T6N1’ in the SCD and MCD designs, respectively. The four proteins were expressed in Escherichia coli as fusions to MBP, purified by amylose affinity chromatography, cleaved from the MBP and further purified by gel filtration chromatography (Supplementary Figure S1A). All four proteins bound in vitro to the RNA for which they were designed with a KD of approximately 20 nM and did not bind detectably to an RNA of similar length but with a different sequence (Figure 1C and Supplementary Figure S1B). Interestingly, the proteins with 14 and 11 repeats bound RNA with similar affinity despite the potential for the longer proteins to make more contacts with RNA.
RNA-binding specificity landscapes of artificial PPR proteins are accounted for by the PPR Code but show high tolerance for mismatches near 3′-ends of binding sites
We used a ‘bind-n-seq’ approach similar to that used previously with PPR10 (10) to comprehensively analyze the sequence specificity of the four artificial PPR proteins. Bind-n-seq assays use deep sequencing to analyze the population of sequences bound by a protein from a large pool of randomized oligonucleotide sequences. Each protein was incubated with a pool of synthetic RNA oligonucleotides whose sequences were either fully or partially randomized with respect to the predicted binding site (see Figure 1D). The assays used proteins at three concentrations, with the RNA in substantial molar excess in each case (see Materials and Methods). The bound and unbound RNAs were separated by native gel electrophoresis, and RNAs in the bound and input pools were analyzed by deep sequencing. The nucleotide frequencies of the input libraries demonstrated minimal bias at randomized positions, and ∼95% of the sequences expected in the partially randomized input pools were detected in the aliquot that was sequenced (Supplementary Figure S2).
In one set of experiments, the three partially randomized oligonucleotides were combined in equimolar amounts and used for binding reactions with either SCD14 or MCD14. Enrichment values were calculated for all possible 7-mers as the frequency of the 7-mer at a specific nucleotide position in the bound fraction divided by its frequency in the input library. The frequency distributions of enrichment values are shown in Figure 2A (SCD14) and Supplementary Figure S3A (MCD14). Highly enriched 7-mers were defined as those that were enriched more than 5 standard deviations above the mean. 7-mers drawn from the 3′-randomized RNA pool dominate the highly-enriched population for both proteins (Figure 2A and Supplementary Figure S3A). The greater diversity of 3′ sequence motifs in the enriched fractions imply a greater tolerance for mismatches between the protein and RNA at the 3′-end than at the 5′-end of the binding site. The native protein PPR10 showed a similar pattern in this regard (10).
Figure 2.
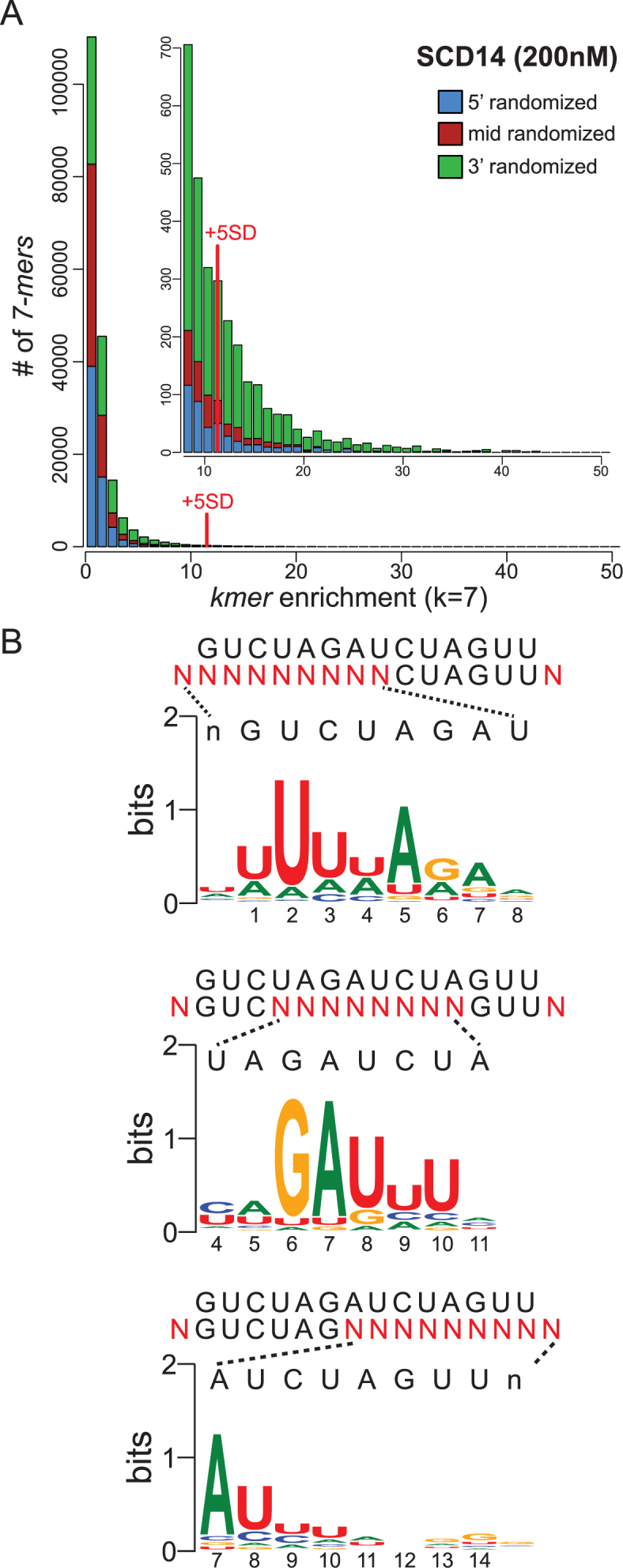
Sequences enriched by SCD14 from pooled 5′-, middle- and 3′-randomized RNAs. (A) Frequency distribution of enrichment values of all 7-mers for the bind-n-seq assay using SCD14 (200 nM) and equimolar amounts of the three partially randomized RNA pools. The graph shows the number of different 7-mers (Y-axis) at each enrichment value (X-axis). The inset shows an expansion of the data from the tail of the distribution. 7-mers that were enriched more than 5 standard deviations above the mean were defined as the highly enriched fraction for subsequent analyses. The analogous plot for MCD14 is shown in Supplementary Figure S3A. (B) Sequence logos derived from sequences harboring highly enriched 7-mers. The randomized nucleotides in each oligonucleotide pool (N) are displayed beneath the intended binding site. Sequences harboring highly enriched 7-mers (>5 SD above the mean) were used to generate each logo, after weighting the input sequences according to their enrichment values. The logo from the 5′-randomized RNA was calculated from 3162 sequences harboring 173 7-mers. The logo from the middle-randomized RNA was drawn from 2992 sequences containing 124 7-mers. The logo from the 3′-randomized RNA was drawn from 17368 sequences harboring 1022 7-mers. Analogous logos for MCD14 are shown in Supplementary Figure S3B.
Sequence logos (27) generated from sequences harboring highly-enriched 7-mers are shown in Figure 2B (SCD14) and Supplementary Figure S3B (MCD14). The logos obtained for the two proteins are similar, indicating that the differences in the sequences of their PPR scaffolds have little effect on motif specificity. The logos confirmed that these proteins have much less nucleotide selectivity near the 3′-end of the binding site than in the middle and 5′-regions. The logos derived from the 5′- and middle-randomized RNAs resemble the predicted binding site, with two exceptions. First, repeats with the N6S1’ code, which were expected to bind both U and C with slight preference for C (5,24), instead preferentially enriched U. By contrast, bind-n-seq assays employing fully randomized oligonucleotides yielded the expected enrichment of C by N6S1’ motifs (see below), and single C-to-U substitutions in the intended RNA target had no apparent effect on binding affinity in gel mobility shift assays (Supplementary Figure S4). It is unclear why the assays involving partially-randomized RNAs preferentially enriched RNAs with U at these positions.
The second deviation from the expected nucleotide specificity involved the first PPR motif, which was expected to bind G but instead selected U. This is a special case, however, in that this motif is derived from PPR10 and it has the unusual amino acid code T6T1’ (see Figure 1A and B). This repeat aligns with G in PPR10’s native ligands and it selected G in bind-n-seq assays with PPR10 (10,30). Its selection of U in these experiments could result either from the context of this motif in the artificial proteins or from the context of the aligning nucleotide in the RNA ligand. Because PPR tracts bind nucleotides via their Watson–Crick face (20), nucleotide substitutions that decrease RNA secondary structure can increase apparent binding affinity and thereby masquerade as sequence specificity (10,21). However, a G→U substitution at position 1 has no impact on the predicted stability of RNA structure formed by the targeted RNA ligand (Supplementary Table S1). These results suggest that the nucleotide specificity of this PPR10-derived repeat is influenced by its protein context.
To reveal nucleotide specificities that may have been masked when the three partially randomized RNA pools were in competition, a second set of bind-n-seq assays used either the 5′-, middle, or 3′- randomized pool individually. The results are displayed as both position-based specificity scores (28) and as sequence logos in Supplementary Figure S5. The results for SCD14 and MCD14 were nearly identical, although the motifs tailored to bind purines in MCD14 appear to be somewhat more selective than the generic motifs in SCD14. Overall, the results were similar to those from the experiments that pooled the three partially randomized RNAs. The experiment involving the 3′-randomized RNA pool revealed, in addition, that nucleotides at positions 11 and 12 are selected as predicted by the PPR code, albeit weakly. These data also suggested weak selection for G at positions 13 and 14, rather than the predicted U. However, this may be an artifact of the very low nucleotide selectivity at these positions (see Figure 2B), as substitution of G for U at position 14 slightly reduced binding affinity in gel mobility shift assays (Supplementary Figure S4A) and nucleotides 13 and 14 can be deleted entirely with no loss of binding affinity (see below).
Taken together, these results show that the PPR code largely accounts for the sequence specificity of both SCD14 and MCD14. Therefore, the non-canonical sequence-specific RNA interactions displayed by PPR10 (10) are not an intrinsic property of PPR tracts. Still, the nucleotide selectivity of identical PPR motifs at different locations varied considerably in several instances. For example, repeat 2 is more highly selective for U than are the identical motifs at positions 4, 8, 10, 13 and 14 (see selectivity scores in Supplementary Figure S5). We considered the possibility that effects of alternative nucleotides on RNA secondary structure contribute to these apparent differences in repeat selectivity. In accord with this view, U to purine (R) substitutions at position 2 substantially increase the propensity for RNA structure whereas Y to R substitutions at positions 4, 8, 10 or 14 do not (Supplementary Table S1). On the other hand, the low selectivity of motif 13 cannot be explained in this way, as the ‘expected’ U at this position results in the least structure of any of the four nucleotides. Thus, these results suggest that identical PPR motifs at different positions can exhibit different degrees of selectivity but the scope and magnitude of these differences are difficult to assess from this analysis due to the influence of nucleotide identities on RNA structure. Experiments described below clarify this issue.
The length of the PPR tract, the position of a PPR motif in the tract and the identity of its specificity determining amino acids impact tolerance for mismatches along a PPR–RNA interface
A third set of experiments used fully randomized RNA oligonucleotides together with each of the four artificial proteins: the 14-repeat proteins SCD14 or MCD14 and the 11-repeat proteins SCD11A or SCD11B (see Figure 1A). Highly-enriched 9-mers were identified independent of their position in the RNA, weighted according to their enrichment value, and aligned with MUSCLE. This multiple sequence alignment was then used to generate sequence logos (Figure 3). The results show selection for motifs that closely resemble the intended target sites for SCD11A, SCD11B and SCD14. These data revealed the expected preference for C-over-U by N6S1’ motifs (5,24), unlike the experiments involving partially randomized oligonucleotides (see Figure 2B).
Figure 3.
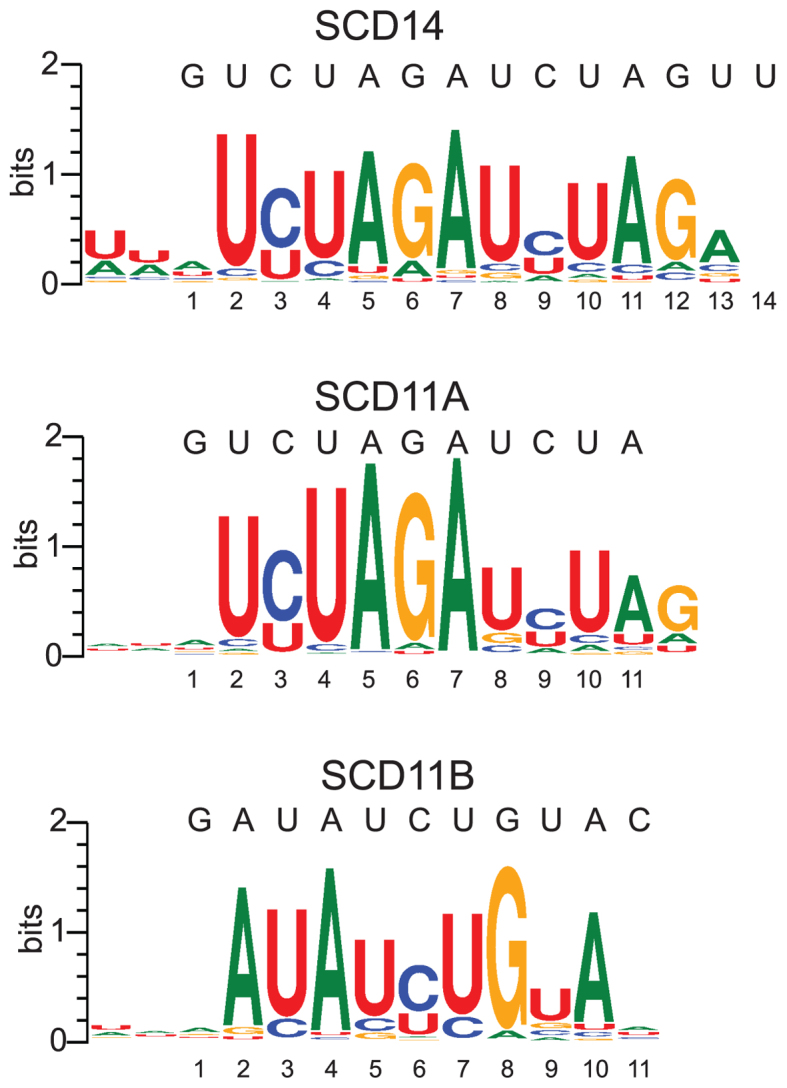
Logos illustrating sequences enriched by SCD11A, SCD11B and SCD14 from fully-randomized RNAs. Enrichment values of all 9-mers in the bound fraction were calculated in a position independent manner. Highly enriched 9-mers (>10 SD above the mean) were used to generate logos after weighting each 9-mer according to its enrichment value and aligning with MUSCLE. The SCD11A, SCD11B and SCD14 logos were generated from 466, 488 and 302 9-mers, respectively.
To assess the contribution of each PPR motif-nucleotide pair to binding affinity, we analyzed the degree to which mismatches at each position decreased sequence enrichment in the binding reactions. To that end, we first calculated the enrichment of sequences matching a degenerate version of each expected binding site (see query sequences in Figure 4 and Supplementary Figure S3C): the degenerate sites allowed either pyrimidine (Y) at positions with C or U, allowed all possible registers of the intact site within the 16-nucleotide RNA, and allowed any nucleotide (N) at positions flanking the site and at the first position in the site, which aligns with the PPR10-derived motif. We then compared the enrichment of these ‘wild-type’ sequences to the enrichment of sequences with a single transversion at each position (Y→R, A→Y, G→Y). The data are plotted in Figure 4 and Supplementary Figure S3C as the ratio of enrichment of sequences matching the mutant degenerate site to that of sequences matching the wild-type degenerate site. This ratio is less than one for transversions at most positions, indicating that the identities of most nucleotides have an impact on binding affinity. However, the 14-repeat and 11-repeat proteins exhibited striking differences in the magnitude and position-dependence of mismatch effects. In binding reactions with SCD14 (Figure 4A) and MCD14 (Supplementary Figure S3C), the maximal effects of mismatches were relatively small, transversions near the 5′ end caused the largest decrease in binding, and transversions at the 3′ positions (13 and 14) had little impact. By contrast, SCD11A, which is identical to SCD14 except that it lacks the three C-terminal PPR motifs, was less tolerant of transversions at most positions and was highly sensitive to transversions at positions 5, 6 or 7 (Figure 4B). Given that SCD11A and SCD14 bind the same target RNA with similar affinity (Figure 1C), these results imply that the 11-repeat protein is more selective for its intended target than is the 14-repeat protein.
Figure 4.

Effects of repeat tract length, repeat position and motif type on tolerance for mismatches. The data come from bind-n-seq experiments performed with fully randomized 16-mer RNA and either SCD14 (A), SCD11A (B) or SCD11B (C). The results for MCD14 are shown in Supplementary Figure S3C. The intended target sequence of each protein and the degenerate version of that sequence used as the query for this analysis are shown below each graph. The degenerate sites substituted Y for either U or C, and substituted N for the 5′ G, which is bound by an atypical repeat from PPR10. The enrichment of sequences matching the degenerate version of each binding site was compared to that of sequences with a single transversion (Y→R, A→Y and G→Y) at the position indicated on the X axis. Read counts for all registers harboring the full site were combined, with any nucleotide allowed at positions flanking the site. The ratio of enrichment of each mutant sequence with respect to that of the wild-type sequence is plotted on the Y-axis. The wild-type consensus sequences were enriched ∼27-fold for SCD14 and ∼40-fold for SCD11A, and SCD11B when the proteins were at 200 nM.
The binding of SCD11A was particularly sensitive to mismatches at the center of its binding site, in a segment harboring three consecutive purines (Figure 4B). The low tolerance for mismatches in this region might be due to its central position, the fact that it involves PPR-purine interactions, or both. Gel mobility shift assays showed that an A-to-U transversion in the center of the SCD11A binding site was more disruptive to binding affinity than was the same transversion at the 3′-end (Supplementary Figure S4B), supporting the view that a central position sensitizes a repeat to mismatches. To thoroughly distinguish between these possibilities, we compared the results from SCD11A to those from SCD11B, whose repeats are identical to those in SCD11A but in a different order (Figure 4C). SCD11B, like SCD11A, was less tolerant of mismatches than were SCD14 and MCD14, supporting the view that the shorter PPR tract is less prone to off-target binding. However, the position-dependence of the mismatch effects differed for SCD11A and SCD11B. Transversion mismatches with purine-binding repeats at positions 4, 8 and 10 had more severe effects than did transversion mismatches with their adjacent pyrimidine-binding repeats. That said, mismatches toward the center of the binding site generally had a more severe impact than did the same type of mismatch toward the periphery. Transversion mismatches with the single consensus G-binding motif in both SCD11A and SCD11B had particularly severe effects on sequence enrichment, suggesting that the interaction between G and its cognate PPR motif contributes more to binding affinity than do other PPR-nucleotide pairings.
Taken together, these results show that the length of the repeat tract, the position of a motif within the tract and the identity of the matched nucleotide can have a major impact on the degree to which PPR-nucleotide mismatches decrease binding affinity. Fourteen-repeat proteins show considerable tolerance for mismatches along their length, albeit with greatest tolerance near the C-terminus. Eleven-repeat proteins are much more sensitive to mismatches, especially near the center of the binding site. Furthermore, purine-binding repeats are less tolerant of transversion mismatches than are pyrimidine-binding motifs, with position within the repeat tract modulating this effect.
High tolerance for binding site truncations from the 3′-end
Results above indicated that the binding of synthetic PPR proteins is highly permissive to mismatches near the 3′ end of the binding site. To address whether the complete absence of nucleotides aligning with C-terminal repeats is similarly tolerated, we took advantage of the fact that the degenerate binding sites will be represented in various registers within the pool of fully randomized 16-nucleotide RNA, including registers that truncate the sites at either the 5′- or 3′-end (see Figure 5A). We used a strategy analogous to the mismatch analysis above: the enrichment of sequences harboring each intact degenerate binding site was compared to that of sequences harboring truncated versions of the degenerate binding site. The data are presented as the ratio of enrichment of sequences harboring each truncated degenerate binding site with respect to sequences harboring the intact degenerate site (Figure 5A). The results suggest that truncation of up to two nucleotides at the 3′-end does not decrease, and might even increase sequence enrichment by SCD14. Sequence enrichment by SCD11A and SCD11B was more sensitive to 3′ truncations, but loss of two nucleotides at the 3′ end of their binding sites still had little impact (Figure 5A). By contrast, truncation of even one nucleotide from the 5′ end had a clear effect, and all 5′ truncations were much more deleterious than were 3′ truncations of the same length (Figure 5A). Gel mobility shift assays confirmed that truncating the SCD14 binding site by two nucleotides at the 3′-end increased binding affinity, whereas truncating two nucleotides at the 5′ end decreased affinity (Figure 5B). Similar results were obtained with MCD14 (Supplementary Figure S6B). These observations suggest that neither sequence-specific nor non-specific contacts at the 3′-end of a 14-nucleotide binding site have a substantive effect on binding affinity, assuming a fully matched sequence upstream.
Figure 5.

Effects of binding site truncations on affinity for SCD11A, SCD11B and SCD14. (A) Enrichment of RNAs harboring truncated binding sites in bind-n-seq assays. Degenerate binding sites were defined as in Figure 4, and are diagrammed below in various registers within the randomized 16-mer used in the bind-n-seq assays. The graph shows the ratio of enrichment of sequences harboring the indicated truncated site, with respect to the enrichment of sequences harboring the intact (degenerate) site. The results were similar at two other protein concentrations (Supplementary Figure S6A). (B) Gel mobility shift assays validating the differential effects of 5′ and 3′ truncations on affinity for SCD14. SCD14 was assayed at 0, 0.5, 2, 8, 32 and 128 nM. Each assay was performed twice and data for both replicates (rep1 and rep2) are shown. Analogous experiments for MCD14 are shown in Supplementary Figure S6B.
DISCUSSION
Assessing the ability of designer nucleic acid binding proteins to discriminate among the sequences they might encounter in vivo presents an ongoing challenge. Some native P-type PPR proteins bind just one or two organellar RNAs in vivo (reviewed in 3), but the ability of PPR proteins to selectively bind intended targets in the much larger sequence space in the nuclear/cytosolic compartment has not been addressed. Furthermore, an in-depth analysis of the sequence specificity of the native protein PPR10 demonstrated that both code-based nucleotide selection and non-canonical nucleotide recognition mechanisms make important contributions to binding affinity (10). In this study, we sought to determine whether artificial PPR tracts built from a consensus scaffold likewise exhibit alternative binding modes, and to advance understanding of the nuances of code-based nucleotide recognition. We found that modular, code-based nucleotide recognition can account for the sequence specificities of the artificial PPR proteins we analyzed. Therefore, the non-canonical nucleotide recognition exhibited by PPR10 (10,17) does not result from features that are intrinsic to PPR tracts, and artificial PPR proteins can be designed without concern for the possibility of alternative binding modes. Our results also provide evidence that the contribution of individual nucleotide-PPR motif pairings toward binding affinity varies according to the length of the PPR–RNA interface, position along that interface and whether the interaction involves a purine or pyrimidine. The data suggest further that extending a consensus PPR tract beyond approximately 11 repeats does not increase affinity for the intended target site, but does increase the likelihood of off-target interactions.
Effects of repeat tract length on the propensity for off-target binding
Our results show that the length of the repeat tract has a strong impact on the balance of on-target to off-target binding. SCD14 and SCD11A bound with similar affinity to an identical RNA harboring their predicted binding sites, despite SCD14’s potential for three additional modular contacts (Figure 1C). However, these proteins responded very differently to RNAs with mismatches. Single transversion mismatches at most positions had only a modest effect on the binding of the 14-repeat proteins, whereas single mismatches at many positions severely compromised the binding of the 11-repeat proteins (Figure 4, Supplementary Figure S3C). Furthermore, binding site truncations from each end had a more severe effect on enrichment by the 11-repeat proteins than the 14-repeat proteins (Figure 5, Supplementary Figure S6).
These results show that the 14-repeat proteins are more prone to off-target binding than the 11-repeat proteins, and suggest that there is some optimal PPR tract length that maximizes affinity for a specific RNA sequence while minimizing tolerance for mismatches. Several aspects of our data suggest that this optimal length is roughly 10 PPR motifs. First, the two 3′-most nucleotides in the binding site of SCD14/MCD14 have little effect on binding affinity but mismatches elsewhere can compromise binding (Figures 4 and 5, Supplementary Figure S3); this suggests that the first eleven artificial repeats make substantive contributions to binding affinity but the last two do not. The binding behavior of SCD11B leads to a similar conclusion: mismatch or truncation of a single nucleotide at the 3′-end of its binding site had no apparent effect on sequence enrichment, but mismatches elsewhere compromised binding (Figures 4C and 5A). Furthermore, transversion mismatches at each position in the SCD11A binding site caused at least a modest loss of sequence enrichment (Figure 4B), suggesting that reducing the number of PPR-nucleotide matches below ten brings the risk of reduced binding affinity. These results are consistent with previous reports that a minimum of six consensus PPR motifs is required for detectable RNA binding activity in vitro and that RNA affinity increases as repeat number is increased to eight (23,24). Our results suggest that increasing the number of repeats to ten or eleven further increases RNA affinity, and that additional repeats beyond eleven have little impact on RNA affinity.
Similar observations have been made in studies of DNA binding by TALE proteins and CRISPR-Cas9. For example, increasing the length of TALE repeat tracts and CRISPR guide RNAs increases tolerance to mismatches (31–34). This has been suggested to result from binding energy in excess of that needed to occupy the intended target (28,34). Furthermore, increasing the length of TALE repeat tracts increases binding affinity up to a plateau, beyond which additional repeats have little effect (32); this has been suggested to arise from structural constraints that prevent optimal spacing between TALE repeats and cognate nucleotides past some number of contiguous pairs (32). Both of these factors may contribute to the differences we observed in the RNA binding properties of artificial PPR proteins with 11 or 14 repeats. The higher tolerance of the 14-repeat proteins to mismatches is consistent with an ‘excess binding energy’ mechanism. That said, gel mobility shift assays showed that increasing PPR repeat number from 11 to 14 adds little to RNA binding affinity (Figure 1C), and that deleting two nucleotides from the 3′-end of the SCD14/MCD14 binding site did not decrease binding affinity (Figure 5B, Supplementary Figure S6B). Based on these observations, we favor a model in which a maximum of approximately 10 contiguous PPR motifs can align in a manner that optimizes contact between the PPR motif and its cognate base, and that additional PPR motifs primarily increase non-specific interactions, possibly via electrostatic interactions between the backbone phosphates and lysine 14 in the consensus scaffold (20,23) (see Figure 1A). Indeed, the PPR superhelix becomes compressed upon binding RNA (17,20) and the distance between the nucleotide binding residues in unbound PPR tracts is larger than that between consecutive nucleotides in unbound RNA (23).
Several features of native P-type PPR proteins are relevant in this context. First, a functional dissection of PGR3, which has 27 PPR motifs and plays dual roles in RNA stabilization and translational activation, showed that it's N-terminal 16 PPR motifs are sufficient for its RNA stabilization function, whereas its C-terminal 11 motifs are required for its translation activation function (9). Thus, PGR3’s long PPR tract is divided into two functional units whose lengths are consistent with the maximal number of contiguous PPR-nucleotide pairings suggested by our results. Second, native P-type PPR proteins often have irregularities toward the center of their PPR array, as well as nucleotide ‘insertions’ toward the center of their binding sites that appear not to align with PPR motifs (5,9). Such discontinuities may be required to allow simultaneous engagement of nucleotides near both ends of long PPR binding sites. Incorporating amino acids with high conformational flexibility toward the center of synthetic PPR proteins may allow for better recognition of longer binding sites, and may increase the optimal scaffold length that maximizes sequence specificity.
Polarity in the tolerance for mismatches suggests that PPR–RNA interactions are seeded near the 5′-end of the binding site
We observed a 5′-to-3′ polarity in the degree to which SCD14 and MCD14 select specific nucleotides in the bind-n-seq assays. In fact, truncating two nucleotides from the 3′-end of the SCD14 binding site actually increased binding affinity (Figure 5). This polarity suggests that PPR–RNA interactions nucleate near the N-terminus/5′-end and propagate downstream, that misalignment between repeats and nucleotides increases toward the C-terminus, and that non-specific interactions begin to dominate after a distance of roughly 10 repeat-nucleotide pairs. In accord with this view, crystal structures of proteins with 10 designer PPR motifs bound to RNA demonstrated poor electron density of the 3′ nucleotides (20), indicating that the 3′-end of the RNA is more dynamic than the 5′-end. These results are reminiscent of findings with both TALE and PUF proteins, which show less sequence specificity toward the 3′ ends of their binding sites (8,11,35,36). This phenomenon led to the notion of an N-terminal ‘organizing center’ in TALE proteins, with decreasing contributions downstream due to increasing mismatch in the spatial positioning of the repeats and recognition protein helices (8).
Implications of the position- and nucleotide-dependence of mismatch tolerance exhibited by 11-repeat artificial PPR proteins
The binding of SCD11A and SCD11B to their expected RNA targets is particularly sensitive to transversion mismatches at several positions (Figure 4). This sensitivity correlates with both position along the binding site and the identity of the nucleotide at that position: mismatches toward the center of the binding site were generally more disruptive than were those near the periphery, R→Y mutations were more disruptive than were Y→R mutations at analogous positions, and G→Y mutations had the most severe impact.
The differing effects of nucleotide identity on mismatch tolerance can be rationalized by the recently elucidated structures of designer PPR proteins bound to each of the four nucleotides (20). These structures support the view that purine-PPR interactions, and particularly interactions with G, contribute more to binding energy than do pyrimidine-PPR interactions. First, purines share a larger surface area with the valines that intercalate between adjacent nucleotides, allowing for more van der Waal's contacts. Second, the ‘code’ amino acids form two or three direct hydrogen bonds with cognate A or G residues, respectively, whereas they form just one direct and one water-mediated hydrogen bond with pyrimidines (20).
Mismatches near the center of the 11-nucleotide binding site were particularly disruptive, suggesting that some number of contiguous PPR-nucleotide matches helps establish an interaction with a specific RNA target. This inference is supported by position-dependent effects of small insertions on sequence enrichment in SCD11A and SCD11B bind-n-seq assays (Supplemental Figure S7): insertions of one or two nucleotides toward the center of the binding sites were much more deleterious than were insertions toward the periphery.
CONCLUSION
Taken together, our results suggest a model in which PPR–RNA interactions generally initiate toward the N-terminus/5′-end, and that the interaction then propagates in the 3′ direction (Figure 6). Non-specific interactions predominate after roughly 10 contiguous matches, likely due to increasing misalignment between nucleotides and PPR motifs. Thus, approximately ten contiguous repeat/nucleotide pairs are sufficient to achieve maximal binding affinity. Because longer PPR tracts allow for additional specific or non-specific contacts that compensate for mismatches elsewhere, they are more prone to off-target action. Our results suggest further that failure to satisfy purine-PPR matches is more deleterious to binding affinity than is failure to satisfy pyrimidine-PPR matches. Thus, purine matches toward the 5′-end may be particularly important for maximizing affinity, while limiting PPR tract length to roughly 10 repeats may be important to maximize specificity. These features have strong parallels with TALE-DNA interactions, which is perhaps unsurprising given the similar protein architectures and modular nucleotide contacts. Additional synthetic PPR proteins could be tailored to test various aspects of this model in the future. However, in vivo assays that address these issues will be essential to realize the potential of designer PPR proteins as in vivo tools.
Figure 6.
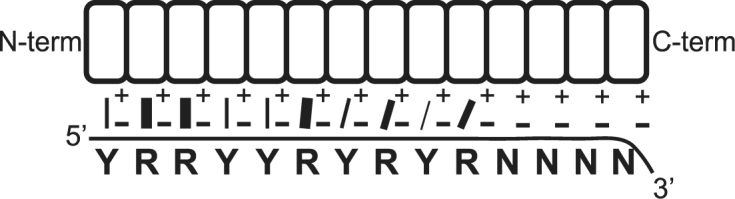
Model for interaction of artificial PPR tracts with RNA. A 14-repeat protein is shown aligned to an RNA ligand. Matches between the specificity-determining amino acids and aligned nucleotide are indicated with lines whose thickness reflects the relative contribution to binding affinity. The cartoon illustrates the inferences that PPR–RNA interactions are established in a 5′-to-3′ direction, that sequence-specific interactions make little contribution to binding affinity past approximately 10 repeats, that purine-PPR interactions make more contribution to binding affinity than do pyrimidine-PPR interactions at analogous positions, and that nucleotides and PPR motifs become increasingly misaligned toward the C-terminus/3′ end until they can no longer make sequence-specific contacts. The model posits that additional repeats beyond the maximum that can be accommodated with modular code contacts contribute to binding affinity via non-specific electrostatic interactions between RNA backbone phosphates (-) and a lysine (+) in the PPR consensus. However, other types of non-specific interaction in this region are also compatible with our results.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Margarita Rojas for assistance with protein purification and Rosalind Williams-Carrier for help with figure preparation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
US National Science Foundation [MCB-1243641 to A.B.]; National Institutes of Health [T32-GM007759 to R.G.M. and J.J.M.]. Funding for open access charge: Internal Academic Support Funds, University of Oregon.
Conflict of interest statement. None declared.
REFERENCES
- 1. Chen Y., Varani G.. Engineering RNA-binding proteins for biology. FEBS J. 2013; 280:3734–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hall T.M. De-coding and re-coding RNA recognition by PUF and PPR repeat proteins. Curr. Opin. Struct. Biol. 2016; 36:116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barkan A., Small I.. Pentatricopeptide repeat proteins in plants. Annu. Rev. Plant Biol. 2014; 65:415–442. [DOI] [PubMed] [Google Scholar]
- 4. Wang X., McLachlan J., Zamore P.D., Hall T.M.. Modular recognition of RNA by a human pumilio-homology domain. Cell. 2002; 110:501–512. [DOI] [PubMed] [Google Scholar]
- 5. Barkan A., Rojas M., Fujii S., Yap A., Chong Y.S., Bond C.S., Small I.. A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet. 2012; 8:e1002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boch J., Scholze H., Schornack S., Landgraf A., Hahn S., Kay S., Lahaye T., Nickstadt A., Bonas U.. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009; 326:1509–1512. [DOI] [PubMed] [Google Scholar]
- 7. Moscou M.J., Bogdanove A.J.. A simple cipher governs DNA recognition by TAL effectors. Science. 2009; 326:1501. [DOI] [PubMed] [Google Scholar]
- 8. Meckler J.F., Bhakta M.S., Kim M.S., Ovadia R., Habrian C.H., Zykovich A., Yu A., Lockwood S.H., Morbitzer R., Elsaesser J. et al. . Quantitative analysis of TALE-DNA interactions suggests polarity effects. Nucleic Acids Res. 2013; 41:4118–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fujii S., Sato N., Shikanai T.. Mutagenesis of individual pentatricopeptide repeat motifs affects RNA binding activity and reveals functional partitioning of Arabidopsis PROTON gradient regulation3. Plant Cell. 2013; 25:3079–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miranda R.G., Rojas M., Montgomery M.P., Gribbin K.P., Barkan A.. RNA-binding specificity landscape of the pentatricopeptide repeat protein PPR10. RNA. 2017; 23:586–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Campbell Z.T., Bhimsaria D., Valley C.T., Rodriguez-Martinez J.A., Menichelli E., Williamson J.R., Ansari A.Z., Wickens M.. Cooperativity in RNA–protein interactions: global analysis of RNA binding specificity. Cell Rep. 2012; 1:570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lurin C., Andres C., Aubourg S., Bellaoui M., Bitton F., Bruyere C., Caboche M., Debast C., Gualberto J., Hoffmann B. et al. . Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell. 2004; 16:2089–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rackham O., Filipovska A.. The role of mammalian PPR domain proteins in the regulation of mitochondrial gene expression. Biochim. Biophys. Acta. 2012; 1819:1008–1016. [DOI] [PubMed] [Google Scholar]
- 14. Small I., Peeters N.. The PPR motif - a TPR-related motif prevalent in plant organellar proteins. Trends Biochem. Sci. 2000; 25:46–47. [DOI] [PubMed] [Google Scholar]
- 15. Cheng S., Gutmann B., Zhong X., Ye Y., Fisher M.F., Bai F., Castleden I., Song Y., Song B., Huang J. et al. . Redefining the structural motifs that determine RNA binding and RNA editing by pentatricopeptide repeat proteins in land plants. Plant J. 2016; 85:532–547. [DOI] [PubMed] [Google Scholar]
- 16. Howard M.J., Lim W.H., Fierke C.A., Koutmos M.. Mitochondrial ribonuclease P structure provides insight into the evolution of catalytic strategies for precursor-tRNA 5′ processing. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:16149–16154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yin P., Li Q., Yan C., Liu Y., Liu J., Yu F., Wang Z., Long J., He J., Wang H.W. et al. . Structural basis for the modular recognition of single-stranded RNA by PPR proteins. Nature. 2013; 504:168–171. [DOI] [PubMed] [Google Scholar]
- 18. Yagi Y., Hayashi S., Kobayashi K., Hirayama T., Nakamura T.. Elucidation of the RNA recognition code for pentatricopeptide repeat proteins involved in organelle RNA editing in plants. PLoS One. 2013; 8:e57286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takenaka M., Zehrmann A., Brennicke A., Graichen K.. Improved computational target site prediction for pentatricopeptide repeat RNA editing factors. PLoS One. 2013; 8:e65343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shen C., Zhang D., Guan Z., Liu Y., Yang Z., Yang Y., Wang X., Wang Q., Zhang Q., Fan S. et al. . Structural basis for specific single-stranded RNA recognition by designer pentatricopeptide repeat proteins. Nat. Commun. 2016; 7:11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kindgren P., Yap A., Bond C.S., Small I.. Predictable alteration of sequence recognition by RNA editing factors from Arabidopsis. Plant Cell. 2015; 27:403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Okuda K., Shoki H., Arai M., Shikanai T., Small I., Nakamura T.. Quantitative analysis of motifs contributing to the interaction between PLS-subfamily members and their target RNA sequences in plastid RNA editing. Plant J. 2014; 80:870–882. [DOI] [PubMed] [Google Scholar]
- 23. Coquille S., Filipovska A., Chia T., Rajappa L., Lingford J.P., Razif M.F., Thore S., Rackham O.. An artificial PPR scaffold for programmable RNA recognition. Nat. Commun. 2014; 5:5729. [DOI] [PubMed] [Google Scholar]
- 24. Shen C., Wang X., Liu Y., Li Q., Yang Z., Yan N., Zou T., Yin P.. Specific RNA recognition by designer pentatricopeptide repeat protein. Mol. Plant. 2015; 8:667–670. [DOI] [PubMed] [Google Scholar]
- 25. Gully B.S., Shah K.R., Lee M., Shearston K., Smith N.M., Sadowska A., Blythe A.J., Bernath-Levin K., Stanley W.A., Small I.D. et al. . The design and structural characterization of a synthetic pentatricopeptide repeat protein. Acta Crystallogr. D Biol. Crystallogr. 2015; 71:196–208. [DOI] [PubMed] [Google Scholar]
- 26. Pfalz J., Bayraktar O., Prikryl J., Barkan A.. Site-specific binding of a PPR protein defines and stabilizes 5′ and 3′ mRNA termini in chloroplasts. EMBO J. 2009; 28:2042–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pattanayak V., Ramirez C.L., Joung J.K., Liu D.R.. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat. Methods. 2011; 8:765–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Williams-Carrier R., Kroeger T., Barkan A.. Sequence-specific binding of a chloroplast pentatricopeptide repeat protein to its native group II intron ligand. RNA. 2008; 14:1930–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prikryl J., Rojas M., Schuster G., Barkan A.. Mechanism of RNA stabilization and translational activation by a pentatricopeptide repeat protein. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fu Y., Sander J.D., Reyon D., Cascio V.M., Joung J.K.. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014; 32:279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rinaldi F.C., Doyle L.A., Stoddard B.L., Bogdanove A.J.. The effect of increasing numbers of repeats on TAL effector DNA binding specificity. Nucleic Acids Res. 2017; 45:6960–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rogers J.M., Barrera L.A., Reyon D., Sander J.D., Kellis M., Joung J.K., Bulyk M.L.. Context influences on TALE-DNA binding revealed by quantitative profiling. Nat. Commun. 2015; 6:7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guilinger J.P., Pattanayak V., Reyon D., Tsai S.Q., Sander J.D., Joung J.K., Liu D.R.. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat. Methods. 2014; 11:429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Garg A., Lohmueller J.J., Silver P.A., Armel T.Z.. Engineering synthetic TAL effectors with orthogonal target sites. Nucleic Acids Res. 2012; 40:7584–7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Valley C.T., Porter D.F., Qiu C., Campbell Z.T., Hall T.M., Wickens M.. Patterns and plasticity in RNA–protein interactions enable recruitment of multiple proteins through a single site. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:6054–6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.