

ABSTRACT
Influenza vaccine composition is reviewed before every flu season because influenza viruses constantly evolve through antigenic changes. To inform vaccine updates, laboratories that contribute to the World Health Organization Global Influenza Surveillance and Response System monitor the antigenic phenotypes of circulating viruses all year round. Vaccine strains are selected in anticipation of the upcoming influenza season to allow adequate time for production. A mismatch between vaccine strains and predominant strains in the flu season can significantly reduce vaccine effectiveness. Models for predicting the evolution of influenza based on the relationship of genetic mutations and antigenic characteristics of circulating viruses may inform vaccine strain selection decisions. We review the literature on state-of-the-art tools and prediction methodologies utilized in modeling the evolution of influenza to inform vaccine strain selection. We then discuss areas that are open for improvement and need further research.
KEYWORDS: influenza evolution, prediction models, influenza vaccine, strain selection, antigenic difference, influenza
Introduction
Influenza (flu) is a highly contagious, acute, respiratory viral disease. Seasonal flu epidemics impact 5–15% of the world's population, resulting in 3–5 million cases of severe illnesses and up to 500,000 deaths annually.1 There are three serotypes of the flu virus: influenza A, B, and C. Influenza A and B viruses are mainly responsible for seasonal flu epidemics, whereas influenza C viruses are less common and usually cause mild upper respiratory illnesses.2 Influenza A viruses are subtyped on the basis of their two surface proteins: hemagglutinin and neuraminidase. Influenza A subtypes and influenza B viruses are further classified into strains based on their antigenic properties.
The first line of defense against seasonal epidemics is the flu shot, which contains two strains of the A virus (H1N1 and H3N2) and one or two strains of the B virus. Since 1970s, influenza B viruses have diverged into two antigenically distinct lineages.3 Therefore, in addition to the trivalent vaccine, manufacturers also produce the quadrivalent vaccine with two influenza B strains to cover both lineages.4 Most individuals have some level of prior immunity. However, new strains with mutations in their epitopes (protein regions that are recognized by human antibodies) frequently arise. These new strains have a fitness advantage over existing dominant strains because they can more effectively escape from host immunity.5 This continuous process of evolution – also known as antigenic drift – results in rapid turnover of the viral population.
The flu shot is unique in that it is annually reformulated and prepared at least six months in advance of the upcoming flu season due to rapid emergence of new strains and the time consuming nature of vaccine production (see Fig. 1).6 In current practice, the flu shot compositions of the Northern and Southern Hemispheres are reviewed and updated as necessary by the World Health Organization (WHO) through a global surveillance and response system.7 Besides surveillance on emerging virus variants, antigenic characterization of circulating viruses by standard ferret antisera is the main determinant in vaccine strain selection. Lesser determinants include genetic variations, prevalence rates, and geographic distributions of virus variants.8 The accurate prediction of emerging strains, however, is a complex problem because of the stochastic nature of the antigenic drift process. Predicting the fate of strains currently circulating in the population is also not easy for two reasons. First, multiple strains carrying different combinations of mutations co-circulate and to some extent compete with one another for susceptible hosts.5 Second, antigenic characterization by ferret antisera is different from that by human post-vaccination antisera because humans and ferrets have different immune systems as well as very different prior exposure histories to influenza virus.8
Figure 1.
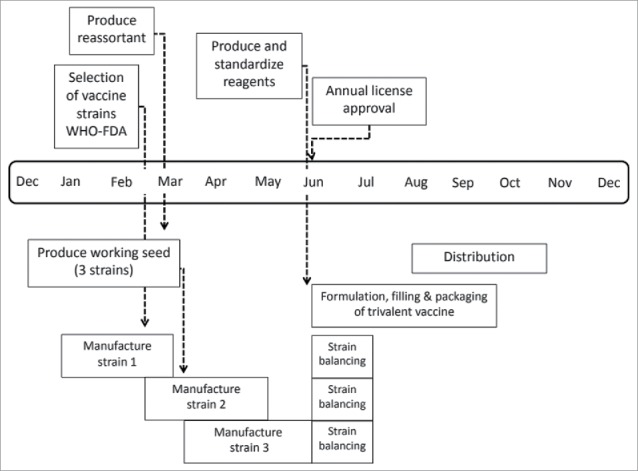
Influenza vaccine manufacturing process and timeline.11 It takes at least six months for the first supplies of approved vaccine to become available once the vaccine composition is decided. This lead time is needed because the vaccine production process involves many sequential steps, and these steps are strictly controlled by government health agencies.
Seasonal influenza vaccine effectiveness mainly depends on how well vaccine strains represent prevalent viruses circulating in the community. The vaccine effectiveness estimates for years without antigenic mismatch between the flu shot strains and circulating strains ranges between 49% and 60%. As recently as the 2014–2015 flu season, however, a poor match between the selected A (H3N2) vaccine strain and the ones that predominantly circulated in that season reduced the vaccine effectiveness to 19% in the US.9 Note that the vaccine effectiveness data only becomes available towards the end of the flu season. For instance, the WHO made the decision on the 2014–2015 Northern Hemisphere vaccine composition in Feb 2014, while vaccine effectiveness data came out in early 2015. Thus, vaccine effectiveness provides only a retrospective review on seasonal vaccine performance, but never plays a role in the selection process for vaccine strains.
A serologic assay, i.e., hemagglutinin inhibition (HI), is used for antigenic characterization of the circulating strains in the flu shot design process.2 The HI assay, however, does not explain the association between antigenic difference and genetic mutations. To accomplish this and model the evolution of influenza, genetic data of previous viruses should be analyzed. The WHO has accumulated a multitude of data for this purpose. Since this data can now be collected rapidly and economically, antigenic characterization of the influenza virus based on its genetic material can enable early detection of emerging strains and increase influenza surveillance efficiency, thus enhancing influenza vaccine strain selection.10
Our goal in this paper is to review the current tools and methods employed in predicting the evolution of influenza which might aid when determining the composition of the seasonal influenza vaccine. In Section2, we discuss the relevant literature about tools developed for visualizing the evolution of influenza. In Section3, we discuss the models proposed for predicting the evolution of influenza. Finally, in Section 4, we provide insights and identify areas in modeling the evolution of influenza that are open for improvement.
Tools for visualizing the evolution of influenza
In any given year, the particular choice of vaccine strain plays a major role in determining vaccine efficacy and so it is of critical importance to develop tools to analyze the ongoing evolution of influenza.12 A widely used tool to visualize the evolution of influenza is called a phylogenetic tree or evolutionary tree. In general, these diagrams show inferred evolutionary relationships among biological entities based on similarities and differences in their genetic characteristics. Every leaf node in the tree represents a species, each edge denotes a relationship between two neighboring species, and the length of an edge indicates the evolutionary distance between species.13
There have been many tools developed to generate and analyze phylogenetic trees,14-19 With respect to influenza, Neher and Bedford12 designed an online visualization tool entitled nextflu that displays a phylogenetic tree of the most recent influenza virus sequences (see Fig. 2). This tool allows users to visualize many genetic and epidemiological features of influenza strains and aids in the current vaccine strain selection process. Bedford and Neher20 demonstrated the use of nextflu to analyze seasonal influenza circulation patterns and provided projections for the 2017–2018 flu season. In a similar vein, Steinbrück and McHardy21 introduced allele dynamics plots (AD plots) as a method for visualizing the evolutionary dynamics of a gene. The AD plot of a population-level sequence sample combines information from phylogenetic inference and ancestral character state reconstruction to identify the alleles that might have selective advantage. Using this tool, Steinbrück and McHardy21 identified emerging strains of influenza A (H3N2) and 2009 A (H1N1) pdm viruses.
Figure 2.

nextFlu display of influenza phylogenetic tree.12
Another method to visualize the evolution of influenza is through antigenic cartography or antigenic maps.22 Antigenic cartography is currently used to analyze the global data from the WHO influenza surveillance network as part of the influenza vaccine strain selection process (see Fig. 3). Antigenic maps differ from genetic trees in that they are based on serology data that reflect the antigenic properties of pathogens (in this case as revealed by HI assay based on ferret antisera). These maps can reveal long-term trends in the antigenic space leading to improved understanding of genetic and antigenic evolution.22
Figure 3.
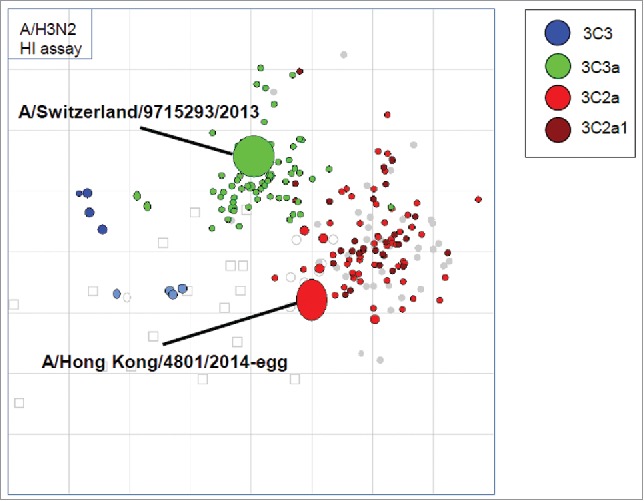
Antigenic cartography of circulating A (H3N2) viruses considered by the WHO in the 2017–18 Northern hemisphere influenza vaccine strain selection meeting.24 Virus clades are color-coded. There is no significant antigenic difference between the 3C2a clade in the brighter red color and the 3C2a1 clade in the darker red color as they cluster. The Hong Kong/4801/2014-egg virus is on the edge of that cluster, and the cluster is becoming more distinguishable from the earlier 3C3a viruses represented by A/Switzerland/9715293/2013 strain, which was a former vaccine strain. The WHO recommended the Hong Kong/4801/2014-like virus for the 2017–18 influenza vaccine. (No change from 2016–17 recommendation).
Antigenic characterization based on the HI assay is a routine procedure for influenza vaccine strain selection. However, the HI assay only reveals the cross reaction among test strains (antigens) and reference antisera (antibodies). Furthermore, antigenic characterization is usually based on multiple HI assays performed by different WHO collaborating centers. The combination of these datasets results in an incomplete HI matrix with many unobserved entries. Cai et al.23 developed a computational tool entitled AntigenMap for antigenic cartography construction from this incomplete matrix. Their approach first reconstructs the HI tables using matrix completion techniques, and then generates the two-dimensional antigenic cartography using multidimensional scaling. By applying this method to HI datasets containing influenza A (H3N2) viruses isolated between 1968 and 2003, Cai et al.23 identified eleven clusters of antigenic variants, representing all major antigenic drift events during these 36 years.
Predicting the evolution of influenza
Through the use of tools mapping the evolution of influenza, such as phylogenetic trees and antigenic cartographs, statistical learning models have been proposed to predict the next evolutionary step of influenza. These models aim to combine serology data and genetic mutation information to explain antigenic difference. We review some of the most recent studies in the literature that propose methods to inform strain selection for the seasonal influenza vaccine.
He and Deem25 constructed protein distance maps for the HA1 surface glycoprotein of the influenza 2009 A (H1N1) pdm virus. In particular, they applied multi-dimensional scaling26 to project the 329-residue long amino acid sequence of the HA1 protein onto two dimensions. This mapping technique was also used by Lapedes and Farber27 to project HI assay data onto lower dimensions. He and Deem25 then used kernel density estimation to detect the incipient clusters on the protein distance map.
Steinbrück and McHardy28 used nonnegative least-squares optimization to map pairwise antigenic distances onto the branches of a phylogenetic tree. This resulted in the inference of antigenic weights for the individual branches of the tree and allowed antigenic weights to be determined for sets of coding changes on the surface glycoprotein hemagglutinin (HA). These weights contribute to identifying the antigenic impact of HA alleles. Steinbrück et al.29 combined phylogenetic trees and AD plots to identify the HA alleles that are most likely to become predominant in the future seasons. They demonstrated how to predict the evolution of the influenza A (H3N2) virus using genetic and antigenic data from isolates sampled between 2002 and 2007. In this retrospective study, they identified the most suitable vaccine strain for the A (H3N2) virus by detecting antigenically novel HA alleles.
Neher et al.30 predicted the fitness of a virus from its genetic information. In their method, first, a genealogical tree for the virus population is constructed. The next step is based on the intuition that as long as differences in fitness values arise from the accumulation of multiple mutations (i.e., antigenic drifts), the branching structure of the genealogical tree should exhibit an observable imprint of the natural selection process as it unfolds. Using this insight and methods borrowed from statistical physics, Neher et al.30 analyzed the shape and branching pattern of the tree to identify the fitness of different strains relative to each other. They tested the proposed method using historical influenza A virus data. In 16 of the 19 years studied, the genealogical tree approach made meaningful predictions about which viruses were most likely to give rise to future epidemics.
Neher et al.31 proposed a phylogenetic tree-based model and a substitution model to explain antigenic difference. The tree based model describes the HI titer between a test and a reference influenza strain as a sum of antigenic changes along the path connecting them in a phylogenetic tree. The substitution model explains HI titers as a sum of contributions associated with amino acid substitutions between the reference and test viruses. Through numerical experiments using data from isolates sampled between 2002 and 2015, Neher et al.31 demonstrated that both the tree based model and the substitution model perform similarly in terms of prediction accuracy.
Łuksza and Lässig32 developed a fitness model to predict the evolution of influenza by identifying changes in the frequencies of strain groups referred to as clades. They considered two major groups of mutations at the epitope and non-epitope regions of the virus' surface protein. Mutations at epitopes are likely to be beneficial to the virus, because they alter the structural features targeted by host antibodies. Thus, a strain can have better fitness than its competitors by being antigenically distinct. In contrast, mutations outside epitope regions are often deleterious because they reduce protein stability or upset evolutionarily conserved viral functions Łuksza and Lässig32 used their model to predict frequencies of clades one year in the future with considerable accuracy. Their observations of how high and low fitness clades evolve provide new perspectives on the evolution of influenza and can potentially contribute to the vaccine selection process.
Wilson and Cox33 suggested that a drift variant of epidemiologic importance usually contains at least four amino acid substitutions located at more than two of the epitope regions on the HA1 polypeptide. Lee and Chen34 showed that the number of amino acid changes in the 131 amino acid positions around the epitope sites had the highest correlation with the antigenic distance and the best performance for predicting antigenic difference. Presumably not all 131 amino acid positions around the epitope regions play a critical role in determining antigenicity, and thus immunodominant positions (i.e., major antibody-binding sites) should be identified using bioinformatics models.35 Liao et al.36 explored the use of scoring methods, such as the construction of similarity classes and substitution matrices, to explain the antigenic differences of viruses' using genetic information. These methods considered polarity, charge and molecular structure of the amino acids. Liao et al.36 employed statistical machine learning methods including iterative filtering, multiple and logistic regression, and support vector machines to quantify the antigenic effect of amino acid substitutions and identify immunodominant positions on the HA1 polypeptide. Similarly, Sun et al.10 utilized bootstrapped ridge regression and antigenic mapping to quantify antigenic difference between influenza strains based on the HA1 sequence data. Recently, Lee et al.37 developed a general purpose computational framework called DAMIP to discover gene signatures that can predict vaccine immunity and efficacy.
The literature on predicting the evolution of influenza is rich and fast-growing. Seasons characterized by low vaccine effectiveness due to a mismatch between the vaccine strains and the circulating strains, such as the 2014–15 influenza season,6 highlight the need to use advanced tools and innovative approaches to improve the accuracy of the strain selection decisions. There is room for development in this field as we discuss in the next section.
Discussion
We have presented a detailed review of the most recent and influential studies developing innovative methods for predicting the long-term evolution of influenza. The overarching goal of these studies is to inform and improve strain selection for the seasonal influenza vaccine. While much progress has been made, there are several areas that are still open for improvement.
When building statistical models for predicting the evolution of influenza, out-of-sample performance is assessed to prevent overfitting. However, the number of candidate model features is usually large. For example, in an amino acid substitution model,31,34 there are 329 amino acid residues on the HA1 polypeptide, and mutation in any one of these residues can potentially cause antigenic difference. Selecting a minimal set of model features (e.g., amino acid residues) to ensure adequate out-of-sample performance is a formidable task that can be addressed via feature selection methods developed in the machine learning literature.38–40
Many of the models in the literature assume rather simple relationships between genetic differences of influenza strains (e.g., the number of amino acid substitutions around the epitope regions) and the extent of cross-immunity that they can induce in a host.5 However, antigenic analysis has shown that only certain amino-acid changes near the epitope regions cause antigenic difference.22 Current efforts to predict the evolution of influenza try to map the serology data onto genetic sequences and phylogenetic trees.31,32,34,37 Occasionally, new strains, which are antigenically different from the circulating dominant strains, still fail to spread in the population although they have a higher probability of fixation.31 Therefore, incorporating serology data to consider empirically informed associations between viral genotypes and their antigenic characteristics in addition to genetic mutations may improve the predictive power of statistical models in detecting emerging strains.
Another direction that is open for improvement pertains to the generation of serology data. Currently, the ferret post-infection antisera are used in antigenic characterization for influenza surveillance because ferrets have human-like respiratory tracts.41,42 Despite this similarity, however, the process of ferret immunity is different from that of human.8 In particular, the human and ferret antibodies elicited via the same process may target different epitopes of the influenza HA.43 Furthermore, ferrets with no prior influenza infections are used to produce reference antisera. In contrast, human immunity is largely shaped by previous influenza infection and/or vaccination history.44 For example, many recent studies have shown that people with repeated annual vaccination had lower vaccine effectiveness than those who were not vaccinated in the prior season, suggesting that immunological education and pre-existing immunity affects seasonal vaccine effectiveness.45-48 Xie et al.8 have clearly demonstrated that human post-vaccination antisera responded differently than ferret post-infection antisera to H3 viruses. Therefore, more emphasis should be placed on human serologic testing in assessing vaccination-induced cross reactivity to improve influenza vaccine strain selection.
In summary, the seasonal influenza is a serious public health concern, causing significant human suffering and economic burden. Due to dynamic evolution of the virus, the influenza vaccine composition is reviewed annually by the WHO to ensure vaccine effectiveness. The overarching goal of this review process is to predict which strains will provide a thorough coverage as vaccine strains against prevalent circulating strains during the upcoming flu season. Predicting virus evolution is an essential interim step towards achieving this goal because once new variants are predicted to emerge, their representative strains will be considered as candidate vaccine strains. If an antigenic mismatch happens to occur between the vaccine strains and the ones dominantly circulating in the population during the flu season, then a vaccine with significantly reduced effectiveness is still distributed because the vaccine production is extremely time consuming. We have discussed tools and methods used in the development of prediction models to aid influenza vaccine strain selection. There are many opportunities for new innovative directions or for improvement of current methodologies. Such developments can potentially lead to more effective prevention of influenza worldwide saving lives, time and money.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Carrat F, Flahault A. Influenza vaccine: The challenge of antigenic drift. Vaccine. 2007;25:6852–62. doi: 10.1016/j.vaccine.2007.07.027 [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization Seasonal Influenza. Available at http://www.who.int/mediacentre/factsheets/fs211/en, 2016. [Google Scholar]
- 3.Beire B, Bauer B, Schweiger B. Differentiation of influenza B virus lineages Yamagata and Victoria by real-time PCR. J Clin Microbiol. 2010;48(4):1425–27. doi: 10.1128/JCM.02116-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grohskopf LA, Olsen SJ, Sokolow LZ, Bresee JS, Cox NJ, Broder KR, Karron RA, Walter EB. Prevention and control of seasonal influenza with vaccines: Recommendations of the advisory committee on immunization practices (ACIP) – United States, 2014–15 influenza season. MMWR Morb Mortal Wkly Rep. 2014;63(32):691–7. [PMC free article] [PubMed] [Google Scholar]
- 5.Koelle K, Rasmussen DA. Influenza: Prediction is worth a shot. Nature. 2014;506(7490):47–49. doi: 10.1038/nature13054 [DOI] [PubMed] [Google Scholar]
- 6.Dos Santos G, Neumeier E, Bekkat-Berkani R. Influenza: Can we cope better with the unpredictable?. Hum Vaccin Immunother. 2016;12(3):699–708. doi: 10.1080/21645515.2015.1086047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.World Health Organization Manual for the laboratory diagnosis and virological surveillance of influenza. Available at http://www.who.int/influenza/gisrs_laboratory/manual_diagnosis_surveillance_influenza/en, 2011. [Google Scholar]
- 8.Xie H, Wan X-F, Ye Z, Plant EP, Zhao Y, Xu Y, Li X, Finch C, Zhao N, Kawano T, et al.. H3N2 mismatch of 2014–15 Northern Hemisphere influenza vaccines and head-to-head comparison between human and ferret antisera derived antigenic maps. Sci Rep. 2015;5:15279. doi: 10.1038/srep15279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Centers for Disease Control and Prevention Seasonal Influenza Vaccine Effectiveness, 2005–2015. Available at http://www.cdc.gov/flu/professionals/vaccination/effectiveness-studies.htm, 2015. [Google Scholar]
- 10.Sun H, Yang J, Zhang T, Long P, Jia K, Yang G, Webby R, Wan X. Using sequence data to infer the antigenicity of influenza virus. mBio. 2013;4(4):e00230–13. doi: 10.1128/mBio.00230-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Özaltın OY, Prokopyev OA, Schaefer AJ. Optimal design of the seasonal influenza vaccine with manufacturing autonomy. Forthcoming at INFORMS Journal on Computing. 2017. [Google Scholar]
- 12.Neher R, Bedford T. nextflu: Real-time tracking of seasonal influenza virus evolution in humans. Bioinformatics. 2015;31(21):3546–48. doi: 10.1093/bioinformatics/btv381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi JH, Jung HY, Kim HS, Cho HG. PhyloDraw: A phylogenetic tree drawing system. Bioinformatics. 2000;16(11):1056–58. [DOI] [PubMed] [Google Scholar]
- 14.Gouy M, Guidon S, Gascuel O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27(2):221–4. [DOI] [PubMed] [Google Scholar]
- 15.Letunic I, Bork P. Interactive tree of life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics. 2006;23(1):127–8. [DOI] [PubMed] [Google Scholar]
- 16.Price MN, Dehal PS, Arkin AP. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26(7):1641–50. doi: 10.1093/molbev/msp077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimodaira H, Hasegawa M. CONSEL: For assessing the confidence of phylogenetic tree selection. Bioinformatics. 2001;17(12):1246–7. [DOI] [PubMed] [Google Scholar]
- 18.Thorley JL, Page RD. RadCon: Phylogenetic tree comparison and consensus. Bioinformatics. 2000;16(5):486–7. [DOI] [PubMed] [Google Scholar]
- 19.Yang Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Bioinformatics. 1997;13(5):555–6. [DOI] [PubMed] [Google Scholar]
- 20.Bedford T, Neher R. Seasonal Influenza Circulation Patterns and Projections for 2017–2018. Available at http://www.biorxiv.org/content/early/2017/03/02/113035, 2017. [Google Scholar]
- 21.Steinbrück L, McHardy AC. Allele dynamics plots for the study of evolutionary dynamics in viral populations. Nucleic Acids Res. 2011;39(1):e4. doi: 10.1093/nar/gkq909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith DJ, Lapedes AS, Jong J, Bestebroer TM, Rimmelzwaan GF, Osterhaus A, Fouchier R. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305(5682):371–6. [DOI] [PubMed] [Google Scholar]
- 23.Cai Z, Zhang T, Wan X-F. A computational framework for influenza antigenic cartography. PLoS Comput Biol. 2010;6(10):e1000949. doi: 10.1371/journal.pcbi.1000949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Food and Drug Administration Vaccines and Related Biological Products Advisory Committee. Available at http://www.fda.gov/cber/advisory/vrbp/vrbpmain.htm, 2017. [Google Scholar]
- 25.He J, Deem MW. Low-dimensional clustering detects incipient dominant influenza strain clusters. Protein Eng Des Sel. 2010;23(12):935–46. doi: 10.1093/protein/gzq078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Everitt BS, Landau S, Leese M. Cluster Analysis, 5th ed. London: John Wiley & Sons, Ltd; 2011. [Google Scholar]
- 27.Lapedes A, Farber R. The geometry of shape space: Application to influenza. J Theor Biol. 2001;212(1):57–69. doi: 10.1006/jtbi.2001.2347 [DOI] [PubMed] [Google Scholar]
- 28.Steinbrück L, McHardy AC. Inference of genotype-phenotype relationships in the antigenic evolution of human influenza A (H3N2) viruses. PLoS Comput Biol. 2012;8(4):e1002492. doi: 10.1371/journal.pcbi.1002492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steinbrück L, Klingen TR, McHardy AC. Computational prediction of vaccine strains for human influenza A (H3N2) viruses. J Virol. 2014;88(20):12123–32. doi: 10.1128/JVI.01861-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neher R, Russell C, Shraiman B. Predicting evolution from the shape of genealogical trees. eLife. 2014;3:e03568. doi: 10.7554/eLife.03568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neher R, Bedford T, Daniels R, Russell C, Shraiman B. Prediction, dynamics, and visualization of antigenic phenotypes of seasonal influenza viruses. Proc Natl Acad Sci. 2016;113(12):E1701–E1709. doi: 10.1073/pnas.1525578113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Łuksza M, Lässig M. A predictive fitness model for influenza. Nature. 2014;507(7490):57–61. doi: 10.1038/nature13087 [DOI] [PubMed] [Google Scholar]
- 33.Wilson IA, Cox NJ. Structural basis of immune recognition of influenza virus hemagglutinin. Annu Rev Immunol. 1990;8(1):737–87. doi: 10.1146/annurev.iy.08.040190.003513 [DOI] [PubMed] [Google Scholar]
- 34.Lee M-S, Chen J. Predicting antigenic variants of influenza A/H3N2 viruses. Emerg Infect Dis. 2004;10(8):1385. doi: 10.3201/eid1008.040107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee M-S, Chen M-C, Liao Y-C, Hsiung CA. Identifying potential immunodominant positions and predicting antigenic variants of influenza A/H3N2 viruses. Vaccine. 2007;25(48):8133–39. doi: 10.1016/j.vaccine.2007.09.039 [DOI] [PubMed] [Google Scholar]
- 36.Liao Y-C, Lee M-S, Ko C-Y, Hsiung CA. Bioinformatics models for predicting antigenic variants of influenza A/ H3N2 virus. Bioinformatics. 2008;24(4):505–12. [DOI] [PubMed] [Google Scholar]
- 37.Lee EK, Nakaya HI, Yuan F, Querec TD, Burel G, Pietz FH, Benecke BA, Pulendran B. Machine learning for predicting vaccine immunogenicity. Interfaces. 2016;46(5):368–90. [Google Scholar]
- 38.Guyon I, Elisseeff A. An introduction to variable and feature selection. J Mach Learn Res. 2003;3(Mar):1157–82. [Google Scholar]
- 39.Huang C-L, Wang C-J. A GA-based feature selection and parameters optimization for support vector machines. Expert Syst Appl. 2006;31(2):231–40. [Google Scholar]
- 40.Siedlecki W, Sklansky J. A note on genetic algorithms for large-scale feature selection. Pattern Recognit Lett. 1989;10(5):335–47. [Google Scholar]
- 41.Belser JA, Katz JM, Tumpey TM. The ferret as a model organism to study influenza A virus infection. Dis Model Mech. 2011;4(5):575–9. doi: 10.1242/dmm.007823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Russell CA, Jones TC, Barr IG, Cox NJ, Garten RJ, Gregory V, Gust ID, Hampson AW, Hay AJ, Hurt AC, et al.. Influenza vaccine strain selection and recent studies on the global migration of seasonal influenza viruses. Vaccine. 2008;26:D31–D34. [DOI] [PubMed] [Google Scholar]
- 43.Lee M-S, Yang C-F. Cross-reactive H1N1 antibody responses to a live attenuated influenza vaccine in children: implication for selection of vaccine strains. J Infect Dis. 2003;188(9):1362–6. doi: 10.1086/379045 [DOI] [PubMed] [Google Scholar]
- 44.Li Y, Myers JL, Bostick DL, Sullivan CB, Madara J, Linderman SL, Liu Q, Carter DM, Wrammert J, Esposito S, et al.. Immune history shapes specificity of pandemic H1N1 influenza antibody responses. J Exp Med. 2013;210(8):1493–1500. doi: 10.1084/jem.20130212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie H, Li L, Ye Z, Li X, Plant EP, Zoueva O, Zhao Y, Jing X, Lin Z, Kawano T, et al.. Differential effects of prior influenza exposures on H3N2 cross-reactivity of human postvaccination sera. Clin Infect Dis. 2017;65(2):259–67. doi: 10.1093/cid/cix269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McLean HQ, Thompson MG, Sundaram ME, Meece JK, McClure DL, Friedrich TC, Belongia EA. Impact of repeated vaccination on vaccine effectiveness against influenza A (H3N2) and B during 8 seasons. Clin Infect Dis. 2014;59(10):1375–85. doi: 10.1093/cid/ciu680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohmit SE, Petrie JG, Malosh RE, Fry AM, Thompson MG, Monto AS. Influenza vaccine effectiveness in households with children during the 2012–2013 season: assessments of prior vaccination and serologic susceptibility. J Infect Dis. 2014;211(10):1519–28. doi: 10.1093/infdis/jiu650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saito N, Komori K, Suzuki M, Morimoto K, Kishikawa T, Yasaka T, Ariyoshi K. Negative impact of prior influenza vaccination on current influenza vaccination among people infected and not infected in prior season: A test-negative case-control study in Japan. Vaccine. 2017;35(4):687–93. [DOI] [PubMed] [Google Scholar]