

Abstract
Impaired blood flow to peripheral tissues during advanced age is associated with endothelial dysfunction and diminished bioavailability of nitric oxide (NO). However, it is unknown whether aging impacts coupling between intracellular calcium ([Ca2+]i) signaling and small- and intermediate K+ channel (SKCa/IKCa) activity during endothelium-derived hyperpolarization (EDH), a signaling pathway integral to dilation of the resistance vasculature. To address the potential impact of aging on EDH, Fura-2 photometry and intracellular recording were applied to evaluate [Ca2+]i and membrane potential of intact endothelial tubes (width, 60 µm; length, 1–3 mm) freshly isolated from superior epigastric arteries of young (4–6 mo) and old (24–26 mo) male C57BL/6 mice. In response to acetylcholine, intracellular release of Ca2+ from the endoplasmic reticulum (ER) was enhanced with aging. Further, treatment with the mitochondrial uncoupler FCCP evoked a significant increase of [Ca2+]i with membrane hyperpolarization in an SKCa/IKCa-dependent manner in the endothelium of old but not young mice. We conclude that the ability of resistance artery endothelium to release Ca2+ from intracellular stores (ie, ER and mitochondria) and hyperpolarize Vm via SKCa/IKCa activation is augmented as compensation for reduced NO bioavailability during advanced age.
Keywords: Artery, Cardiovascular, Endothelial Cell, Mitochondria, Biology of Aging
Gerontologists predict that individuals of advanced age (≥65 years) will compose approximately 25% of the United States population by 2030 (1). Aging is the key risk factor for cardiovascular disease (2,3) with endothelial dysfunction a prime indicator (4,5). This disorder is typically manifested by impaired vasodilation to acetylcholine (ACh) (6,7) or the restriction of blood flow during physical activity (6,8). Nevertheless, recent findings indicate that aging may be beneficial to endothelial cell (EC) survival (9). In the present study, our goal was to determine whether, and if so, how the endothelium of resistance arteries may adapt to maintain its role in effecting vasodilation during advanced age.
During the local control of tissue blood flow, vasodilation is coordinated along the branches of resistance networks, including the proximal feed arteries and the arteriolar networks they supply (10). An integral mechanism of vasodilation in the resistance vasculature entails endothelium-derived hyperpolarization, “EDH” (11,12). Thus, in response to a rise in intracellular Ca2+ concentration ([Ca2+]i), the opening of small- and intermediate- calcium-activated K+ channels (SKCa/IKCa) in ECs generates hyperpolarization, which spreads from cell to cell through gap junctions between ECs and into surrounding smooth muscle cells (SMCs) through myoendothelial gap junctions (13,14). In resistance arteries, the endothelium serves as the principal cellular pathway for electrical signal transmission (15–18) which can be modulated by affecting gap junctions coupling neighboring cells or altering the activity of ion channels expressed in the plasma membrane (11,19). In contrast to the lack of selective pharmacological interventions for gap junctions (20,21), SKCa/IKCa are effective targets for pharmacological treatment of endothelial dysfunction (22–25). Whereas advanced age diminishes blood flow to key organs and tissue in association with a decrease in the bioavailability of nitric oxide (NO) (26,27), little is known of how the aging process impacts coupling between [Ca2+]i signaling and SKCa/IKCa activity underlying EDH. Because these signaling events effect vasodilation in the small resistance arteries that control tissue blood flow (28,29), greater insight is needed to understand how EDH may be affected during advanced age, where tissue perfusion may be compromised.
The principal organelles that govern the intracellular storage, sequestration, and release of [Ca2+]i are the endoplasmic reticulum (ER) and mitochondrion (30–32). Muscarinic receptor (M3) stimulation (eg, by ACh) increases the availability of cytoplasmic inositol trisphosphate (IP3), thereby triggering a rise in [Ca2+]i via the activation of inositol 1,4, trisphosphate receptors (IP3Rs) in the ER (12,33). The rise in [Ca2+]i is sustained by the influx of extracellular Ca2+ through open transient receptor potential channels (33–35) and this response is enhanced during advanced age (9). Because ECs rely primarily on glycolysis rather than oxidative phosphorylation for generating adenosine triphosphate (ATP) (36,37), key roles for endothelial mitochondria entail the regulation of [Ca2+]i and the production of reactive oxygen species (31,38). Moreover, the role of mitochondria in buffering [Ca2+]i can increase with advanced age, as shown for intestinal SMCs (39) and peripheral sympathetic neurons (40). In turn, release of Ca2+ from mitochondria effects membrane hyperpolarization via SKCa/IKCa activation and these ion channels are highly expressed in the endothelium of resistance arteries (41). The proton ionophore carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) dissipates the inner mitochondria membrane potential (≈ −180 mV relative to cytosol) with consequences of releasing mitochondrial Ca2+ (42–47) leading to activation of KCa channels (46). Further, the ability of FCCP to uncouple mitochondria and depolarize the inner mitochondrial membrane potential (∆Ψmt; 42–44) may also reflect the physiological actions of uncoupling proteins (48) and mitochondrial K+ channels (49,50). Thus, FCCP may be used as a tool to evaluate the role of mitochondria as a Ca2+ source for governing membrane potential (Vm) of ECs. However, the respective roles of ER and mitochondria in shaping [Ca2+]i signaling and activating endothelial SKCa/IKCa to evoke hyperpolarization has received little attention.
The effect of aging and associated diseases (eg, obesity, type II diabetes, hypertension) on SKCa/IKCa function in generating EDH-dependent vasodilation during aging lacks consensus. Some have suggested a loss of SKCa/IKCa-dependent vasodilation (25,51–53), whereas others have observed enhanced function of SKCa/IKCa in both intact arteries (54–60) and freshly isolated endothelium (41). The latter finding supports the hypothesis that an increase in EDH signaling serves as a compensatory mechanism in response to diminished NO bioavailability for endothelium-dependent vasodilation (61,62). By eliminating external influences (eg, perivascular nerves, SMCs, blood flow, hormones), the study of intact, freshly isolated endothelium from resistance arteries provides a powerful experimental approach to evaluate how aging affects the intrinsic ability of the ER and mitochondria within ECs to intrinsically activate SKCa/IKCa and to thereby engage in transducing [Ca2+]i dynamics into changes in Vm.
In accord with impaired tissue blood flow during cardiovascular aging (27,61,63), we questioned how EDH as a vasodilator signal was affected by advanced age. A specific aim of this study was to resolve the interplay between mitochondria-associated [Ca2+]i signaling and hyperpolarization in endothelium freshly isolated from resistance arteries of mouse skeletal muscle with advanced age. Using mice at ages that correspond to humans in their early 20’s and mid-60s (64), we tested the hypothesis that enhanced SKCa/IKCa activation effected through increased [Ca2+]i signaling following mitochondrial uncoupling is integral to EDH during advanced age.
Methods
Animal Care and Use
All animal care and experimental procedures were approved by the Animal Care and Use Committee of the University of Missouri and performed in accord with the National Research Council’s Guide for the Care and Use of Laboratory Animals (8th ed., 2011). Experiments were performed on young (4–6 months; n = 31) and old (24–26 months; n = 43) male C57BL/6 mice obtained from the National Institute on Aging (NIA) colonies at Charles River Laboratories (Wilmington, MA). Prior to use in experiments, mice were housed at the University Missouri for at least one week on a 12 hours: 12 hours light: dark cycle at approximately 23°C with fresh tap water and standard chow available ad libitum. On the morning of an experiment, a mouse was anesthetized using pentobarbital sodium (60 mg/kg, intraperitoneal injection) and abdominal fur was removed by shaving. Following removal of tissues for experiments, the anesthetized mouse was killed by exsanguination.
Surgery and Microdissection
A ventral midline incision was made through the skin from the sternum to the pubis to expose the abdominal musculature. While viewing through a stereo microscope (SMZ800, Nikon; Tokyo, Japan), fat and connective tissue superficial to the sternum were removed to expose the proximal end of superior epigastric artery (SEA) bilaterally; each SEA was ligated together with its adjacent vein (6-0 silk suture; Ethicon; Somerville, NJ) to maintain blood in the lumen and thereby facilitate visualization during dissection. The abdominal musculature was removed from the mouse and each half was pinned onto transparent silicone rubber (Sylgard 184, Dow Corning; Midland, MI) in physiological salt solution (PSS) maintained at 4°C. Each artery was dissected from its proximal end to the first branch point (segment length: ~2 cm) and then cannulated at one end to flush blood the vessel lumen with PSS. Cannulae (tip outer diameter (OD), 50–80 µm) were made from heat-polished borosilicate glass capillaries (G150T-4, Warner Instruments; Hamden, CT).
Solutions
Control PSS contained the following: (in mmol/L): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 N-2-Hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 10 glucose. For “0 [Ca2+]o”, CaCl2 was eliminated and EGTA (10–3 M) was added to sequester any free Ca2+ in the PSS (65). The pH of all solutions was adjusted to 7.4 using NaOH prior to use. During vessel dissection, CaCl2 was absent from the PSS to relax SMCs. During dissociation of SMCs to obtain endothelial tubes (hereafter referred to as “tubes”, which are effectively collapsed under these conditions), PSS contained 0.62 mg/mL papain (≥6 units), 1.5 mg/mL collagenase (≥15 units), 1.0 mg/mL dithioerythritol, 0.1% bovine serum albumin (USB Corp.; Cleveland, OH), and 0.1 mmol/L CaCl2. Reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless indicated otherwise.
Endothelial Tube Isolation and Superfusion
Endothelial tubes were prepared as described (11,66). Briefly, each SEA was cut into segments 3–5 mm long, incubated in dissociation PSS for 30 minutes at 37°C, then transferred to a tissue chamber (RC-27N, Warner) containing dissociation PSS at room temperature. To dissociate SMCs, a vessel segment was gently triturated using aspiration and ejection from a micropipette during visual inspection at 200X. Dissociation pipettes were prepared from borosilicate glass capillary tubes [1.0 mm OD/0.58 mm ID; World Precision Instruments (WPI), Sarasota, FL] that were pulled (P-97; Sutter Instruments; Novato, CA) and heat-polished at one end (tip internal diameter: 80–120 µm). Following dissociation, the tissue chamber containing an endothelial tube (width: ~60 µm, length: ≤1 mm) was secured to an aluminum platform (width: 14.5 cm, length: 24 cm, thickness: 0.4 cm). A micromanipulator (DT3-100, Siskiyou Corp.; Grants Pass, OR) mounted at each end of the platform held a blunt ended heat-polished micropipette (OD, 60–100 µm) that was used to position and secure the tube against the bottom (coverslip) of the tissue chamber. The tube was superfused at approximately 4 mL/min with control PSS; flow through the chamber was parallel to the axis of the tube. The aluminum platform was mounted on an inverted microscope (Eclipse TS100, Nikon) positioned on a vibration-isolated table (Technical Manufacturing Corp., Peabody, MA). Throughout experiments, temperature of the chamber was maintained at 32°C using an in-line heater (SH-27B, Warner) and heating platform (PH6, Warner) coupled to a temperature controller (TC-344B, Warner). These preparations are stable for about 5 hours (33,66) and the present experiments were typically completed within 2 hours.
Dye Tracking of the ER and Mitochondria
Mitochondria and ER were labeled simultaneously in endothelial tubes using MitoTracker Deep Red (Excitation: 644/Emission: 665) and ER Tracker Green (Excitation: 504/Emission: 511), respectively, according to the manufacturer’s instructions (Molecular Probes, Eugene, OR). Briefly, Mitotracker Deep Red (10–7 M) and ER tracker (10–7 M) were applied to freshly isolated tubes for 30 minutes at 37°C before images were captured using a 63× glycerol immersion objective (numerical aperture = 1.3) on a Leica SP5 confocal microscope using LAS Software (Leica Microsystems, Wetzlar, Germany).
Ca2+ Photometry
Ca2+ photometry was performed using an IonOptix system (Milford, MA) as described (33,34,67). Briefly, prior to loading Fura-2 dye, the endothelial tube was maintained at room temperature for 10 minutes, whereas autofluorescence was recorded at 510 nm during alternate excitation at 340 and 380 nm (10 Hz). Fura-2 AM dye (5 µM; F14185, Life Technologies, Eugene, OR) was loaded for 20 minutes followed by 20 minutes of washout to allow for intracellular de-esterification. Temperature was raised to 32°C during the final 10 minutes. Autofluorescence during excitation at 340 and 380 nm (average values over 30 s acquisition) were subtracted from respective recordings at 510 nm. The imaging window was 140 µm × 50 µm using a 40X objective (Nikon S Fluor; numerical aperture, 0.90) and encompassed about 50 ECs (34).
Intracellular Recording and Current Microinjection
Microelectrodes were pulled (P-97; Sutter) from glass capillary tubes (GC100F-10, Warner) and backfilled with 2 mol/L KCl (tip resistance, ~150 MΩ). Membrane potential of ECs was measured using an Axoclamp 2B electrometer (Molecular Devices; Sunnyvale, CA) coupled to a function generator (CFG253, Tektronix; Beaverton, OR) and an IE-210 amplifier (Warner). An Ag/AgCl pellet placed in effluent PSS served as a reference electrode. Amplifier outputs were connected to an analog-to-digital converter (Digidata 1322A, Molecular Devices) and data were recorded at 1,000 Hz on a Dell personal computer using Axoscope 10.1 software (Molecular Devices). Individual ECs were penetrated along the midline of the endothelial tube while viewing at 400X magnification (11,34,41). For simultaneous Ca2+ photometry and electrophysiology, the photometric window for acquiring Ca2+ measurements was positioned adjacent to the recording electrode (34).
Pharmacology
To examine the effect of ER Ca2+ release and Ca2+ influx underlying hyperpolarization with advancing age, 3 × 10–6 M ACh was applied for 2 minutes, which we have shown to evoke maximum hyperpolarization and peak [Ca2+]i in preparations of SEA endothelium identical to those studied here (9,33,41). As the role of mitochondrial Ca2+ release and consequential changes in Vm evoked by the mitochondrial uncoupler FCCP have not been tested in the intact endothelium, we evaluated full concentration-response relationships for [Ca2+]i and Vm in response to a cumulative increase in [FCCP], from 10–8 M to 10–5 M. Responses were allowed to stabilize for 3 minutes at each [FCCP]. Based on these data, ensuing experiments were performed using 3 × 10–7 M (submaximal) and 10–6 M (maximal) [FCCP] as these concentrations revealed differences in [Ca2+]i and Vm responses between young vs. old mice. Values for summary data were collected during the peak of F340/F380 and Vm responses. During exposure to 3 × 10–7 M FCCP, 3 × 10–7 M apamin + 10–7 M charybdotoxin were applied to block SKCa and IKCa, respectively, to evaluate the role of respective ion channels in EC hyperpolarization.
Data Analysis
Analyses included: (a) Fura-2 fluorescence emission collected at 510 nm and expressed as the ratio during excitation at 340 nm and 380 nm (F340/F380); (b) change in F340/F380 ratio (∆F340/F380) = peak response F340/F380 − preceding baseline F340/F380; (c) resting baseline Vm (mV); (d) change in Vm (ΔVm) = peak response Vm − preceding baseline Vm. Summary data reflect values averaged over 10 seconds during stable recordings. Statistical analyses (GraphPad Software, Inc.; La Jolla, CA) included paired and unpaired Student’s t-tests, one-way and two-way repeated measures analysis of variance, with Tukey and Bonferroni post-hoc comparisons, respectively. Differences were accepted as statistically significant with p <.05. Summary data are presented as means ± standard error. Values for n reflects the number of independent endothelial tubes each studied from a different mouse for a given experimental protocol.
Results
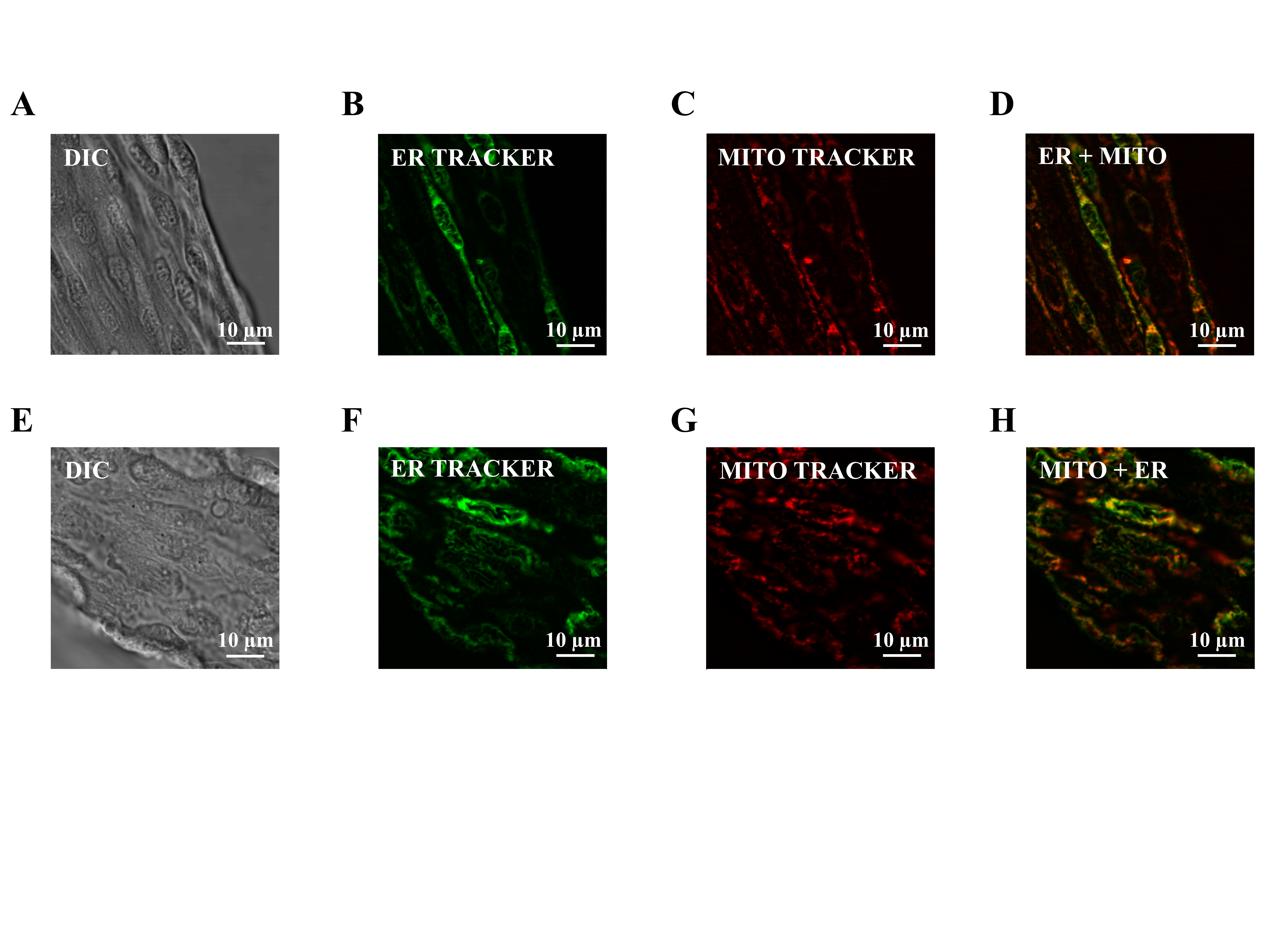
Both ER and mitochondria are present in the endothelium of young and old mice (see Supplementary Figure 1). How Ca2+ and electrical signaling are shaped by respective organelles to initiate EDH is unknown, particularly in the context of advancing age. In the present experiments, we tested how Ca2+ release from the ER or from the mitochondria affected Vm through SKCa/IKCa activation in endothelium of resistance arteries from young and old mice.
Aging Enhances ER-Dependent [Ca2+]i Increases and Hyperpolarization
As illustrated in Figures 1A and 1B (left panels), from resting baseline in the presence of 2 mM extracellular Ca2+ concentration ([Ca2+]o), ACh evoked an initial “peak” rise in F340/F380 attributable to release of Ca2+ from the ER followed by a sustained “plateau” attributable to Ca2+ influx (35,65). Resting values and peak F340/F380 responses to ACh were similar between age groups (Figures 1C and D). Pretreatment with 0 [Ca2+]o PSS containing EGTA (10–3 M) for 1 minute prior to ACh application eliminated the plateau phase leaving only the initial peak phase of the [Ca2+]i response (Figures 1A and B, right panels). A trend for greater plateau [Ca2+]i was observed in old vs. young endothelium under control conditions and was statistically significant at 30 seconds and 60 seconds (≥1.5-fold for ∆F340/F380) following the peak phase of the [Ca2+]i response to ACh during removal of extracellular Ca2+ (Figure 1D). The peak phase of either age group was not altered significantly during 0 [Ca2+]o PSS (Figures 1C and D). The kinetics of Vm in response to ACh mirrored those of [Ca2+]i (Figures 2A and B, left panels). Thus, hyperpolarization exhibited both a peak phase and a plateau phase under control conditions, whereas Vm returned rapidly to resting levels during 0 [Ca2+]o despite sustained exposure to ACh (Figures 2A and B, right panels). However, under control conditions, Vm at rest and in response to ACh were significantly greater in old vs. young endothelium (p < .05) and this difference between age groups persisted during the intermediate period (30 s and 60 s) of the plateau phase during 0 [Ca2+]o PSS (Figure 2C). Though not consistently reaching statistical significance, similar differences between age groups were apparent for the changes in Vm from respective control baselines (Figure 2D). These data illustrate that the rise in [Ca2+]i and hyperpolarization to ACh are enhanced during aging.
Figure 1.

Aging enhances ER-dependent [Ca2+]i increases in response to ACh. Fura-2 recordings of [Ca2+]i in response to 3 × 10–6 M ACh in the presence of 2 × 10–3 M [Ca2+]o (Control, “C”) and with 0 [Ca2+]o in A, Young and B, Old endothelium. C, Summary data for F340/F380 and D, ΔF340/F380 at designated time points (gray dots; refer to traces in A and B). Note initial peak of [Ca2+]i transients (reflecting Ca2+ release from ER) followed by sustained plateau (reflecting Ca2+ entry through the plasma membrane) during 90 s following peak response. Note diminished plateau during 0 [Ca2+]o to eliminate Ca2+ entry (A and B, right vs. left panels). Peak [Ca2+]i responses were similar across groups irrespective of [Ca2+]o (C and D). However, in Old endothelium, increases in [Ca2+]i remain elevated at 30 s and 60 s with 0 [Ca2+]o (D). *p < .05, old vs. young, n = 10 per group. ACh = acetylcholine; ER = endoplasmic reticulum.
Figure 2.

Aging enhances hyperpolarization in response to ACh. Vm recordings (obtained simultaneously with Fura-2 data in Figure 2) in response to 3 × 10–6 M ACh in the presence of 2 × 10–3 M [Ca2+]o (Control, “C”) and with 0 [Ca2+]o in A, Young and B, Old endothelium. Summary data indicate C, Vm and D, ΔVm (treatment − control) for respective age groups. Note initial “peak” of hyperpolarization followed by sustained “plateau” through 90 s (A and B, left panels). Defined time points indicated by gray dots in panels A and B. Note diminished plateau during 0 [Ca2+]o to eliminate Ca2+ entry (A and B, right vs. left panels)]. Peak Vm responses were greater (p < .05) in old vs. young in the presence of 2 × 10–3 M [Ca2+]o (panels C and D). Further, Vm remained hyperpolarized (*p < .05) in old vs. young at 30 s and 60 s during 0 [Ca2+]o, reflecting sustained internal release of Ca2+ (panel D). Traces for Figures 1A and 2A (Young) and Figures 1B and 2B (Old) represent simultaneous [Ca2+]i and electrical measurements. *p < .05, old vs. young, n = 10 per group. ACh = acetylcholine.
Aging Enhances [Ca2+]i and Membrane Hyperpolarization Responses to Mitochondrial Uncoupling
Whether aging affects mitochondrial Ca2+ release and Vm in microvascular endothelium have not been determined. Thus, concentration-response relationships to FCCP (10–8 M to 10–5 M) for [Ca2+]i (Figure 3) and Vm (Figure 4) were determined for young and old endothelium. FCCP is a proton ionophore that collapses the inner mitochondrial membrane potential (typically −180 mV in reference to the cytoplasm) which reduces the affinity for Ca2+ ions within the mitochondrial matrix (42–45). As a result, a slow increase in [Ca2+]i ensues (as compared to rapid [Ca2+]i increases on M3 receptor stimulation; compare Figures 1 and 3). Increases in [Ca2+]i occurred during treatment with 3 × 10–7 M FCCP and were similar in young (Figure 3A, left and Figures 3C and D) vs. old (Figure 3B, left and Figures 3C and D). However, the magnitude of [Ca2+]i increased further during 10–6 M FCCP in old (Figure 3B, right and Figures 3C and D) but not in young (Figure 3A, right and Figures 3C and D). Thus, peak [Ca2+]i reached a higher maximum in old vs. young endothelium in response to FCCP, suggesting a greater capacity for mitochondrial Ca2+ release in old. Net changes in Vm to FCCP were variable in young, being mild hyperpolarization (∆Vm < −10 mV) during 3 × 10–7 M (Figure 4A, left and Figures 4C and D) with a significant depolarization (>+10 mV) in response to 10–6 M (Figure 4A, right and Figures 4C and D). In old, significant hyperpolarization of Vm (>−10 mV) occurred to both 3 × 10–7 M and 10–6 M FCCP (Figures 4B–D). Altogether, peak [Ca2+]i increases to FCCP were higher in old and matched by hyperpolarization, whereas peak [Ca2+]i increases in young were lower in magnitude and typically corresponded to depolarization. Thus, [Ca2+]i responses to FCCP are greater in old vs. young endothelium, with membrane hyperpolarization corresponding to mitochondrial uncoupling during aging.
Figure 3.

Aging elevates [Ca2+]i during mitochondrial uncoupling. Fura-2 recordings during 3 × 10–7 M and 10–6 M FCCP in A, young and B, old endothelium. Note that the F340/F380 ratio did not change from 3 × 10–7 M to 10–6 M FCCP in young endothelium, whereas it increased in old endothelium. Summary data indicate C, peak F340/F380 ratio and D, responses relative to control (“C”) during exposure to 10–8 to 10–5 M FCCP. As the traces for Figures 3A and 4A (young) and Figures 3B and 4B (old) represent simultaneous [Ca2+]i and electrical measurements, the gray dots in A and B indicate peak [Ca2+]i responses that coincide with peak Vm responses in the traces shown for Figures 4A and 4B. *p < .05 for young vs. old (n = 4 per group). Additional experiments (n = 12–22 total) were performed for comparisons for 3 × 10–7 M to 10–6 M FCCP treatments between young and old [(3 × 10–7 M: young, ∆F340/F380: 0.25 ± 0.02, n = 14 and old, ∆F340/F380: 0.23 ± 0.02, n = 12) and (10–6 M: young, ∆F340/F380: 0.23 ± 0.02, n = 12 and old: ∆F340/F380: 0.42 ± 0.04, n = 22; p < .05 vs. young)]. FCCP = carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone.
Figure 4.

Aging reverses Vm responses of endothelium to mitochondrial uncoupling. (A, B) Vm recordings during 3 × 10–7 M and 10–6 M FCCP in (A) young and (B) old endothelial tubes. Note that mild hyperpolarization (<−10 mV) shifts to significant depolarization (>+10 mV) from 3 × 10–7 M to 10–6 M in young, whereas hyperpolarization responses are sustained in Old. Summary data for peak Vm (C) and changes in Vm (D) relative to control (“C”) in response to 10–8 to 10–5 M FCCP. As the traces for Figures 3A and 4A (young) and Figures 3B and 4B (old) represent simultaneous [Ca2+]i and electrical measurements, the gray dots in A and B indicate peak Vm responses that coincide with peak [Ca2+]i responses in the traces shown for Figures 3A and 3B. Endothelial tubes from young reach a maximal depolarization (ΔVm ≈ 15 mV), whereas those from old reach a maximal hyperpolarization (ΔVm ≈ −20 mV) at the same [FCCP] of 10–6 M. *p < .05 for young (n = 10) vs. old (n = 8). Additional experiments (n = 20–22 total) were performed for comparisons for 3 × 10–7 M to 10–6 M FCCP treatments between young and old [(3 × 10–7 M: young, ∆Vm: −5 ± 3 mV, n = 20 and old, ∆Vm: −13 ± 2 mV, n = 21; p < .05 vs. young) and (10–6 M: young, ∆Vm: 14 ± 2 mV, n = 22 and old: ∆Vm: −19 ± 3 mV, n = 22; p < .05 vs. young)]. FCCP = carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone.
Because FCCP evoked hyperpolarization in Old but not Young endothelium and the activation of SKCa/IKCa channels is integral to EC hyperpolarization (14,62,68), we tested whether the effect of FCCP was susceptible to the actions of the SKCa/IKCa channel blockers apamin and charybdotoxin in the endothelium from old mice. As illustrated in Figure 5, blockade of SKCa/IKCa channels in old endothelium converted the response to FCCP (3 × 10–7 M) from hyperpolarization to depolarization. Thus, when SKCa and IKCa were prevented from opening in old endothelium, the Vm response to FCCP approximated that in young endothelium. This difference between age groups suggests that SKCa/IKCa channel activation responds to [Ca2+]i increases following mitochondrial uncoupling as an adaptation to advanced age.
Figure 5.

Blockade of SKCa/IKCa inhibits hyperpolarization in response to FCCP in endothelium of old mice. (A) Representative continuous Vm recording during 3 × 10–7 M FCCP in the absence and presence of SKCa/IKCa blockade (3 × 10–7 M apamin + 10–7 M charybdotoxin; Ap + ChTx). (B) Summary data illustrating that SKCa/IKCa blockade reverses hyperpolarization to depolarization during mitochondrial uncoupling with FCCP. Compared to control Vm, FCCP evoked hyperpolarization (ΔVm ≈ −15 mV). Treatment with Ap + ChTx alone produced ≈10 mV depolarization; in the presence of FCCP, Ap + ChTx depolarized cells by ≈ 25 mV vs. control. *p < .05 (n = 4). FCCP = carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone; SKCa/IKCa, small- and intermediate K+ channel.
Discussion
In accord with impaired tissue blood flow with cardiovascular aging (27,61,63), this study investigated whether advanced age affected EDH signaling and whether mitochondrial regulation of [Ca2+]i was involved. A specific aim was to resolve the interplay between mitochondria-associated [Ca2+]i signaling and membrane hyperpolarization in native intact endothelium freshly isolated from resistance arteries of mouse skeletal muscle at ages that correspond to humans in their early 20’s and mid-60s (64). We tested the hypothesis that enhanced SKCa/IKCa activation effected through increased [Ca2+]i signaling following mitochondrial uncoupling is integral to EDH during advanced age. The present data indicate that aging enhances [Ca2+]i signaling and membrane hyperpolarization of ECs in response to ACh, the “gold standard” stimulus for evoking EDH (12,14,34). A key finding is that, in old but not young endothelium, mitochondrial uncoupling with FCCP evoked robust [Ca2+]i increases and hyperpolarized Vm. In contrast, FCCP stimulation of young endothelium resulted in depolarization during the rise in [Ca2+]i. Further, block of SKCa/IKCa channels with apamin and charybdotoxin in old endothelium reversed FCCP responses from hyperpolarization to depolarization similar to that observed in young endothelium. Our findings collectively demonstrate that cardiovascular aging enhances [Ca2+]i signaling and SKCa/IKCa channel activity that underlies vasodilation through EDH and that Ca2+ release from mitochondria may be integral to this adaptation.
EDH With Aging: ER and Mitochondria-Mediated Ca2+ Release
In response to the classic endothelium-dependent vasodilator ACh, the duration of elevated [Ca2+]i governed the duration of Vm hyperpolarization (Figures 1 and 2). These data illustrate the integral role of [Ca2+]i (ie, via IP3R-mediated Ca2+ signaling from the ER) to initiate membrane hyperpolarization of the endothelium in response to a physiological agonist (68). The ability of resistance artery endothelium to release Ca2+ from the ER in response to ACh (Figure 1) and hyperpolarize Vm (Figure 2) via SKCa/IKCa activation was enhanced in old vs. young endothelium. These findings are consistent with our previous studies (9,41) using the same experimental model and age groups studied here.
Mitochondria can sequester Ca2+ to upregulate the activity of citric acid cycle proteins involved in oxidative phosphorylation, potentially increasing the rate of respiratory chain activity and production of hydrogen peroxide (69). As ECs primarily depend on glycolysis for the generation of ATP (36,37), it has been proposed that mitochondria of vascular endothelium play a prominent role in regulating [Ca2+]i (31,38). Indeed, the mitochondrium is a potent Ca2+ buffering organelle equipped with Ca2+ uniporters, Na+/Ca2+ exchangers, and a negative ∆Ψmt (≈−180 mV) that attract cations (70). Further, mitochondrial Ca2+ signaling can increase during advanced age (39,40) and may thereby provide an alternative source of Ca2+ for activating SKCa/IKCa channels in ECs. We therefore reasoned that dissipation of ∆Ψmt with FCCP would trigger release of mitochondrial Ca2+ into the cytosol and thereby activate KCa channels to evoke hyperpolarization. Indeed, in old but not young endothelium, robust [Ca2+]i increases (Figure 3) and membrane hyperpolarization (Figure 4) due to SKCa/IKCa activation (Figure 5) were observed in response to FCCP. Further, blocking SKCa/IKCa channels in old endothelium prior to FCCP exposure approximated the depolarization to FCCP observed in young endothelium under control conditions (compare young data in Figure 4 with old in Figure 5).
Experimental Advantages and Limitations: Simultaneous Ca2+ Photometry and Intracellular Vm Recordings
An advantage of using Ca2+ photometry is the ability to obtain data from the same ECs throughout protocols of at least 5 minutes duration (see Figures 1–4). In this manner, Fura-2 photometry concomitant with intracellular recording provides insight with respect to the relationship between [Ca2+]i and SKCa/IKCa channel activation during membrane hyperpolarization. However, a limitation of Ca2+ photometry is that it reflects an average [Ca2+]i for ECs within the recording window, whereas intracellular recording using sharp microelectrodes reflects the difference in electrochemical driving force between the cytoplasm and the extracellular fluid. A consequence of this limitation is the inability to detect subcellular events occurring in subcellular signaling microdomains within the vicinity of individual ion channels (71). Nevertheless, the present findings provide a foundation for future studies using techniques that enable such higher resolution.
Use of FCCP to Uncouple Mitochondria and Mobilize Mitochondrial Ca2+
As an agent that generally uncouples mitochondria and depolarizes ∆Ψmt (42–44), FCCP may reflect the broader physiological and pathological functions of native mitochondrial uncoupling proteins (48) and mitochondrial K+ channels (49,50). In this manner, FCCP also targets mitochondrial Ca2+ buffering (43,46,47,72,73) and thereby activates KCa channels (46). However, depolarization to micromolar concentrations FCCP in young endothelium (see Figure 4) may also reflect nonselective actions of H+ and/or Na+ influx into the cell interior across the plasma membrane (43). In old endothelium, the possibility of such depolarization to FCCP may be “masked” by relatively large [Ca2+]i increases vs. young and the predominant effect of SKCa/IKCa activation and hyperpolarization (compare Figures 4 and 5). Earlier findings concluded that FCCP may also promote depletion of ER Ca2+ based on reasoning that mitochondrial production of ATP is reduced, thereby impairing the ability of the smooth ER Ca2+ ATP-ase (commonly referred to as SERCA) pumps to refill the ER with Ca2+ (74). However, those findings were based on a low affinity divalent cation dye (ie, Furaptra) (74) that is nonselective for both Ca2+ vs. Mg2+ and ER vs. mitochondria (75). In addition, because ECs rely primarily on glycolysis for generating ATP (36,37), glucose was maintained at 10–2 M in the PSS used for superfusion throughout experiments. Importantly, the kinetics of [Ca2+]i responses to FCCP found here (ie, stable peak within 3 min) do not align with the earlier study [(74); which required ≥5 minutes for a response with no apparent stability]. Further, the present experiments used freshly isolated endothelium (within 1 hour of being in the animal), whereas previous studies have evaluated mitochondrial Ca2+ buffering in isolated ECs or cultured ECs having altered (ie, “cobblestone”) morphology (76) and ion channel expression (77). Such conditions alter the spatial and functional relationships between ER, mitochondria and the plasma membrane that are inherent to native endothelium. In accord with the present findings, we posit that increased mitochondrial Ca2+ release, and an overall increase in the release of Ca2+ from internal stores, promotes EDH to help maintain vasodilation—and thereby tissue blood flow—during advanced age.
SKCa/IKCa Function With Aging and Vascular Disease
How SKCa/IKCa function conveys EDH-dependent vasodilation during aging has lacked a consensus. Some studies suggest a loss of SKCa/IKCa-dependent vasodilation (51,53,78), whereas others have found enhanced SKCa/IKCa function (41,55,57). Difficulty in interpretation may be attributed to factors confounding EC function such as perivascular nerves, SMCs and the flow of blood (carrying hormones, etc.). In addition, complementary signaling pathways include [Ca2+]i activation of endothelial NO synthase (eNOS) and large conductance calcium-activated K+ channels (BKCa) on SMCs, which can hyperpolarize ECs through myoendothelial coupling (79). These vasodilator pathways can also be governed by endothelial mitochondria and reactive oxygen species (70,71). Indeed, BKCa function is increased with aging in SMCs of the mouse SEA (the source of endothelial tubes used within the current study) (80). Discrepancies between studies may also reflect a lack of awareness for structural changes (eg, arterial stiffening and smooth muscle hypertrophy) of the vasculature that may occur in conjunction with altered ion channel activity during aging (53,80,81). Thus, a key aspect of the experimental design used here was to eliminate other influences to evaluate the intrinsic ability of SKCa/IKCa to engage in the transduction of [Ca2+]i signals to hyperpolarization in the endothelium isolated from resistance arteries of young and old mice. The present findings are consistent with our previous reports indicating that the function of underlying components of EDH signaling (ie, [Ca2+]i and SKCa/IKCa function) is enhanced during aging (9,41).
SKCa/IKCa as Pharmacological Targets: Increasing NO Bioavailability Through Hyperpolarization-Induced Ca2+ Entry
Through pharmacological intervention, SKCa/IKCa can be activated without elevating [Ca2+]i (22–25,82,83). Thus, in the face of impaired vascular NO production with aging, endothelial hyperpolarization via direct SKCa/IKCa activation can promote Ca2+ influx into the cell according to its electrochemical gradient (34) and thereby increase eNOS activation to generate NO (23,84), particularly with concomitant activation of Ca2+-permeant channels in the plasma membrane (34). The activation of SKCa/IKCa may also serve as a feed-forward mechanism, whereby the increase in Ca2+ influx amplifies hyperpolarization via SKCa/IKCa activation (34,85). In such manner, the direct activation of SKCa/IKCa can be used effectively to promote vasodilation and tissue blood flow (86,87) during cardiovascular aging (27,61,63).
Enhanced Endothelial Ca2+ Mobilization and Activation of SKCa/IKCa Channels During Aging: A Potential Compensatory Mechanism for Reduced NO Bioavailability
The current study offers new perspective with regard to ER and mitochondrial mobilization of intracellular Ca2+ to activate endothelial SKCa/IKCa. In particular, the release of Ca2+ from endothelial mitochondria can activate SKCa/IKCa channels to generate hyperpolarization and may thereby compensate for decreased bioavailability of NO in old age (26,27,62). However, only 10 to 15 mV of hyperpolarization from resting Vm (ie, −30 to −40 mV) is sufficient for maximal dilation of arteries and arterioles (16,18). Overactivation of SKCa/IKCa to the extent where Vm >−60 mV can cause “leaky” membranes, whereby the initiation and longitudinal spread of hyperpolarization along electrically coupled ECs is short-circuited (11,41). Thus, a method for preserving endothelial function during old age may be to restrict Ca2+ overload of mitochondria and limit the production of reactive oxygen species (69,88) to prevent overactivation of SKCa/IKCa channels (41). Nevertheless, the ability of endothelial mitochondria to affect SKCa/IKCa channel function via increases in [Ca2+]i and reactive oxygen species may be harnessed to promote vasodilation and maintain blood flow during healthy aging to compensate for reductions in NO bioavailability (89). Thus, a balanced therapeutic approach is implied.
Summary and Conclusions
With advancing age, individuals 65 years or older will comprise approximately 25% of the US population by 2030 (1). This demographic typically manifests morbidity and mortality in the form of cardiovascular disease (2,3) with the hallmark of endothelial dysfunction (4,5) underlying impaired tissue blood flow. Our goal in the present study was to determine whether, and if so, how the endothelium of resistance arteries may adapt to maintain its role in effecting vasodilation during advanced age. We focused on the relationship between Ca2+ release from internal stores and the activation of SKCa/IKCa channels in native endothelium freshly isolated from skeletal muscle of young and old mice with an emphasis on the role of mitochondria. Whereas the increases in both [Ca2+]i and membrane potential in response to ACh (ie, muscarinic receptor activation) were sustained with advanced age, uncoupling mitochondria in the endothelium of old mice enhanced [Ca2+]i responses and augmented membrane hyperpolarization through activation of SKCa/IKCa channels. Thus, despite elevated oxidative signaling (9,41,61) and diminished endothelium-dependent dilation with advancing age (26,27), the ability of endothelium to mediate vasodilation via the EDH electrical signaling axis is preserved. In such manner, mitochondrial [Ca2+]i signaling may effectively govern SKCa/IKCa activity during advanced age. With the integrity of EDH maintained via mitochondria versus the ER, targeting specific intracellular Ca2+ stores that govern SKCa/IKCa activity offers the potential for advancing therapeutic strategies designed to protect and restore tissue blood flow and oxygen delivery.
Supplementary Material
Supplementary data is available at The Journals of Gerontology, Series A: Biological Sciences and Medical Sciences online.
Funding
This work was supported by National Institutes of Health grants K99/R00-AG047198 (EJB), R01-HL086483 (SSS), and R37-HL041026 (SSS). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
None.
Supplementary Material
{kind=link}
Acknowledgments
Rebecca Shaw provided excellent technical assistance obtaining the images in Supplementary Figure 1.
References
- 1. Goldman DP, Cutler D, Rowe JW, et al. Substantial health and economic returns from delayed aging may warrant a new focus for medical research. Health Aff (Millwood). 2013;32:1698–1705. doi:10.1377/hlthaff.2013.0052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abete P, Della-Morte D, Gargiulo G, et al. Cognitive impairment and cardiovascular diseases in the elderly. A heart-brain continuum hypothesis. Ageing Res Rev. 2014;18:41–52. doi:10.1016/j.arr.2014.07.003 [DOI] [PubMed] [Google Scholar]
- 3. Berridge MJ. Vitamin D, reactive oxygen species and calcium signalling in ageing and disease. Philos Trans R Soc Lond B Biol Sci. 2016;371. doi:10.1098/rstb.2015.0434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bugiardini R, Manfrini O, Pizzi C, Fontana F, Morgagni G. Endothelial function predicts future development of coronary artery disease: a study of women with chest pain and normal coronary angiograms. Circulation. 2004;109:2518–2523. doi:10.1161/01.CIR.0000128208.22378.E3 [DOI] [PubMed] [Google Scholar]
- 5. Schächinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation. 2000;101:1899–1906. [DOI] [PubMed] [Google Scholar]
- 6. Kirby BS, Voyles WF, Simpson CB, Carlson RE, Schrage WG, Dinenno FA. Endothelium-dependent vasodilatation and exercise hyperaemia in ageing humans: impact of acute ascorbic acid administration. J Physiol. 2009;587:1989–2003. doi:10.1113/jphysiol.2008.167320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taddei S, Virdis A, Mattei P, et al. Aging and endothelial function in normotensive subjects and patients with essential hypertension. Circulation. 1995;91:1981–1987. [DOI] [PubMed] [Google Scholar]
- 8. Proctor DN, Parker BA. Vasodilation and vascular control in contracting muscle of the aging human. Microcirculation. 2006;13:315–327. doi:10.1080/10739680600618967 [DOI] [PubMed] [Google Scholar]
- 9. Socha MJ, Boerman EM, Behringer EJ, Shaw RL, Domeier TL, Segal SS. Advanced age protects microvascular endothelium from aberrant Ca2+ influx and cell death induced by hydrogen peroxide. J Physiol. 2015;593:2155–2169. doi:10.1113/JP270169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bagher P, Segal SS. Regulation of blood flow in the microcirculation: role of conducted vasodilation. Acta Physiol (Oxf). 2011;202:271–284. doi:10.1111/j.1748-1716.2010.02244.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Behringer EJ, Segal SS. Tuning electrical conduction along endothelial tubes of resistance arteries through Ca2+-activated K+ channels. Circ Res. 2012;110:1311–1321. doi:10.1161/CIRCRESAHA.111.262592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Busse R, Fichtner H, Lückhoff A, Kohlhardt M. Hyperpolarization and increased free calcium in acetylcholine-stimulated endothelial cells. Am J Physiol. 1988;255:H965–H969. [DOI] [PubMed] [Google Scholar]
- 13. Bagher P, Davis MJ, Segal SS. Visualizing calcium responses to acetylcholine convection along endothelium of arteriolar networks in Cx40BAC-GCaMP2 transgenic mice. Am J Physiol Heart Circ Physiol. 2011;301:H794–H802. doi:10.1152/ajpheart.00425.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garland CJ, Hiley CR, Dora KA. EDHF: spreading the influence of the endothelium. Br J Pharmacol. 2011;164:839–852. doi:10.1111/j.1476-5381.2010.01148.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Emerson GG, Segal SS. Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: role in vasomotor control. Circ Res. 2000;87:474–479. [DOI] [PubMed] [Google Scholar]
- 16. Emerson GG, Segal SS. Endothelial cell pathway for conduction of hyperpolarization and vasodilation along hamster feed artery. Circ Res. 2000;86:94–100. [DOI] [PubMed] [Google Scholar]
- 17. Looft-Wilson RC, Payne GW, Segal SS. Connexin expression and conducted vasodilation along arteriolar endothelium in mouse skeletal muscle. J Appl Physiol (1985). 2004;97:1152–8. doi:10.1152/japplphysiol.00133.2004 [DOI] [PubMed] [Google Scholar]
- 18. Wölfle SE, Chaston DJ, Goto K, Sandow SL, Edwards FR, Hill CE. Non-linear relationship between hyperpolarisation and relaxation enables long distance propagation of vasodilatation. J Physiol. 2011;589:2607–2623. doi:10.1113/jphysiol.2010.202580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feher A, Broskova Z, Bagi Z. Age-related impairment of conducted dilation in human coronary arterioles. Am J Physiol Heart Circ Physiol. 2014;306:H1595–H1601. doi:10.1152/ajpheart.00179.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Billaud M, Lohman AW, Johnstone SR, Biwer LA, Mutchler S, Isakson BE. Regulation of cellular communication by signaling microdomains in the blood vessel wall. Pharmacol Rev. 2014;66:513–569. doi:10.1124/pr.112.007351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Juszczak GR, Swiergiel AH. Properties of gap junction blockers and their behavioural, cognitive and electrophysiological effects: animal and human studies. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:181–198. doi:10.1016/j.pnpbp.2008.12.014 [DOI] [PubMed] [Google Scholar]
- 22. Jenkins DP, Yu W, Brown BM, Løjkner LD, Wulff H. Development of a QPatch automated electrophysiology assay for identifying KCa3.1 inhibitors and activators. Assay Drug Dev Technol. 2013;11:551–560. doi:10.1089/adt.2013.543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kerr PM, Tam R, Narang D, et al. Endothelial calcium-activated potassium channels as therapeutic targets to enhance availability of nitric oxide. Can J Physiol Pharmacol. 2012;90:739–752. doi:10.1139/y2012-075 [DOI] [PubMed] [Google Scholar]
- 24. Radtke J, Schmidt K, Wulff H, Köhler R, de Wit C. Activation of KCa3.1 by SKA-31 induces arteriolar dilatation and lowers blood pressure in normo- and hypertensive connexin40-deficient mice. Br J Pharmacol. 2013;170:293–303. doi:10.1111/bph.12267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tajbakhsh N, Sokoya EM. Compromised endothelium-dependent hyperpolarization-mediated dilations can be rescued by NS309 in obese Zucker rats. Microcirculation. 2014;21:747–753. doi:10.1111/micc.12157 [DOI] [PubMed] [Google Scholar]
- 26. Muller-Delp JM. Aging-induced adaptations of microvascular reactivity. Microcirculation. 2006;13:301–314. doi:10.1080/10739680600619023 [DOI] [PubMed] [Google Scholar]
- 27. Seals DR, Jablonski KL, Donato AJ. Aging and vascular endothelial function in humans. Clin Sci (Lond). 2011;120:357–375. doi:10.1042/CS20100476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hill CE, Hickey H, Sandow SL. Role of gap junctions in acetylcholine-induced vasodilation of proximal and distal arteries of the rat mesentery. J Auton Nerv Syst. 2000;81:122–127. [DOI] [PubMed] [Google Scholar]
- 29. Shimokawa H, Godo S. Diverse functions of endothelial NO synthases system: NO and EDH. J Cardiovasc Pharmacol. 2016;67:361–366. doi:10.1097/FJC.0000000000000348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Geiger JE, Magoski NS. Ca2+-induced Ca2+ release in Aplysia bag cell neurons requires interaction between mitochondrial and endoplasmic reticulum stores. J Neurophysiol. 2008;100:24–37. doi:10.1152/jn.90356.2008 [DOI] [PubMed] [Google Scholar]
- 31. Groschner LN, Waldeck-Weiermair M, Malli R, Graier WF. Endothelial mitochondria–less respiration, more integration. Pflugers Arch. 2012;464:63–76. doi:10.1007/s00424-012-1085-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Malli R, Frieden M, Trenker M, Graier WF. The role of mitochondria for Ca2+ refilling of the endoplasmic reticulum. J Biol Chem. 2005;280:12114–12122. doi:10.1074/jbc.M409353200 [DOI] [PubMed] [Google Scholar]
- 33. Behringer EJ, Socha MJ, Polo-Parada L, Segal SS. Electrical conduction along endothelial cell tubes from mouse feed arteries: confounding actions of glycyrrhetinic acid derivatives. Br J Pharmacol. 2012;166:774–787. doi:10.1111/j.1476-5381.2011.01814.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Behringer EJ, Segal SS. Membrane potential governs calcium influx into microvascular endothelium: integral role for muscarinic receptor activation. J Physiol. 2015;593:4531–4548. doi:10.1113/JP271102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cohen KD, Jackson WF. Membrane hyperpolarization is not required for sustained muscarinic agonist-induced increases in intracellular Ca2+ in arteriolar endothelial cells. Microcirculation. 2005;12:169–182. doi:10.1080/10739680590904973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Culic O, Gruwel ML, Schrader J. Energy turnover of vascular endothelial cells. Am J Physiol. 1997;273:C205–C213. [DOI] [PubMed] [Google Scholar]
- 37. Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci USA. 2006;103:5379–5384. doi:10.1073/pnas.0601026103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Davidson SM, Duchen MR. Endothelial mitochondria: contributing to vascular function and disease. Circ Res. 2007;100:1128–1141. doi:10.1161/01.RES.0000261970.18328.1d [DOI] [PubMed] [Google Scholar]
- 39. Lopes GS, Ferreira AT, Oshiro ME, et al. Aging-related changes of intracellular Ca2+ stores and contractile response of intestinal smooth muscle. Exp Gerontol. 2006;41:55–62. doi:10.1016/j.exger.2005.10.004 [DOI] [PubMed] [Google Scholar]
- 40. Buchholz JN, Behringer EJ, Pottorf WJ, Pearce WJ, Vanterpool CK. Age-dependent changes in Ca2+ homeostasis in peripheral neurones: implications for changes in function. Aging Cell. 2007;6:285–296. doi:10.1111/j.1474-9726.2007.00298.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Behringer EJ, Shaw RL, Westcott EB, Socha MJ, Segal SS. Aging impairs electrical conduction along endothelium of resistance arteries through enhanced Ca2+-activated K+ channel activation. Arterioscler Thromb Vasc Biol. 2013;33:1892–1901. doi:10.1161/ATVBAHA.113.301514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heinonen E, Akerman KE, Kaila K. Depolarization of the mitochondrial membrane potential increases free cytosolic calcium in synaptosomes. Neurosci Lett. 1984;49:33–37. [DOI] [PubMed] [Google Scholar]
- 43. Park KS, Jo I, Pak K, et al. FCCP depolarizes plasma membrane potential by activating proton and Na+ currents in bovine aortic endothelial cells. Pflugers Arch. 2002;443:344–352. doi:10.1007/s004240100703 [DOI] [PubMed] [Google Scholar]
- 44. Sandoval ME. Studies on the relationship between Ca2+ efflux from mitochondria and the release of amino acid neurotransmitters. Brain Res. 1980;181:357–367. [DOI] [PubMed] [Google Scholar]
- 45. Villalba M, Pereira R, Martinez-Serrano A, Satrústegui J. Altered cell calcium regulation in synaptosomes and brain cells of the 30-month-old rat: prominent effects in hippocampus. Neurobiol Aging. 1995;16:809–816. [DOI] [PubMed] [Google Scholar]
- 46. Yuan XJ, Sugiyama T, Goldman WF, Rubin LJ, Blaustein MP. A mitochondrial uncoupler increases KCa currents but decreases Kv currents in pulmonary artery myocytes. Am J Physiol. 1996;270:C321–C331. [DOI] [PubMed] [Google Scholar]
- 47. Poburko D, Potter K, van Breemen E, et al. Mitochondria buffer NCX-mediated Ca2+-entry and limit its diffusion into vascular smooth muscle cells. Cell Calcium. 2006;40:359–371. doi:10.1016/j.ceca.2006.04.031 [DOI] [PubMed] [Google Scholar]
- 48. Woyda-Ploszczyca AM, Jarmuszkiewicz W. The conserved regulation of mitochondrial uncoupling proteins: from unicellular eukaryotes to mammals. Biochim Biophys Acta. 2017;1858:21–33. doi:10.1016/j.bbabio.2016.10.003 [DOI] [PubMed] [Google Scholar]
- 49. Katakam PV, Gordon AO, Sure VN, Rutkai I, Busija DW. Diversity of mitochondria-dependent dilator mechanisms in vascular smooth muscle of cerebral arteries from normal and insulin-resistant rats. Am J Physiol Heart Circ Physiol. 2014;307:H493–H503. doi:10.1152/ajpheart.00091.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Katakam PV, Wappler EA, Katz PS, et al. Depolarization of mitochondria in endothelial cells promotes cerebral artery vasodilation by activation of nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2013;33:752–759. doi:10.1161/ATVBAHA.112.300560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chennupati R, Lamers WH, Koehler SE, De Mey JG. Endothelium-dependent hyperpolarization-related relaxations diminish with age in murine saphenous arteries of both sexes. Br J Pharmacol. 2013;169:1486–1499. doi:10.1111/bph.12175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leo CH, Hart JL, Woodman OL. Impairment of both nitric oxide-mediated and EDHF-type relaxation in small mesenteric arteries from rats with streptozotocin-induced diabetes. Br J Pharmacol. 2011;162:365–377. doi:10.1111/j.1476-5381.2010.01023.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Matz RL, Schott C, Stoclet JC, Andriantsitohaina R. Age-related endothelial dysfunction with respect to nitric oxide, endothelium-derived hyperpolarizing factor and cyclooxygenase products. Physiol Res. 2000;49:11–18. [PubMed] [Google Scholar]
- 54. Chadha PS, Haddock RE, Howitt L, et al. Obesity up-regulates intermediate conductance calcium-activated potassium channels and myoendothelial gap junctions to maintain endothelial vasodilator function. J Pharmacol Exp Ther. 2010;335:284–293. doi:10.1124/jpet.110.167593 [DOI] [PubMed] [Google Scholar]
- 55. Choi S, Kim JA, Li HY, et al. KCa3.1 upregulation preserves endothelium-dependent vasorelaxation during aging and oxidative stress. Aging Cell. 2016;15:801–810. doi:10.1111/acel.12502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Climent B, Moreno L, Martínez P, et al. Upregulation of SK3 and IK1 channels contributes to the enhanced endothelial calcium signaling and the preserved coronary relaxation in obese Zucker rats. PLoS One. 2014;9:e109432. doi:10.1371/journal.pone.0109432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Giachini FR, Carneiro FS, Lima VV, et al. Upregulation of intermediate calcium-activated potassium channels counterbalance the impaired endothelium-dependent vasodilation in stroke-prone spontaneously hypertensive rats. Transl Res. 2009;154:183–193. doi:10.1016/j.trsl.2009.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mokhtar SS, Vanhoutte PM, Leung SW, et al. Endothelium dependent hyperpolarization-type relaxation compensates for attenuated nitric oxide-mediated responses in subcutaneous arteries of diabetic patients. Nitric Oxide. 2016;53:35–44. doi:10.1016/j.niox.2015.12.007 [DOI] [PubMed] [Google Scholar]
- 59. Schach C, Resch M, Schmid PM, Riegger GA, Endemann DH. Type 2 diabetes: increased expression and contribution of IKCa channels to vasodilation in small mesenteric arteries of ZDF rats. Am J Physiol Heart Circ Physiol. 2014;307:H1093–H1102. doi:10.1152/ajpheart.00240.2013 [DOI] [PubMed] [Google Scholar]
- 60. Stead R, Musa MG, Bryant CL, et al. Developmental conditioning of endothelium-derived hyperpolarizing factor-mediated vasorelaxation. J Hypertens. 2016;34:452–63; discussion 463. doi:10.1097/HJH.0000000000000833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Muller-Delp JM, Gurovich AN, Christou DD, Leeuwenburgh C. Redox balance in the aging microcirculation: new friends, new foes, and new clinical directions. Microcirculation. 2012;19:19–28. doi:10.1111/j.1549-8719.2011.00139.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Félétou M. Endothelium-dependent hyperpolarization and endothelial dysfunction. J Cardiovasc Pharmacol. 2016;67:373–387. doi:10.1097/FJC.0000000000000346 [DOI] [PubMed] [Google Scholar]
- 63. Hearon CM, Jr, Dinenno FA. Regulation of skeletal muscle blood flow during exercise in ageing humans. J Physiol. 2016;594:2261–2273. doi: 10.1113/JP270593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Flurkey K, Currer J, Harrison D. Mouse Models in Aging Research. In: Fox JG, Davisson MT, Quimby FW, Barthold SW, Newcomer CE, Smith AL, eds. The Mouse in Biomedical Research. 2nd Ed Burlington: American College of Laboratory Animal Medicine (Elsevier); 2007: 637–672. [Google Scholar]
- 65. Socha MJ, Domeier TL, Behringer EJ, Segal SS. Coordination of intercellular Ca(2+) signaling in endothelial cell tubes of mouse resistance arteries. Microcirculation. 2012;19:757–770. doi:10.1111/micc.12000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Socha MJ, Segal SS. Isolation of microvascular endothelial tubes from mouse resistance arteries. J Vis Exp. 2013:e50759. doi:10.3791/50759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Socha MJ, Hakim CH, Jackson WF, Segal SS. Temperature effects on morphological integrity and Ca²⁺ signaling in freshly isolated murine feed artery endothelial cell tubes. Am J Physiol Heart Circ Physiol. 2011;301:H773–H783. doi:10.1152/ajpheart.00214.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Busse R, Edwards G, Félétou M, Fleming I, Vanhoutte PM, Weston AH. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2002;23:374–380. [DOI] [PubMed] [Google Scholar]
- 69. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–C833. doi:10.1152/ajpcell.00139.2004 [DOI] [PubMed] [Google Scholar]
- 70. Olson ML, Chalmers S, McCarron JG. Mitochondrial organization and Ca2+ uptake. Biochem Soc Trans. 2012;40:158–167. doi:10.1042/BST20110705 [DOI] [PubMed] [Google Scholar]
- 71. Sonkusare SK, Bonev AD, Ledoux J, et al. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012;336:597–601. doi:10.1126/science.1216283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dedkova EN, Ji X, Lipsius SL, Blatter LA. Mitochondrial calcium uptake stimulates nitric oxide production in mitochondria of bovine vascular endothelial cells. Am J Physiol Cell Physiol. 2004;286:C406–C415. doi:10.1152/ajpcell.00155.2003 [DOI] [PubMed] [Google Scholar]
- 73. Malli R, Frieden M, Osibow K, Graier WF. Mitochondria efficiently buffer subplasmalemmal Ca2+ elevation during agonist stimulation. J Biol Chem. 2003;278:10807–15. doi:10.1074/jbc.M212971200 [DOI] [PubMed] [Google Scholar]
- 74. Landolfi B, Curci S, Debellis L, Pozzan T, Hofer AM. Ca2+ homeostasis in the agonist-sensitive internal store: functional interactions between mitochondria and the ER measured In situ in intact cells. J Cell Biol. 1998;142:1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Golovina VA, Blaustein MP. Spatially and functionally distinct Ca2+ stores in sarcoplasmic and endoplasmic reticulum. Science. 1997;275:1643–1648. [DOI] [PubMed] [Google Scholar]
- 76. Simmers MB, Pryor AW, Blackman BR. Arterial shear stress regulates endothelial cell-directed migration, polarity, and morphology in confluent monolayers. Am J Physiol Heart Circ Physiol. 2007;293:H1937–H1946. doi:10.1152/ajpheart.00534.2007 [DOI] [PubMed] [Google Scholar]
- 77. Sandow SL, Grayson TH. Limits of isolation and culture: intact vascular endothelium and BKCa. Am J Physiol Heart Circ Physiol. 2009;297:H1–H7. doi:10.1152/ajpheart.00042.2009 [DOI] [PubMed] [Google Scholar]
- 78. Long DA, Newaz MA, Prabhakar SS, et al. Loss of nitric oxide and endothelial-derived hyperpolarizing factor-mediated responses in aging. Kidney Int. 2005;68:2154–2163. doi:10.1111/j.1523-1755.2005.00671.x [DOI] [PubMed] [Google Scholar]
- 79. Sandow SL, Tare M, Coleman HA, Hill CE, Parkington HC. Involvement of myoendothelial gap junctions in the actions of endothelium-derived hyperpolarizing factor. Circ Res. 2002;90:1108–1113. [DOI] [PubMed] [Google Scholar]
- 80. Hayoz S, Bradley V, Boerman EM, Nourian Z, Segal SS, Jackson WF. Aging increases capacitance and spontaneous transient outward current amplitude of smooth muscle cells from murine superior epigastric arteries. Am J Physiol Heart Circ Physiol. 2014;306:H1512–H1524. doi:10.1152/ajpheart.00492.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chaston DJ, Baillie BK, Grayson TH, et al. Polymorphism in endothelial connexin40 enhances sensitivity to intraluminal pressure and increases arterial stiffness. Arterioscler Thromb Vasc Biol. 2013;33:962–970. doi:10.1161/ATVBAHA.112.300957 [DOI] [PubMed] [Google Scholar]
- 82. Strobaek D, Teuber L, Jorgensen TD, et al. Activation of human IK and SK Ca2+ -activated K+ channels by NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime). Biochim Biophys Acta. 2004;1665:1–5. doi:10.1016/j.bbamem.2004.07.006 [DOI] [PubMed] [Google Scholar]
- 83. Li W, Halling DB, Hall AW, Aldrich RW. EF hands at the N-lobe of calmodulin are required for both SK channel gating and stable SK-calmodulin interaction. J Gen Physiol. 2009;134:281–293. doi:10.1085/jgp.200910295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sheng JZ, Ella S, Davis MJ, Hill MA, Braun AP. Openers of SKCa and IKCa channels enhance agonist-evoked endothelial nitric oxide synthesis and arteriolar vasodilation. FASEB J. 2009;23:1138–1145. doi:10.1096/fj.08-120451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Socha MJ, Behringer EJ, Segal SS. Calcium and electrical signalling along endothelium of the resistance vasculature. Basic Clin Pharmacol Toxicol. 2012;110:80–86. doi:10.1111/j.1742-7843.2011.00798.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bellien J, Favre J, Iacob M, et al. Arterial stiffness is regulated by nitric oxide and endothelium-derived hyperpolarizing factor during changes in blood flow in humans. Hypertension. 2010;55:674–680. doi:10.1161/HYPERTENSIONAHA.109.142190 [DOI] [PubMed] [Google Scholar]
- 87. Mishra RC, Belke D, Wulff H, Braun AP. SKA-31, a novel activator of SKCa and IKCa channels, increases coronary flow in male and female rat hearts. Cardiovasc Res. 2013;97:339–348. doi:10.1093/cvr/cvs326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Peng TI, Jou MJ. Oxidative stress caused by mitochondrial calcium overload. Ann N Y Acad Sci. 2010;1201:183–188. doi:10.1111/j.1749-6632. 2010.05634.x [DOI] [PubMed] [Google Scholar]
- 89. Behringer EJ, Segal SS. Spreading the signal for vasodilatation: implications for skeletal muscle blood flow control and the effects of ageing. J Physiol. 2012;590:6277–6284. doi:10.1113/jphysiol.2012.239673 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.