

ABSTRACT
Ophthalmic acid (OA; l-γ-glutamyl-l-2-aminobutyryl-glycine) is an analog of glutathione (GSH; l-γ-glutamyl-l-cysteinyl-glycine) in which the cysteine moiety is replaced by l-2-aminobutyrate. OA is a useful peptide for the pharmaceutical and/or food industries. Herein, we report a method for the production of OA using engineered Escherichia coli cells. yggS-deficient E. coli, which lacks the highly conserved pyridoxal 5′-phosphate-binding protein YggS and naturally accumulates OA, was selected as the starting strain. To increase the production of OA, we overexpressed the OA biosynthetic enzymes glutamate-cysteine ligase (GshA) and glutathione synthase (GshB), desensitized the product inhibition of GshA, and eliminated the OA catabolic enzyme γ-glutamyltranspeptidase. The production of OA was further enhanced by the deletion of miaA and ridA with the aim of increasing the availability of ATP and attenuating the unwanted degradation of amino acids, respectively. The final strain developed in this study successfully produced 277 μmol/liter of OA in 24 h without the formation of by-products in a minimal synthetic medium containing 1 mM each glutamate, 2-aminobutyrate, and glycine.
IMPORTANCE Ophthalmic acid (OA) is a peptide that has the potential for use in the pharmaceutical and/or food industries. An efficient method for the production of OA would allow us to expand our knowledge about its physiological functions and enable the industrial/pharmaceutical application of this compound. We demonstrated the production of OA using Escherichia coli cells in which OA biosynthetic enzymes and degradation enymes were engineered. We also showed that unique approaches, including the use of a ΔyggS mutant as a starting strain, the establishment of an S495F mutation in GshA, and the deletion of ridA or miaA, facilitated the efficient production of OA in E. coli.
KEYWORDS: ophthalmic acid, pyridoxal 5′-phosphate, glutathione, YggS
INTRODUCTION
Ophthalmic acid (OA; l-γ-glutamyl-l-2-aminobutyryl-glycine) was initially identified to be a glutathione (GSH; l-γ-glutamyl-l-cysteinyl-glycine) analog in calf lens (1) and was later detected in the lenses of many higher animals (2) and in microorganisms (3–5). OA is produced through the consecutive reactions of glutamate (Glu)-cysteine (Cys) ligase (GshA) and glutathione synthase (GshB), which are the same enzymes involved in the synthesis of GSH (Fig. 1). Owing to the structural similarity between OA and GSH, OA is used as a non-thiol-containing analog of GSH (6, 7). OA is known to inhibit both the degradation of insulin in sections of rat adipose tissue (8) and the activity of glyoxalase I (9). A recent study that profiled liver metabolites following acetaminophen-induced hepatotoxicity reported that the concentration of OA is closely correlated with GSH consumption in mouse serum and liver. This observation has raised the possibility that OA can be used as an oxidative stress biomarker to indicate hepatic GSH consumption (10). In Synechocystis sp. strain PCC6803, OA was proposed to be a stress-induced marker of l-Cys depletion, which is triggered by the increased demand for GSH biosynthesis under conditions of stress (4). In addition, recent studies have revealed that GSH, OA, l-γ-glutamyl-l-valyl-glycine, and their analogs activate the calcium-sensing receptors of humans and act as kokumi substances that modify the five basic tastes (11, 12).
FIG 1.
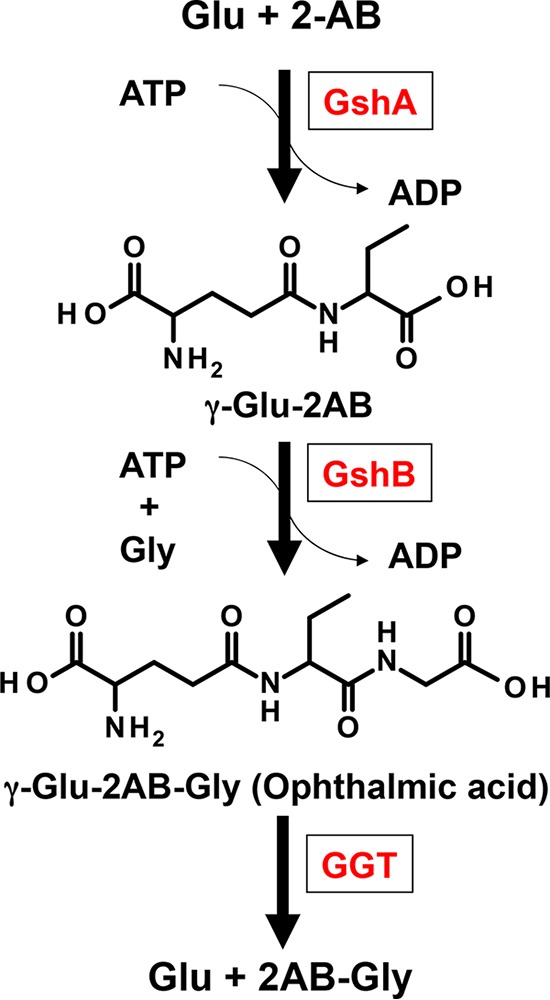
Biosynthesis and degradation pathway of OA. Abbreviations: GshA, glutamate-cysteine ligase; GshB, glutathione synthase; GGT, γ-glutamyltranspeptidase; 2-AB, 2-aminobutyrate.
Although OA has potential applications in the pharmaceutical and/or food industries, it is currently a highly expensive substance. The development of an efficient method to produce OA will enable us to perform further research on this molecule and investigate its potential applications in the pharmaceutical and/or food industries. In the present study, we developed a method for the production of OA using engineered Escherichia coli cells. To this end, we utilized a yggS-deficient E. coli strain (E. coli ΔyggS) reported to accumulate OA as a starting strain (5). YggS is a highly conserved pyridoxal 5′-phosphate (PLP)-binding protein that plays important roles in the homeostasis of amino acids and/or B6 vitamers (13, 14). In addition, we deleted γ-glutamyltranspeptidase (GGT), which is the primary enzyme that degrades GSH, and investigated its contribution to the degradation of OA. To enhance the OA biosynthetic pathway, we cooverexpressed GshA and GshB, eliminated the feedback inhibition ability of GshA, and evaluated their effects on the production of OA. The enamine intermediates 2-aminoacrylate/2-crotonate, which are generated by the action of Ser/Thr dehydratase and transiently accumulate in enamine intermediate deaminase (RidA)-deficient cells, have been reported to inactivate several PLP-dependent enzymes involved in amino acid metabolism (15–17). To attenuate the unwanted catabolism of OA's constituent amino acids by PLP-dependent enzymes, we deleted ridA and investigated the effect on the production of OA. Hara et al., using a single-gene deletion library of E. coli strains (Keio Collection), identified some genes that, when deleted, resulted in a higher efficiency of ATP regeneration and a higher level of production of GSH in the deletion mutant than in the parental strain (18, 19). Among them, we selected two genes, ptsG and miaA, and examined the effects of deleting those genes on the production of OA.
In the present paper, we report the results of an investigation using the above-described strategies to develop an efficient method for the production of OA in E. coli. The engineered E. coli strain constructed by combining these approaches specifically produced ∼80 mg/liter of OA with a yield of 28% in 24 h.
RESULTS AND DISCUSSION
The deletion of GGT effectively reduces the degradation of OA.
With the aim of increasing the production of OA, we sought to identify and disrupt the enzymes responsible for OA degradation in E. coli. Previous studies have demonstrated that both GGT and tripeptidase (PepT) play important roles in GSH degradation and that GGT contributes to GSH degradation to a greater extent than PepT in E. coli (20, 21).
Thus, we constructed a yggS and ggt double-knockout strain (E. coli ΔyggS Δggt) and examined the effect on OA degradation. The deletion of ggt did not significantly affect the growth rate of the E. coli cells in M9 medium (Fig. 2A). With the parental strain (E. coli ΔyggS), the highest concentration of OA during the cultivation period was observed at 28 h (0.6 μmol/liter). The concentration of OA subsequently decreased with prolonged cultivation and was below the detection limit at 86 h. With the ΔyggS Δggt strain, the highest concentration of OA was 1.0 μmol/liter and the OA content did not decrease with prolonged cultivation (Fig. 2B). These results demonstrate that GGT is crucial for the catabolism of OA and that the deletion of ggt both increased the production of OA and suppressed the decrease in the concentration of OA with prolonged cultivation. The reason for the transient decrease in the content of OA during the cultivation period is currently unclear.
FIG 2.

Effect of ggt deletion on growth (A) and cultivation time-dependent changes of the OA content (B). ΔyggS and ΔyggS Δggt cells were cultivated by cultivation method A in the absence of Cam and IPTG (without amino acid supplementation). Samples were collected at the indicated times, and the cultivation time-dependent changes of the OA content were analyzed by amino acid analyses. All results are expressed as means ± standard deviations of the data obtained from at least 3 independent experiments. WT, wild type.
GshA-GshB coexpression increases the production of OA.
OA is synthesized by the actions of GshA and GshB (Fig. 1). Our previous study showed that the overexpression of GshA or GshB effectively increased the content of OA in ΔyggS cells in the log phase (5). We thus overexpressed GshA or GshB in the ggt-deficient strain (E. coli ΔyggS Δggt), which we cultivated in M9 medium until the cells reached the stationary phase of growth (88 h), and examined the effects on the production of OA. As expected, the overexpression of GshA or GshB increased the production of OA by 7.9- or 3.8-fold, respectively, compared to that for the control harboring the empty vector (Fig. 3). This result suggests that the simultaneous expression of these two enzymes may further enhance the production of OA.
FIG 3.
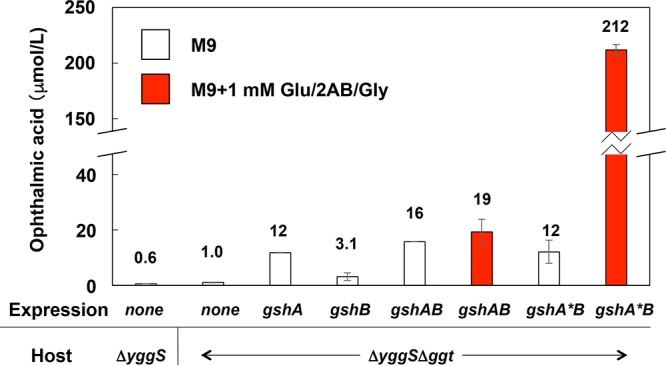
Effect of GshA and/or GshB overexpression on OA production. The ΔyggS and ΔyggS Δggt strains harboring empty vector pCA24N (none), pCA24N-gshA (gshA), pCA24N-gshB (gshB), pGshAB (gshAB), or pGshA*B (gshA*B) were grown to the stationary phase by cultivation method A for 88 h with 1 mM each Glu, 2-AB, and Gly or without amino acids. The total amount of OA in the cultivation system was analyzed by amino acid analysis as described in Materials and Methods. All data are expressed as means ± standard deviations of the data obtained after at least 3 repetitions of each assay.
To test that possibility, we constructed a GshA-GshB coexpression plasmid (pGshAB), which was then introduced into the ΔyggS Δggt strain, thereby generating ΔyggSΔggt-GshAB. The overexpression of the two enzymes was verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting using an anti-His tag antibody (data not shown). ΔyggSΔggt-GshAB cells produced 15.7 μmol/liter of OA, which was an amount 10 times greater than the amount produced by the ΔyggS Δggt strain harboring the empty vector (Fig. 3).
Although ΔyggSΔggt-GshAB cells exhibited the highest levels of production of OA, the concentration was only 30% higher than the amount produced by the GshA-overexpressing strain (ΔyggSΔggt-GshA, 11.8 μmol/liter) (Fig. 3). We speculated that the limited increase in the production of OA by ΔyggSΔggt-GshAB cells was partially due to the limited availability of certain constituent amino acids. However, when we supplemented the cells with three constituent amino acids, namely, Glu, l-2-aminobutyrate (2-AB), and glycine (Gly) (1 mM each), the production of OA was not noticeably altered (Fig. 3). This result suggests that the limited availability of an amino acid(s) was not the primary reason for the limited increase in the concentration of OA in the ΔyggSΔggt-GshAB strain.
The S495F mutation in GshA significantly increases the production of OA in the presence of amino acids.
GshA exhibits strong product inhibition, which is mediated by the reduced form of GSH and limits the biosynthesis of GSH (22–24). The production of OA may also be limited by this product inhibition. The 494th and/or 495th amino acid residue in GshA has been reported to be involved in the product inhibition ability of GSH (25, 26). In addition, the S495F mutation of GshA confers resistance to its product inhibition ability (H. Suzuki, Kyoto Institute of Technology [KIT], unpublished data).
We therefore introduced the S495F mutation into GshA (referred to as GshA*) and examined the effect on product inhibition by analyzing the activities of the Glu–2-AB and Glu-Cys ligases in the cell extracts in the presence or absence of GSH. As shown in Fig. 4, the Glu–2-AB ligase activity catalyzed by wild-type GshA was inhibited by 50% in the presence of GSH. In contrast, the activity of the GshA(S495F) mutant was not significantly affected by the presence of GSH, although its activity decreased by approximately 20%. Similar results regarding Glu-Cys ligase activity were observed. The data validate the suggestion that the S495F mutation of GshA confers desensitization of its product inhibition ability. Considering the structural similarity between GSH and OA, the S495F mutation was also expected to confer resistance to product inhibition by OA.
FIG 4.
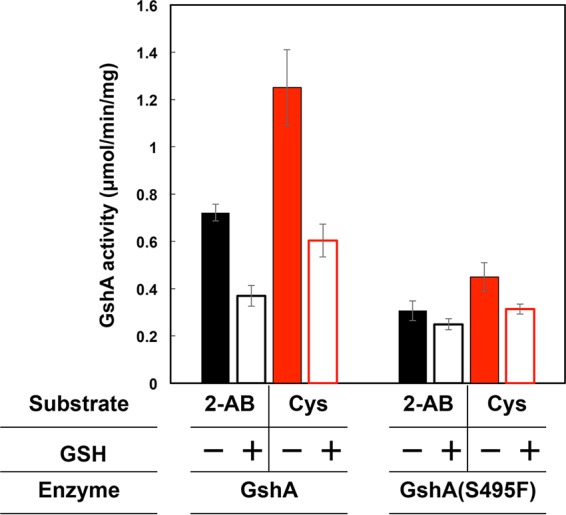
Effect of GSH on GshA or GshA(S495F) activity. The Glu-2-AB (black) or Glu-Cys ligase activity (red) catalyzed by GshA or GshA(S495F) was determined as described in Materials and Methods. The reduced form of GSH significantly inhibited the enzyme activity catalyzed by GshA. All data are expressed as means ± standard deviations of the data obtained after at least 3 repetitions of each assay.
In the absence of amino acid supplements, the ΔyggS Δggt strain harboring pGshA*B (ΔyggSΔggt-GshA*B) produced 12.2 μmol/liter of OA, which was similar to the amount produced by ΔyggSΔggt-GshAB cells (15.7 μmol/liter) (Fig. 3). In contrast, in the presence of 1 mM each Glu, 2-AB, and Gly, the ΔyggSΔggt-GshA*B cells produced 213 μmol/liter of OA, which represented an 11-fold increase in the level of production compared with that by ΔyggSΔggt-GshAB cells (19.2 μmol/liter) (Fig. 3). The production of OA by ΔyggSΔggt-GshA*B cells was significantly stimulated by supplementation with amino acids. Eliminating the product inhibition ability of GshA and supplementing the engineered cells with amino acids was an effective way to increase the production of OA.
Effects of varying the culture conditions.
In the production system described above, we used cultivation method A (see the Materials and Methods section). This method was time-consuming, and the cells took 4 days to reach the stationary phase, probably because the overexpression of GshA and GshB significantly inhibited cell growth. We therefore varied the cultivation conditions to shorten the cultivation time. When we used cultivation method B, in which the cell growth medium was supplemented with 1 mM each constituent amino acid of OA, ΔyggSΔggt-GshA*B cells produced 200 μmol/liter of OA in 24 h, which was almost the same as the level of production under cultivation method A (212 μmol/liter; Table 1). Thus, we subsequently used cultivation method B for the production of OA. In the new cultivation method, the E. coli cells were used like a whole-cell biocatalyst. ΔyggSΔggt-GshA*B cells reached the stationary phase (optical density at 600 nm [OD600], ∼1.8) in 7 h, and the cell density was unchanged by further cultivation (Fig. 5A). A lack of supplementation with 2-AB resulted in a low level of OA production. Culturing of the cells for 24 h without shaking resulted in a low level of production of OA, indicating the importance of the oxygen supply and/or control in OA production. When we used 2.5× M9 medium containing 2.5 mM each Glu, 2-AB, and Gly, the cells produced 377 μmol/liter of OA. In M9 medium containing 5 mM each Glu, 2-AB, and Gly, ΔyggSΔggt-GshA*B cells produced 281 μmol/liter and 331 μmol/liter of OA within 24 h and 64 h, respectively (Table 1). Increasing the concentrations of the amino acids in the medium seemed to increase the production of OA. However, the yield of OA (i.e., the efficiency of OA production) decreased as the concentrations of exogenous amino acids increased (Table 1).
TABLE 1.
Effect of cultivation conditions on OA productiona
| Medium | Cultivation method | Cultivation time (h) | OA concn (μmol/liter) | Yield (%) |
|---|---|---|---|---|
| M9 | A | 88 | 12 ± 5 | |
| M9 plus 1 mM each Glu, 2-AB, and Gly | A | 88 | 212 ± 5 | 21 |
| M9 plus 1 mM each Gln, 2-AB, and Gly | A | 88 | 186 ± 7 | 19 |
| M9 plus 1 mM each Glu, 2-AB, and Gly | B | 24 | 200 ± 3 | 20 |
| M9 plus 1 mM each Glu, 2-AB, and Gly (without shaking) | B | 24 | 39 ± 3 | 3.9 |
| M9 plus 1 mM each Glu and Gly | B | 24 | 10 ± 1 | 1.0 |
| 2.5× M9 plus 2.5 mM each Glu, 2-AB, and Gly | B | 24 | 376 ± 60 | 15 |
| M9 plus 5 mM each Glu, 2-AB, and Gly | B | 24 | 281 ± 36 | 5.6 |
| M9 plus 5 mM each Glu, 2-AB, and Gly | B | 64 | 331 ± 17 | 6.6 |
ΔyggSΔggt-GshA*B cells were grown by cultivation method A or B with M9 medium (or 2.5× M9 medium) containing Cam, IPTG, and the indicated concentrations of amino acids. The total amount of OA in the cultivation system was determined at the indicated cultivation times. Yield indicates the level of OA production based on the concentrations of the added amino acids. All results are expressed as means ± standard deviation of the data obtained from at least 3 independent experiments.
FIG 5.
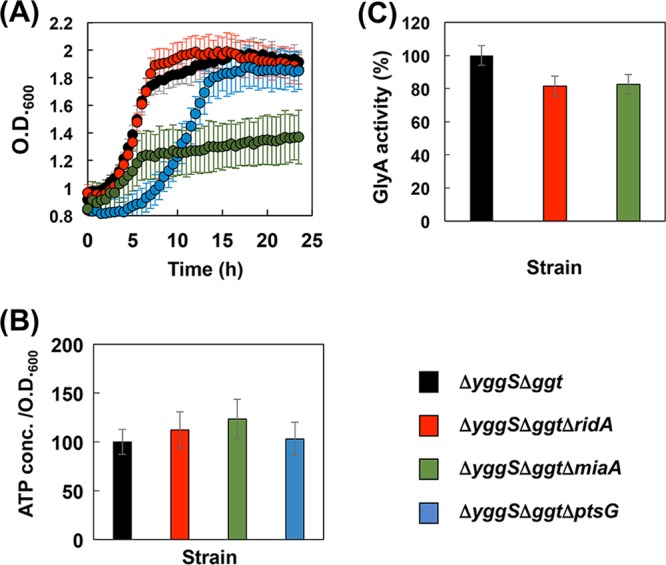
Effect of the ptsG, miaA, or ridA deletion on growth, ATP level, and GlyA activity. (A) The ΔyggS, Δggt, ΔyggS Δggt ΔptsG, ΔyggS Δggt ΔmiaA, or ΔyggS Δggt ΔridA strain harboring pGshA*B was grown for 24 h by cultivation method B with 1 mM each Glu, 2-AB, and Gly. The OD600 of the cultures was monitored every 30 min using an OD-Monitor C&T apparatus (Taitec Co., Ltd.). The ΔyggS Δggt, ΔyggS Δggt ΔptsG, ΔyggS Δggt ΔmiaA, or ΔyggS Δggt ΔridA strain harboring pGshA*B was grown for 4 h by cultivation method B with 1 mM each Glu, 2-AB, and Gly. (B and C) The ATP concentrations in the culture medium (B) and the GlyA activity in the cell extract (C) were determined as described in Materials and Methods. Data are expressed as the means ± standard deviations of the data obtained after 3 repetitions of each assay.
The RidA mutation alleviates the unwanted metabolism of amino acids.
Table 2 shows the concentrations of amino acids after the cultivation of ΔyggSΔggt-GshA*B cells for 24 h by cultivation method B with 1 mM each Glu, 2-AB, and Gly. After the 24-h cultivation period, the system contained significant amounts of OA and 2-AB but less than 10 μmol/liter each of Glu and Gly. This result indicates that approximately 80% of the supplemented Glu and Gly was consumed by pathways other than those involved in OA production. The reduction of this unwanted catabolism of the constituent amino acids will be required to obtain further increases in the production of OA.
TABLE 2.
Concentrations of OA and amino acids after cultivationa
| Component | Concn (μmol/liter) |
|||
|---|---|---|---|---|
| ΔyggS Δggt strain | ΔyggS Δggt ΔridA strain | ΔyggS Δggt ΔptsG strain | ΔyggS Δggt ΔmiaA strain | |
| OA | 200 ± 3 | 231 ± 4 | 182 ± 18 | 277 ± 7 |
| Glu | 8.4 ± 0.2 | 16 ± 3 | 0.6 ± 0.8 | ND |
| 2-AB | 210 ± 24 | 485 ± 29 | 3.4 ± 0.7 | 321 ± 21 |
| Gly | 5.6 ± 0.8 | 525 ± 7 | 6.3 ± 0.7 | 583 ± 11 |
The ΔyggS Δggt, ΔyggS Δggt ΔridA, ΔyggS Δggt ΔptsG, or ΔyggS Δggt ΔmiaA strains, all of which harbored pGshA*B, were grown by cultivation method B with 1 mM each Glu, 2-AB, and Gly. After the 24-h cultivation, the concentrations of OA and amino acids in the cultivation system were determined. Data are expressed as the means ± standard deviations obtained from at least 3 independent experiments. ND, not detected.
In E. coli, Glu is metabolized by several PLP-dependent aminotransferases. Branched-chain amino acid aminotransferase (IlvE) and alanine-valine transaminase metabolize 2-AB, while Gly is degraded by the actions of serine hydroxymethyltransferase (GlyA) and the glycine cleavage system (27, 28). Thus, PLP-dependent enzymes play primary roles in the catabolism of the constituent amino acids of OA.
In this study, we focused on RidA (reactive intermediate/imine deaminase A), which deaminates the 3- and 4-carbon enamines (2-aminoacrylate and 2-aminocapronate, respectively) that are generated as intermediates by the l-Ser/l-Thr dehydratases. In ridA-deficient Salmonella enterica, 2-aminoacrylate inactivates certain PLP enzymes, such as IlvE (29), GlyA (15, 16), and alanine racemase (17), by forming a covalent adduct in the active site. GlyA is suggested to be the most physiologically sensitive target of inactivation by 2-aminoacrylate in S. enterica (16). We hypothesized that the deletion of ridA would lead to the accumulation of 2-aminoacrylate/2-aminocapronate, resulting in the inactivation of various PLP enzymes and the attenuation of the unwanted degradation of amino acids in E. coli cells.
The ΔyggS Δggt ΔridA strain was generated, and the effect on the production of OA was examined. ΔyggSΔggtΔridA-pGshA*B cells produced 231 μmol/liter of OA in M9 medium containing 1 mM each Glu, 2-AB, and Gly, which represented an approximately 15% increase in the level of production compared with that of the parental strain (ΔyggSΔggt-GshA*B). Amino acid analysis revealed that the cultivation medium of the ridA-deficient strain retained a significant amount of Gly after fermentation (525 μmol/liter). The residual amounts of 2-AB or Glu were also slightly increased in the ridA-deficient mutant compared to those in the parental strain (Table 2). These data suggest that the deletion of ridA especially affects the enzyme activities of GlyA, as observed in S. enterica (15, 16). We therefore determined the GlyA activity in the cell extracts of ΔyggSΔggt-GshA*B and ΔyggSΔggtΔridA-GshA*B and confirmed that GlyA activity was ∼20% lower in the cell extract of ΔyggSΔggtΔridA-pGshA*B than that in the cell extract of ΔyggSΔggt-pGshA*B. The inactivation of RidA was a useful strategy for the suppression of Gly catabolism.
Exogenous Ser/Thr may further elevate the production of 2-aminoacrylate/2-aminocrotonate in ridA-deficient cells and further increase the production of OA. We examined this possibility, but no clear correlation between the addition of exogenous Ser/Thr and the production of OA was observed (data not shown).
The deletion of miaA increases the production of OA.
OA is produced by the sequential action of two ATP-requiring enzymes (Fig. 1). The elevation of the availability of ATP probably enables a further increase in the production of OA. Previously, Hara et al. (18) investigated the efficiency of cellular ATP regeneration in permeable E. coli cells using a genome-wide single-gene-deletion E. coli K-12 library (the Keio Collection [30]). They identified several genes to be suppressors of ATP generation, since their deletion resulted in an increase in the rate of cellular ATP synthetic activity (18). Importantly, most of the mutant strains exhibited a higher rate of ATP-driven GSH synthesis than the parental strain (19).
We selected two genes, miaA and ptsG, from the list of genes whose mutation increases ATP-driven GSH synthesis (19) and investigated whether the deletion of those two genes could similarly increase the amount of OA synthesized. The deletion of miaA or ptsG was previously reported to increase total cellular ATP synthetic activities by 371% or 218%, respectively. Moreover, ΔptsG or ΔmiaA cells expressing γ-glutamylcysteine synthase (GSH I) and glutathione synthase (GSH II) exhibited ∼400% more GSH production than the corresponding wild-type cells (19). ptsG encodes a subunit of a glucose phosphotransferase system permease and participates in the transport of glucose (31). miaA encodes dimethylallyl diphosphate:tRNA dimethylallyltransferase, which catalyzes the addition of a 5-carbon isopentenyl moiety to the exocyclic amine of A37 to yield N6-isopentenyladenosine (32, 33).
As shown in Table 2, ΔyggSΔggtΔptsG-GshA*B produced 182 μmol/liter of OA when using cultivation method B, in which the growth medium was supplemented with 1 mM each of the constituent amino acids of OA. This value was slightly lower than that observed for the parental strain (ΔyggSΔggt-GshA*B). The ATP levels in the cultivation system of ΔyggSΔggtΔptsG-GshA*B was comparable to that in the cultivation system of ΔyggSΔggt-GshA*B under the conditions examined (Fig. 5B). In addition, we found that the strains with the ptsG deletion showed significantly lower growth rates and OD600 values in the stationary phase than the parental strain (Fig. 5A). The deletion of ptsG may hamper the glucose uptake of the cells and suppress their ability to consume glucose, and the ptsG mutant cells might consume other nutrients, such as Glu, 2-AB, and Gly, at a higher rate.
Under similar conditions, ΔyggSΔggtΔmiaA-GshA*B cells produced 277 μmol/liter of OA, which was a 39% increase compared with the amount produced by the parental strain (Table 2). The yield of OA on the basis of the amounts of exogenous amino acids added was 28%, which was the highest yield of OA production detected in the present study. The ATP levels in the cultivation system of ΔyggSΔggtΔmiaA-GshA*B were ∼20% higher than those in the cultivation system of ΔyggSΔggt-GshA*B under the cultivation conditions examined (Fig. 5B), which probably contributed to the increase in the level of the OA production in the miaA-deficient strain. Interestingly, we noticed after 24 h of cultivation of ΔyggSΔggtΔmiaA-GshA*B that the culture medium contained substantial amounts of Gly (Table 2). Assays of GlyA activity identified a decrease in GlyA activity in the cell-free extract of ΔyggSΔggtΔmiaA-GshA*B, which probably explains the reduced consumption of Gly in the cultivation system of ΔyggSΔggtΔmiaA-GshA*B. The deletion of miaA is known to affect the expression of several genes, including operons of phenylalanine biosynthesis (34), tryptophanase (35), and phenylalanine-tRNA synthase (36). The expression of glyA may be decreased in the miaA background. In addition, ΔyggSΔggtΔmiaA-GshA*B exhibited slow growth under the cultivation conditions examined (Fig. 5A). All these effects that appeared with the miaA deletion may contribute to the increase in the level of OA production.
OA specifically accumulates in the culture media of the engineered E. coli strains.
Using the four engineered E. coli strains (ΔyggSΔggt-GshA*B, ΔyggSΔggtΔridA-GshA*B, ΔyggSΔggtΔmiaA-GshA*B, and ΔyggSΔggtΔptsG-GshA*B), the OA contents of the culture media and the cells were separately determined. The intracellular concentrations of OA in ΔyggSΔggt-GshA*B, ΔyggSΔggtΔridA-GshA*B, ΔyggSΔggtΔmiaA-GshA*B, and ΔyggSΔggtΔptsG-GshA*B were 5.3, 4.8, 3.5, and 5.0 nmol/mg cell, respectively, indicating that the intracellular concentrations of OA did not significantly differ among these strains. The determination of the OA content in the culture medium confirmed that most of the OA was present in the medium (Fig. 6A). Importantly, as shown in Fig. 6B and C, OA was the major amino acid and/or amine in the medium of the ΔyggSΔggtΔmiaA-GshA*B cells. Similar results were obtained using the other three strains (data not shown). These results indicate that our engineered E. coli system is suitable for the production and purification of OA.
FIG 6.

OA is specifically accumulated in the medium. (A) Ratio of the OA content in cells and medium. The ΔyggS Δggt, ΔyggS Δggt ΔridA, ΔyggS Δggt ΔmiaA, or ΔyggS Δggt ΔptsG strain harboring pGshA*B was grown for 24 h by cultivation method B with 1 mM each Glu, 2-AB, and Gly, and the OA contents in the cells and the medium were separately determined. The data represent the extracellular levels of OA. (B, C) Elution profiles of amino acids in the medium after cultivation. ΔyggS Δggt ΔmiaA-GshA*B was grown by cultivation method B with 1 mM each Glu, 2-AB, and Gly for 24 h (B) or 48 h (C).
Conclusions.
This study reports a method for the production of OA using engineered E. coli, in which E. coli ΔyggS was used as the starting strain. The deletion of ggt, which encodes the enzyme responsible for the degradation of OA, as well as the cooverexpression of the OA synthetic enzymes GshA and GshB and the reduction of the product inhibition ability of GshA by the S495F mutation, all effectively increased the level of production of OA by E. coli. Additionally, we propose a new finding that the inactivation of ridA may be an effective strategy to attenuate the unwanted catabolism of certain amino acids in E. coli. The deletion of miaA may also further elevate the production of OA. The strain developed in the present study specifically produced ∼80 mg/liter of OA in 24 h with a yield of 28%.
MATERIALS AND METHODS
Bacterial strains and media.
The bacterial strains used in this study were generated from Escherichia coli BW25113 (Table 3). The M9 medium consisted of 0.6% Na2HPO4, 0.3% KH2PO4, 0.1% NH4Cl, 0.05% NaCl, 0.2% glucose, 1 mM MgSO4, 10 μM FeSO4, 10 μM CaCl2, and 0.001% thiamine-HCl. The 2.5× M9 medium contained 1.5% Na2HPO4, 0.75% KH2PO4, 0.25% NH4Cl, 0.125% NaCl, 0.5% glucose, 2.5 mM MgSO4, 25 μM FeSO4, 25 μM CaCl2, and 0.025% thiamine-HCl. LB medium was 1% tryptone, 0.5% yeast extract, and 1% NaCl. Solid medium was made by adding 1.5% agar. Antibiotics were added as needed at the following concentrations: kanamycin (Kan), 30 μg/ml; chloramphenicol (Cam), 30 μg/ml; ampicillin (Amp), 100 μg/ml. Isopropyl-β-d-thiogalactoside (IPTG) was added to the M9 medium at a final concentration of 0.1 mM. l-Glutamic acid, dl-2-aminobutyric acid, and glycine were purchased from Wako or Kanto Chemical and supplemented into the M9 medium at the final concentrations indicated above and below.
TABLE 3.
Strains and plasmids used in this study
| Strain or plasmid | Parental strain | Characteristic | Source or reference |
|---|---|---|---|
| Strains | |||
| KRX | [F′ traD36 ΔompP proA+B+ lacIq Δ(lacZ)M15] ΔompT endA1 recA1 gyrA96 (Nalr), thi-1 hsdR17(rK− mK+) e14 (mcrA) relA1 supE44 Δ(lac-proAB) Δ(rhaBAD)::T7 RNA polymerase | Promega | |
| BW25113 | Wild type of E. coli [Δ(araD-araB)567 Δ(rhaD-rhaB)568 ΔlacZ4787 (::rrnB3) hsdR514 rph-1] | Keio Collection | |
| ΔyggS | BW25113 | BW25113 ΔyggS::kan (Keio collection JW2918-KC) | Keio Collection |
| Δggt | BW25113 | BW25113 Δggt::kan (Keio Collection JW3412-KC) | Keio Collection |
| Δggt(kan) | JW3412-KC | BW25113 Δggt | This study |
| ΔyggS Δggt | Δggt(kan) | BW25113 Δggt ΔyggS::kan | This study |
| ΔyggS Δggt-GshA | ΔyggS Δggt | ΔyggS Δggt/pCA24N-gshA | This study |
| ΔyggS Δggt-GshB | ΔyggS Δggt | ΔyggS Δggt/pCA24N-gshB | This study |
| ΔyggSΔggt-GshAB | ΔyggS Δggt | ΔyggS Δggt/pGshAB | This study |
| ΔyggSΔggt-GshA*B | ΔyggS Δggt | ΔyggS Δggt/pGshA*B | This study |
| ΔyggS Δggt(kan) | ΔyggS Δggt | BW25113 ΔyggS Δggt | This study |
| ΔyggS Δggt ΔptsG | ΔyggS Δggt(kan) | BW25113 ΔyggS Δggt ΔptsG::kan | This study |
| ΔyggSΔggtΔptsG-GshA*B | ΔyggS Δggt ΔptsG | ΔyggS Δggt ΔptsG/pGshA*B | This study |
| ΔyggS Δggt ΔridA | ΔyggS Δggt(kan) | BW25113 ΔyggS Δggt ΔridA::kan | This study |
| ΔyggSΔggtΔridA-GshA*B | ΔyggS Δggt ΔridA | ΔyggS Δggt ΔridA/pGshA*B | This study |
| ΔyggS Δggt ΔmiaA | ΔyggS Δggt(kan) | BW25113/ΔyggS Δggt ΔmiaA::kan | This study |
| ΔyggSΔggtΔmiaA-GshA*B | ΔyggS Δggt ΔmiaA | ΔyggS Δggt ΔmiaA/pGshA*B | This study |
| Plasmids | |||
| pCP20 | Expresses FLP recombinase, ori(Tsa) Ampr Camr | 38 | |
| pKD13 | Kanr gene flanking FLP recombination target sequence, Ampr | 37 | |
| pKD46 | Expresses bacteriophage λ Red recombinase by arabinose-inducible promoter, ori(Ts) Ampr | 37 | |
| pCA24N-gshA | Expresses His-tagged GshA (ASKA library, JW2663-AM) | 40 | |
| pCA24N-gshB | Expresses His-tagged GshB (ASKA library, JW2914-AM) | 40 | |
| pCA24N-folD | Expresses His-tagged FolD (ASKA library, JW0518-AM) | 40 | |
| pGshAB | Expresses His-tagged GshA and GshB, PT5-lac promoter | This study | |
| pGshA*B | Expresses His-tagged GshA(S495F) and GshB, PT5-lac promoter | This study |
Ts, temperature sensitive.
Molecular methods.
Deletion of target genes was performed using the bacteriophage λ Red recombinase system described by Datsenko and Wanner (37). For the construction of the ΔyggS Δggt strain, a PCR product was generated with Tks Gflex DNA polymerase (TaKaRa), primers yggS-H1 and yggS-H2 (Table 4), and pKD13 as a template. The purified PCR product (500 ng) was electroporated into the Δggt(kan) strain, which lacks the Kan resistance gene (kan). The Δggt(kan) mutant was generated from the Δggt strain using plasmid pCP20 expressing FLP recombinase (38). The resultant colonies that appeared on LB solid medium were picked up, and the gene knockout was confirmed by direct colony PCR with primers yggS-300up and yggS-200dwn. Deletion of the genes ridA, miaA, and ptsG was performed in a similar way using the ΔyggS Δggt(kan) mutant as the parental strain. The ΔyggS Δggt(kan) strain was generated from the ΔyggS Δggt strain using the pCP20 plasmid.
TABLE 4.
Primers used in this study

The construction of pGshAB and pGshA*B was as follows. The gshB gene from pCA24N-gshB was amplified by PCR with primers pCA24N-gshB-F and pCA24N-gshB-R. The PCR product was digested with SalI and HindIII, gel purified, and ligated into pCA24N-gshA, which had previously been digested with the same restriction enzymes, forming pGshAB. The constructs were introduced into E. coli KRX (Promega), and the sequence of the insert was confirmed by sequence analysis. The gshA gene carried by pGshAB was mutated by site-directed mutagenic PCR using the primers gshA-S495F-F and gshA-S495F-R and KOD FX neo DNA polymerase (Toyobo), forming pGshA*B. The pGshAB in the reaction mixture was eliminated by DpnI treatment. In pGshA*B, the Ser495 codon (TCT) was altered to Phe (TTT).
Cultivation methods. (i) Cultivation method A.
E. coli cells were precultured in LB medium containing Cam at 30°C with shaking (160 rpm) for 12 to 16 h. Cells were collected by centrifugation (3,500 × g, 5 min) and washed twice with phosphate-buffered saline (PBS; 10 mM Na2HPO4, 1.8 mM KH2PO4, 140 mM NaCl, 2.7 mM KCl, pH 7.5). The cell suspension in PBS was seeded into 5 ml M9 medium containing Cam and IPTG at an initial optical density at 600 nm (OD600) of 0.01. The cells were grown at 30°C with shaking (160 rpm) using glass test tubes (16.5 mm in diameter by 165 mm in height) for several days. If required, amino acids were added to the culture media at the concentrations indicated above.
(ii) Cultivation method B.
E. coli cells were precultured in LB medium containing Cam at 30°C with shaking (160 rpm) for 12 to 16 h. Cell pellets were washed twice with PBS and resuspended in PBS. The cell suspension in PBS was seeded into 5 ml M9 medium containing Cam and IPTG at an initial OD600 of 1.0. The cells were cultivated at 30°C with shaking (160 rpm) for 24 h using glass test tubes (16.5 mm in diameter by 165 mm in height). If required, the culture media were supplemented with amino acids at the concentrations indicated above.
Determination of OA and amino acid concentrations.
The concentrations of OA and some amino acids were determined by high-performance liquid chromatography (HPLC) as described previously (5, 13), with slight modification. For analysis of the OA content in the cell culture (cells and medium), medium was collected and the same volume of 10% trichloroacetic acid (TCA) was added. Samples were vortexed for 10 min, incubated for 30 min at 4°C, and centrifuged for 20 min at 20,000 × g. The TCA was removed from the supernatant by extraction three times with water-saturated diethyl ether. Samples were dried with a centrifugal evaporator and resuspended in water. They were diluted with water, and a 10-μl portion was mixed with 25 μl of the o-phthalaldehyde (OPA)–N-acetyl-l-cysteine (NAC)–borate reagent (a 1:9 mixture of the NAC-OPA reagent and 0.4 M borate buffer [pH 9.5]). The NAC-OPA reagent contains 10 mg of NAC and 20 mg of OPA in 1 ml methanol. After incubation for 15 min at 4°C, 215 μl of distilled water was added. The mixture was centrifuged for 15 min at 20,000 × g at 4°C, and the supernatant (15 μl) was analyzed using a Shimadzu HPLC system. A Mightysil RP-18 GPII column (4.6 [inside diameter {i.d.}] by 250 mm; particle size, 5 μm; Kanto Chemical) and a Mightysil RP-18 GPII guard column (4.6 by 5 mm [i.d.]; particle size, 5 μm; Kanto Chemical) preequilibrated with mobile phase A (10 mM Na2HPO4, 10% methanol, pH 6.5) were used for the analyses. The columns were maintained at 24°C, and the flow rate was 0.8 ml/min. Separation was achieved by increasing the concentration of mobile phase B (10 mM Na2HPO4, 60% methanol, pH 6.5) in mobile phase A from 0% to 30% over 34 min. Fluorescence was monitored with a Shimadzu RF-20A fluorescence detector (excitation wavelength, 340 nm; emission wavelength, 450 nm). The ophthalmic acid standard was purchased from PH Japan.
Determination of GshA and GlyA activities.
ΔyggSΔggt-GshAB, ΔyggSΔggt-GshA*B, ΔyggS ΔggtΔridA-GshA*B, or ΔyggS ΔggtΔmiaA-GshA*B cells were cultivated for 4 h by cultivation method B with 1 mM each Glu, 2-AB, and Gly. The cells were collected by centrifugation (3,500 × g, 5 min) and washed once with PBS. The cell pellet was stored at −80°C until use.
(i) GshA activity.
The cell pellet was suspended in a buffer containing 50 mM Tris-HCl, 5 mM MgCl, and 20% glycerol, pH 7.4, and disrupted by sonication. The cell debris was removed by centrifugation (20 min, 20,000 × g, 4°C), and the supernatants were used for analyses. The Glu–2-AB or Glu-Cys ligase activity was determined at 30°C by a coupling assay with pyruvate kinase and lactic dehydrogenase as described previously (24). The reaction mixture (1 ml) was 100 mM Tris-HCl (pH 8.2), 1 mM each Glu, 1 mM 2-AB, or Cys, 20 mM MgCl2, 5 mM ATP, 2 mM phosphoenol pyruvate, 150 mM KCl, 0.3 mM NADH, 9.4 U pyruvate kinase, 10 U lactic dehydrogenase, and the cell extract. When required, the reduced form of GSH was added at a final concentration of 10 mM. Protein concentrations were determined using a Bio-Rad protein assay with bovine serum albumin as the standard.
(ii) GlyA activity.
GlyA activity was determined as described by Schirch and Gross (39), with slight modification. The cell pellet was suspended in PBS containing 10% glycerol, sonicated, and centrifuged (20,000 × g, 4°C) for 30 min. The resultant cell extract (50 μl) was added to a reaction mixture (1 ml) containing 50 mM HEPES-NaOH, pH 7.5, 0.2 mM tetrahydrofolate, 0.2 mM NADP+, 5 mM l-Ser, 20 μM PLP, 0.1% 2-mercaptoethanol and incubated at 37°C for 10 min. After termination of the reaction by adding 100 μl of 1 M K2CO3 (pH 9.5), purified FolD (2 mg/ml, 10 μl) was added to the reaction mixture, and the change in the absorbance at 340 nm was determined. FolD was purified from the E. coli cells harboring pCA24N-folD by Ni-affinity chromatography and stored in a buffer consisting of 12.5 mM Tris-HCl, pH 7.5, 75 mM KCl, 12.5 mM 2-mercaptoethanol, and 50% glycerol at −80°C until it was used.
Determination of ATP levels.
ΔyggSΔggt-GshA*B, ΔyggSΔggtΔridA-GshA*B, ΔyggSΔggtΔmiaA-GshA*B, and ΔyggSΔggtΔptsG-GshA*B cells were cultivated for 4 h by cultivation method B with 1 mM each Glu, 2-AB, and Gly. Twelve microliters of the culture medium (cells and medium) was withdrawn, and ATP levels were measured by use of a Lucifer 250 kit (Kikkoman Co., Japan) according to the manufacturer's protocol.
ACKNOWLEDGMENTS
This work was supported in part by grants from the Mishima Kaiun Foundation, an NISR young investigator research grant from the Noda Institute for Scientific Research, and JSPS KAKENHI grant number 16K18686 (to T.I.).
REFERENCES
- 1.Waley SG. 1958. Acidic peptides of the lens. 3. The structure of ophthalmic acid. Biochem J 68:189–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsuboi S, Hirota K, Ogata K, Ohmori S. 1984. Ophthalmic and norophthalmic acid in lens, liver, and brain of higher animals. Anal Biochem 136:520–524. doi: 10.1016/0003-2697(84)90255-0. [DOI] [PubMed] [Google Scholar]
- 3.Sajiki K, Pluskal T, Shimanuki M, Yanagida M. 2013. Metabolomic analysis of fission yeast at the onset of nitrogen starvation. Metabolites 3:1118–1129. doi: 10.3390/metabo3041118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Narainsamy K, Farci S, Braun E, Junot C, Cassier-Chauvat C, Chauvat F. 2016. Oxidative-stress detoxification and signalling in cyanobacteria: the crucial glutathione synthesis pathway supports the production of ergothioneine and ophthalmate. Mol Microbiol 100:15–24. doi: 10.1111/mmi.13296. [DOI] [PubMed] [Google Scholar]
- 5.Ito T, Yamauchi A, Hemmi H, Yoshimura T. 2016. Ophthalmic acid accumulation in an Escherichia coli mutant lacking the conserved pyridoxal 5′-phosphate-binding protein YggS. J Biosci Bioeng 122:689–693. doi: 10.1016/j.jbiosc.2016.06.010. [DOI] [PubMed] [Google Scholar]
- 6.Herson PS, Ashford ML. 1999. Reduced glutathione inhibits beta-NAD+-activated non-selective cation currents in the CRI-G1 rat insulin-secreting cell line. J Physiol 514:47–57. doi: 10.1111/j.1469-7793.1999.047af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leslie EM, Ito K, Upadhyaya P, Hecht SS, Deeley RG, Cole SP. 2001. Transport of the beta-O-glucuronide conjugate of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) by the multidrug resistance protein 1 (MRP1). Requirement for glutathione or a non-sulfur-containing analog. J Biol Chem 276:27846–27854. [DOI] [PubMed] [Google Scholar]
- 8.Offord RE, Philippe J, Davis JG, Halban PA, Berger M. 1979. Inhibition of degradation of insulin by ophthalamic acid and by a bovine pancreatic proteinase inhibitor. Biochem J 182:249–251. doi: 10.1042/bj1820249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cliffe EE, Waley SG. 1961. The mechanism of the glyoxalase I reaction, and the effect of ophthalmic acid as an inhibitor. Biochem J 79:475–482. doi: 10.1042/bj0790475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soga T, Baran R, Suematsu M, Ueno Y, Ikeda S, Sakurakawa T, Kakazu Y, Ishikawa T, Robert M, Nishioka T, Tomita M. 2006. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J Biol Chem 281:16768–16776. doi: 10.1074/jbc.M601876200. [DOI] [PubMed] [Google Scholar]
- 11.Ohsu T, Amino Y, Nagasaki H, Yamanaka T, Takeshita S, Hatanaka T, Maruyama Y, Miyamura N, Eto Y. 2010. Involvement of the calcium-sensing receptor in human taste perception. J Biol Chem 285:1016–1022. doi: 10.1074/jbc.M109.029165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuroda M, Kato Y, Yamazaki J, Kai Y, Mizukoshi T, Miyano H, Eto Y. 2013. Determination and quantification of the kokumi peptide, γ-glutamyl-valyl-glycine, in commercial soy sauces. Food Chem 141:823–828. doi: 10.1016/j.foodchem.2013.03.070. [DOI] [PubMed] [Google Scholar]
- 13.Ito T, Iimori J, Takayama S, Moriyama A, Yamauchi A, Hemmi H, Yoshimura T. 2013. Conserved pyridoxal protein that regulates Ile and Val metabolism. J Bacteriol 195:5439–5449. doi: 10.1128/JB.00593-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prunetti L, El Yacoubi B, Schiavon CR, Kirkpatrick E, Huang L, Bailly M, ElBadawi-Sidhu M, Harrison K, Gregory JF, Fiehn O, Hanson AD, de Crécy-Lagard V. 12 February 2016. Evidence that COG0325 proteins are involved in PLP homeostasis. Microbiology doi: 10.1099/mic.0.000255. [DOI] [PubMed] [Google Scholar]
- 15.Flynn JM, Christopherson MR, Downs DM. 2013. Decreased coenzyme A levels in ridA mutant strains of Salmonella enterica result from inactivated serine hydroxymethyltransferase. Mol Microbiol 89:751–759. doi: 10.1111/mmi.12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ernst DC, Downs DM. 2015. 2-Aminoacrylate stress induces a context-dependent glycine requirement in ridA strains of Salmonella enterica. J Bacteriol 198:536–543. doi: 10.1128/JB.00804-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flynn JM, Downs DM. 2013. In the absence of RidA, endogenous 2-aminoacrylate inactivates alanine racemases by modifying the pyridoxal 5′-phosphate cofactor. J Bacteriol 195:3603–3609. doi: 10.1128/JB.00463-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hara KY, Shimodate N, Ito M, Baba T, Mori H, Mori H. 2009. Systematic genome-wide scanning for genes involved in ATP generation in Escherichia coli. Metab Eng 11:1–7. doi: 10.1016/j.ymben.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Hara KY, Shimodate N, Hirokawa Y, Ito M, Baba T, Mori H, Mori H. 2009. Glutathione production by efficient ATP-regenerating Escherichia coli mutants. FEMS Microbiol Lett 297:217–224. doi: 10.1111/j.1574-6968.2009.01682.x. [DOI] [PubMed] [Google Scholar]
- 20.Lin J, Liao X, Zhang J, Du G, Chen J. 2009. Enhancement of glutathione production with a tripeptidase-deficient recombinant Escherichia coli. J Ind Microbiol Biotechnol 36:1447–1452. doi: 10.1007/s10295-009-0631-y. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Quan C, Wang C, Wu H, Li Z, Ye Q. 2016. Systematic manipulation of glutathione metabolism in Escherichia coli for improved glutathione production. Microb Cell Fact 15:38. doi: 10.1186/s12934-016-0439-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Apontoweil P, Berends W. 1975. Glutathione biosynthesis in Escherichia coli K-12. Properties of the enzymes and regulation. Biochim Biophys Acta 399:1–9. doi: 10.1016/0304-4165(75)90205-6. [DOI] [PubMed] [Google Scholar]
- 23.Janowiak BE, Hayward MA, Peterson FC, Volkman BF, Griffith OW. 2006. Gamma-glutamylcysteine synthetase-glutathione synthetase: domain structure and identification of residues important in substrate and glutathione binding. Biochemistry 45:10461–10473. doi: 10.1021/bi052483v. [DOI] [PubMed] [Google Scholar]
- 24.Huang CS, Moore WR, Meister A. 1988. On the active site thiol of gamma-glutamylcysteine synthetase: relationships to catalysis, inhibition, and regulation. Proc Natl Acad Sci U S A 85:2464–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murata K, Kimura A. 1982. Cloning of a gene responsible for the biosynthesis of glutathione in Escherichia coli B. Appl Environ Microbiol 44:1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwak J-H, Nam Y-S, Lee S-Y. 1998. Site-specific mutagenesis of the gshI gene for increasing the activity of γ-glutamylcysteine synthetase in Escherichia coli K-12. BMB Rep 31:254–257. [Google Scholar]
- 27.Newman EB, Batist G, Fraser J, Isenberg S, Weyman P, Kapoor V. 1976. The use of glycine as nitrogen source by Escherichia coli K12. Biochim Biophys Acta 421:97–105. doi: 10.1016/0304-4165(76)90173-2. [DOI] [PubMed] [Google Scholar]
- 28.Plamann MD, Rapp WD, Stauffer GV. 1983. Escherichia coli K12 mutants defective in the glycine cleavage enzyme system. Mol Gen Genet 192:15–20. doi: 10.1007/BF00327641. [DOI] [PubMed] [Google Scholar]
- 29.Schmitz G, Downs DM. 2004. Reduced transaminase B (IlvE) activity caused by the lack of yjgF is dependent on the status of threonine deaminase (IlvA) in Salmonella enterica serovar Typhimurium. J Bacteriol 186:803–810. doi: 10.1128/JB.186.3.803-810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio Collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meins M, Zanolari B, Rosenbusch JP, Erni B. 1988. Glucose permease of Escherichia coli. Purification of the IIGlc subunit and functional characterization of its oligomeric forms. J Biol Chem 263:12986–12993. [PubMed] [Google Scholar]
- 32.Bartz JK, Kline LK, Söll D. 1970. N6-(Delta 2-isopentenyl)adenosine: biosynthesis in vitro in transfer RNA by an enzyme purified from Escherichia coli. Biochem Biophys Res Commun 40:1481–1487. doi: 10.1016/0006-291X(70)90035-5. [DOI] [PubMed] [Google Scholar]
- 33.Rosenbaum N, Gefter ML. 1972. Delta 2-isopentenylpyrophosphate: transfer ribonucleic acid 2-isopentenyltransferase from Escherichia coli. Purification and properties of the enzyme. J Biol Chem 247:5675–5680. [PubMed] [Google Scholar]
- 34.Gowrishankar J, Pittard J. 1982. Regulation of phenylalanine biosynthesis in Escherichia coli K-12: control of transcription of the pheA operon. J Bacteriol 150:1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gollnick P, Yanofsky C. 1990. tRNA(Trp) translation of leader peptide codon 12 and other factors that regulate expression of the tryptophanase operon. J Bacteriol 172:3100–3107. doi: 10.1128/jb.172.6.3100-3107.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Springer M, Trudel M, Graffe M, Plumbridge J, Fayat G, Mayaux JF, Sacerdot C, Blanquet S, Grunberg-Manago M. 1983. Escherichia coli phenylalanyl-tRNA synthetase operon is controlled by attenuation in vivo. J Mol Biol 171:263–279. doi: 10.1016/0022-2836(83)90093-1. [DOI] [PubMed] [Google Scholar]
- 37.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. doi: 10.1016/0378-1119(95)00193-A. [DOI] [PubMed] [Google Scholar]
- 39.Schirch L, Gross T. 1968. Serine transhydroxymethylase. Identification as the threonine and allothreonine aldolases. J Biol Chem 243:5651–5655. [PubMed] [Google Scholar]
- 40.Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. 2005. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res 12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]