

Abstract
Alzheimer’s disease (AD) is rarely addressed in the context of aging even though there is an overlap in pathology. We previously used a phenotypic screening platform based on old age–associated brain toxicities to identify the flavonol fisetin as a potential therapeutic for AD and other age-related neurodegenerative diseases. Based on earlier results with fisetin in transgenic AD mice, we hypothesized that fisetin would be effective against brain aging and cognitive dysfunction in rapidly aging senescence-accelerated prone 8 (SAMP8) mice, a model for sporadic AD and dementia. An integrative approach was used to correlate protein expression and metabolite levels in the brain with cognition. It was found that fisetin reduced cognitive deficits in old SAMP8 mice while restoring multiple markers associated with impaired synaptic function, stress, and inflammation. These results provide further evidence for the potential benefits of fisetin for the treatment of age-related neurodegenerative diseases.
Keywords: Alzheimer’s, disease, Cognition, Metabolome, Inflammation, DHA
Currently, there are no disease-modifying drugs for any age-associated neurological diseases. This is at least in part due to the fact that the vast majority of these diseases arise from a confluence of multiple toxic insults to the brain that accumulate during normal aging and interact with lifestyle, environmental, and genetic risk factors with varying degrees of penetrance. For example, although Alzheimer’s disease (AD) is defined in terms of plaque and tangle pathology, it is frequently associated with other detrimental events such as microvascular damage and inflammation (1). Therefore, it is unlikely that hitting a single target will result in significant benefits to patients with AD (2). This conclusion has been largely born out by the failure of numerous clinical trials for single target drugs in AD (3). Nevertheless, current drug research efforts continue to be almost exclusively focused on single protein targets and the identification of small molecules that can modulate these targets with high affinity (1). Our laboratory uses a drug discovery paradigm based on a set of cell-based screening assays that mimic numerous aspects of old age–associated neurodegeneration and AD pathology (4). This approach has led to the identification of fisetin, a neuroprotective flavonol that enhances memory in normal animals (5) and in AD transgenic mice (6). Fisetin is also protective in animal models of other age-associated neurological disorders including Huntington’s disease (7) and stroke (8,9).
Although AD transgenic rodents have been used as state-of-the-art models for AD drug candidate testing, the vast majority rely on familial AD (FAD) mutations. Unfortunately, none of the drug candidates identified using these models has translated to the clinic. Given that FAD accounts for less than 1% of all AD cases (10), animal models that more accurately reflect the predominant sporadic forms of AD and other dementias are needed (11).
Age is by far the greatest risk factor for AD (11). One model of aging is the senescence-accelerated prone 8 (SAMP8) mouse that exhibits a progressive, age-associated decline in brain function similar to human AD patients (reviewed in (12, 13)). As they age, SAMP8 mice develop an early deterioration in learning and memory as well as a number of pathophysiological alterations in the brain including increased oxidative stress, inflammation, vascular impairment, gliosis, Aβ accumulation, and tau hyperphosphorylation. Using an integrative multiomics approach, we recently identified a number of behavioral and physiological changes that are altered with aging in these mice (14). Moreover, we showed that the AD drug candidate, J147, reduced AD-associated pathology, and many metabolic aspects of aging in old SAMP8 mice (14).
Since fisetin and J147 were both selected based on their ability to prevent old age–related brain toxicities and both are effective in transgenic mouse models of AD, we wanted to determine if fisetin was also protective in the SAMP8 model of aging. Changes in behavior, protein expression, and the levels of metabolites in old SAMP8 mice fed with control or fisetin diets were compared with young SAMP8 control mice. These data demonstrate the ability of fisetin to suppress many of these changes while reducing some aspects of aging.
Methods
Study Design
The aim of this project was to investigate whether the flavonol fisetin protects SAMP8 mice from aging and AD-associated pathology and to assay the associated metabolic changes. Seventeen 3-month-old male SAMP8 mice were fed with control diet (LabDiet 5015, TestDiet, Richmond, IN) and eighteen 3-month-old male SAMP8 mice were fed with fisetin diet (LabDiet 5015 + 500 ppm fisetin, TestDiet) until they reached 10 months old. At this age, SAMP8 mice present a strong phenotype (15). The dose of fisetin used was 500 ppm (~25 mg/kg/day), which previously proved effective in AD transgenic mice (6). Fourteen 3-month-old male SAMP8 mice were used as the young control group. The SAMP8 mice are an inbred strain and, as such, young SAMP8 mice were chosen as controls for young age. Given the 7-month duration of the feeding paradigm, the effect of fisetin diet could only be assessed in old SAMP8 mice, and any age-related changes defined by the comparison to the young SAMP8 animals. All mice were randomly assigned to experimental groups. The number of mice per group was determined based on previous experiments (15) and was sufficient to attain statistical power. Six old SAMP8 mice fed with control diet and five old SAMP8 mice fed with fisetin diet died throughout the course of this study including during the behavioral testing. Behavioral testing was carried out 1 month prior to sacrifice and collection of biological material. Data were analyzed by blinded researchers when appropriate.
SAMP8 Mice
The SAMP8 line was acquired from Harlan Laboratories (UK). Mouse body weights were measured regularly and no significant differences were found between the groups (Supplementary Figure S1).
Behavioral Assays
Open field
The open field test was performed using MED Associates hardware and the Activity Monitor software according to the manufacturer’s instructions (MED Associates Inc, St. Albans, VT). Animals were individually placed into clear Plexiglas boxes (40.6 × 40.6 × 38.1 cm) surrounded by multiple bands of photo beams and optical sensors that measure horizontal and vertical activity. Their movement was detected as breaks within the beam matrices and automatically recorded for 30 minutes.
Elevated plus maze
The maze consisted of four arms (two open without walls and two enclosed by 15.25-cm high walls) 30-cm long and 5-cm wide in the shape of a plus. A video-tracking system (Noldus EthoVision) was used to automatically collect behavioral data. The software was installed on a PC with a digital video camera mounted overhead on the ceiling, which automatically detected and recorded when mice entered the open or closed arms of the maze and the time spent in each. Mice were habituated to the room 24 hours before testing and habituated to the maze for 1 minute before testing by placing them in the center of the maze and blocking entry to the arms. Mice were then tested for a 5-minute period and their behavior recorded. Disinhibition was measured by comparing the time spent on the open arms to time spent on the closed arms.
Object recognition
Mice were tested in a standard home cage. Phase 1 (Habituation): Each mouse was placed into the apparatus (no objects present) for two 10-minute sessions separated by 1–4 hours to habituate to testing environment. Phase 2 (Training): Two identical Velcro-backed objects (object “A”) were attached to designated corners of the apparatus. The mouse was placed into the apparatus opposite to the objects and recorded by a camera for 10 minutes. Phase 3 (Test): One hour after training, the test phase began. Only one of the objects was replaced with a new object (object “B”). The mouse was placed into the apparatus opposite to the objects and recorded for 5 minutes. The apparatus was wiped and objects cleaned with 70% alcohol to remove odors between mice. “object recognition index” was calculated by dividing the amount of time spent with object B by the total time spent with objects A + B and multiplied by 100.
Barnes maze
The maze consisted of a flat circular surface (36” diameter) with 20 equally spaced holes (2” diameter) along the outer edge. One of the holes led to a dark hide box whereas the other 19 led to false boxes that were too small to be entered. The latency to enter the hide box was recorded. The test was conducted in three phases. Phase 1 (Training): A hide box was placed under one of the holes. Animals were placed into an opaque cylinder in the center of the maze for 30 seconds to promote spatial disorientation at the start of the test. After 30 seconds, the cylinder was removed and the animal explored the maze until it found and entered the hide box. The number of incorrect entries was scored. If the mouse failed to enter the box within 3 minutes, it was gently led into the box. The animal remained in the box for an additional 20 seconds before it was removed from the box and gently placed into the home cage. Training is repeated three times a day for 4 days. The location of the hide box remained the same during every trial, but it was shifted between subjects to reduce the potential for unintended intramaze cues. Phase 2 (Retention): This phase measures retention of spatial memory following a delay. After a 2-day break from training, each animal was retested for a 1-day, three-trial session using the same hide box location as before. Phase 3 (Reversal): This phase examines memory reversal. On the day following the retention phase, a new hide box location was established 180 degrees to the original location. The same method as before was used and trials were repeated three times a day over two consecutive days.
Tissue Preparation
Mice were anesthetized and their blood collected by cardiac puncture. After perfusing with phosphate-buffered saline, their brains were removed. Half of the brain was fixed and processed for histology and the other half was dissected (to collect cortex and hippocampus) and prepared for Western blot (WB), eicosanoid, and global metabolomic analysis as described (14).
Western Blotting
WBs were carried out as described previously (14). The primary antibodies used were: HRP-conjugated rabbit anti-actin (#5125, 1/20,000), HSP40 (#4871, 1/1000), HSP60 (#4870, 1/5000), HSP70 (#4872, 1/5000), HSP90 (#4877, 1/5000), SAPK/JNK (#9252, 1/1000), Phospho-SAPK/JNK Thr183/Tyr185 (#4668, 1/1000), p25/35 (#2680, 1/1000) from Cell Signaling Technology; Arc (#sc-15325, 1/2000), GFAP (#AB5804, 1/5000), SAP102 (#AB5170, 1/400) from Millipore; Homer 1 (#SAB1411661, 1/1000) from Sigma. Horseradish peroxidase-conjugated secondaries goat anti-rabbit, goat anti-mouse, or rabbit anti-goat (Biorad) diluted 1/5000 were used.
Immunohistochemistry
Immunohistochemistry was carried out as described previously (6). Anti-Iba-1 (#019-19741, 1/4000, from Wako) and biotinylated rabbit secondary antibody (#BA1000, 1/400 from Vector Laboratories) were used. The number of microglia per mm2 of hippocampus was quantified using the Image J software (NIH). Total counts in 2–4 sections per eight mouse brains of each group were determined in an unbiased fashion.
Eicosanoid Analysis
Eicosanoids were prepared and analyzed as described previously (6).
Metabolomic Analysis
Metabolomic analyses were conducted at Metabolon as described previously (14). For statistical analyses and data display, any missing values were assumed to be below the limits of detection and imputed with the compound minimum (minimum value imputation). An estimate of the false discovery rate (Q-value) was calculated to take into account the multiple comparisons that normally occur in metabolomic-based studies, with Q <0.05 used as an indication of high confidence in a result. Welch’s two-sample t test was used to identify biochemicals that differed significantly between experimental groups.
Bioinformatics and Statistics
Data in figures are presented as group mean ± SD or as box-and-whisker plots indicating the group minimum, lower quartile, median, upper quartile, and group maximum.
Statistical analysis of the three groups was carried out by one-way analysis of variance (ANOVA) followed by Tukey–Kramer multiple comparison post-hoc test was used. For data regarding multiple time points, two-way repeated-measures ANOVA and post-hoc Bonferroni-corrected t tests were applied. GraphPad Prism 6 was used and significance of difference was indicated as *p <.05, **p <.01, and ***p <.001.
Study Approval
All experiments were performed in accordance with the U.S. Public Health Service Guide for Care and Use of Laboratory Animals and protocols approved by the IACUC at the Salk Institute.
Results
To test the effects of fisetin on the development of the SAMP8 phenotype, two groups of 3-month-old mice were fed with control or fisetin diet for an additional 7 months, while another group of 3-month-old mice was used as a young control group. Since the SAMP8 mice are an inbred strain, young SAMP8 mice were chosen as the most appropriate control. Given the 7-month duration of the feeding paradigm, the effects of the fisetin diet could only be assessed in old SAMP8 mice, and age-related changes were defined by comparison to the young SAMP8 animals.
Fisetin Prevents Cognitive and Locomotor Deficits With Age in SAMP8 Mice
We started by investigating whether fisetin could prevent age-associated cognitive decline in old SAMP8 mice using the Barnes maze (Figure 1A), the object recognition test (Figure 1B), and the elevated plus maze (Figure 1C). The Barnes maze is used to analyze spatial learning and hippocampal-dependent memory. In this assay, mice use visual cues to locate a hidden box. With repeated trials, animals with an intact memory show a significant reduction in the time (latency) to locate the box. If the box is moved to another location in the maze (reversal test), normal animals rapidly disengage from the previously learned information and relearn the new location. No changes between the three groups were found in the escape latencies during the learning and the retention phases (data not shown). However, when tested during the reversal phase, which is more sensitive to smaller deficits in learning and memory, the old SAMP8 mice showed a deficit relative to the young mice in their ability to relearn the new location of the escape box (Figure 1A). Importantly, the ability of the old fisetin-fed mice to relearn the new location was essentially identical to that of the young mice.
Figure 1.

Fisetin improves cognitive function and locomotor activity in old SAMP8 mice. Spatial learning/memory and recognition memory were evaluated by the Barnes maze (A) and object recognition (B) assays, respectively. The elevated plus maze (C) was used to measure disinhibition. Average velocity (D), number of vertical events (E), and vertical time (F) were assessed in young mice and old SAMP8 mice fed with control or fisetin diets with the open-field test. One-way ANOVA followed by Tukey–Kramer post-hoc test and two-way repeated-measures ANOVA and post-hoc Bonferroni-corrected t test (n = 12–13/group). All data are mean ± SD. *p < .05, ***p < .001. ANOVA = analysis of variance; SAMP8 = senescence-accelerated prone 8.
The object recognition test evaluates short-term recognition memory and is based on the spontaneous tendency of mice to spend more time exploring a novel object than a familiar one. The choice to explore the novel object reflects the use of learning and recognition memory. There was a significant decrease in the recognition index with age in SAMP8 mice, which was prevented by fisetin (Figure 1B).
The elevated plus maze examines disinhibition behavior based on the aversion of normal mice to open spaces. Dementia is clinically associated with disinhibition and AD mouse models tend to exhibit increased disinhibition (6,16). Accordingly, old SAMP8 mice spent significantly more time in the open arms compared to the young SAMP8 mice (Figure 1C). The time in the open arms was decreased modestly but nonsignificantly by fisetin. Altogether these data show that fisetin prevents the deterioration of several aspects of behavior and memory that are altered in old SAMP8 mice.
By monitoring the spontaneous behavior of mice in the open field assay, we found a decline in activity parameters between the young and the old SAMP8 mice (Figure 1D–F and (14)). Fisetin had a positive effect on locomotor activity as it slightly improved the average velocity (Figure 1D) and significantly increased the number of vertical counts (Figure 1E) and time vertical (Figure 1F) in the old SAMP8 mice. Fisetin had no effect on the body weights (Supplementary Figure S1A) or food consumption (Supplementary Figure S1B).
Behavioral Deficits in Old SAMP8 Are Associated With Dysregulation of Homeostatic Responses in the Brain That Are Restored by Fisetin
We next investigated if the decline in the cognitive performance of old SAMP8 mice and the therapeutic effects of fisetin were associated with changes in specific proteins relevant to neuronal function and stress. The expression of three proteins associated with synaptic function, activity-regulated cytoskeleton-associated protein (Arc), Homer, and synapse-associated protein 102 (SAP102), all decreased in old mice compared to young mice (Figure 2A–C and (14)), and treatment with fisetin partly to almost fully prevented these decreases. It was then asked if these changes were accompanied by alterations in the levels of proteins involved in the cellular responses to stress relevant to aging and AD (Figure 2D–F). Although the levels of heat shock protein 70 (HSP70) were not significantly altered between groups (not shown), changes in HSP40, HSP60, and HSP90 were detected in the old SAMP8 mice (Figure 2D–F and (14)). Fisetin significantly increased HSP90 levels to those found in the young control mice and partially increased the levels of HSP40 (Figure 2D and F). In contrast, HSP60 levels increased with aging and this increase was significantly prevented by fisetin (Figure 2E).
Figure 2.
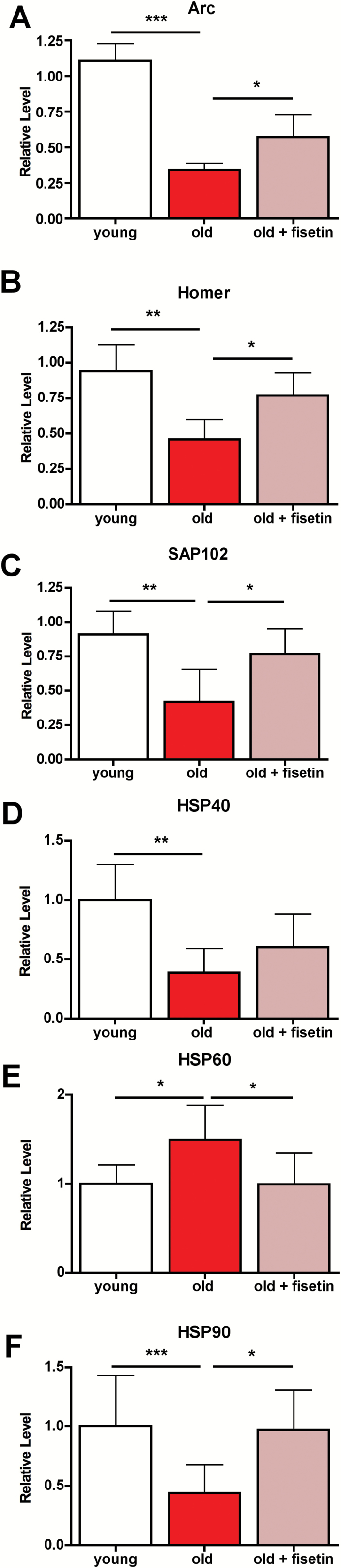
Dysregulation of neuronal homeostasis and stress responses in the hippocampus of old SAMP8 mice is partially restored by fisetin. RIPA-soluble fractions from hippocampal tissue were analyzed by Western blotting for relevant markers of neuronal homeostasis and stress and are presented relative to actin. Arc (A), Homer 1 (B), SAP102 (C), HSP40 (D), HSP60 (E), and HSP90 (F). One-way ANOVA followed by Tukey–Kramer post-hoc test (n = 6/group). All data are mean ± SD. *p < .05, **p < .01, ***p < .001. ANOVA = analysis of variance; SAMP8 = senescence-accelerated prone 8.
Fisetin Decreases Brain Inflammation in old SAMP8 Mice
Given the relevance of inflammation to aging and AD, a characterization of the effects of fisetin on the inflammatory status of the aged SAMP8 brain was carried out. We previously reported an increased expression of glial fibrillary acidic protein (GFAP), a marker for astrocyte activation (17), in the hippocampus of old SAMP8 mice (14,15). We found (15) that there was a direct correlation between the levels of GFAP as determined by immunohistochemistry and the levels as determined by Western blotting. In the current study, GFAP expression in the hippocampus of the old SAMP8 mice, as determined by Western blotting, was significantly reduced by fisetin (Figure 3A).
Figure 3.

Markers of increased inflammation in the hippocampus of old SAMP8 mice are partially prevented by fisetin. (A) Astrocyte activation measured by Western blot of GFAP levels. (B) Ratio of the p25 and p35 forms of the Cdk5 activator was measured by Western blotting. (C) Activation of the stress/inflammation-associated SAPK/JNK was measured by Western blot analysis of its phosphorylation at Thr183/Tyr185. (D) Quantification of microglia in the hippocampus. One-way ANOVA followed by Tukey–Kramer post-hoc test (n = 6/group). All data are mean ± SD. *p < .05, **p < .01, ***p < .001. ANOVA = analysis of variance; GFAP = glial fibrillary acidic protein; SAMP8 = senescence-accelerated prone 8; APK/JNK = stress-activated protein kinase/Jun-amino-terminal kinase.
In transgenic AD mice, it has been suggested that p25, the cleavage product of the Cdk5 activator p35, drives astrocyte activation (18). However, in the old SAMP8 mice, the p25/p35 ratio was decreased relative to the young mice and fisetin had no significant effect relative to either the young or old mice (Figure 3B). In contrast, WB analysis revealed an activation of the stress-activated protein kinase/Jun-amino-terminal kinase (SAPK/JNK), determined by its phosphorylation, in the hippocampus of old SAMP8 mice (Figure 3C and (14)). Previously, we showed that SAPK/JNK activation was a good marker of microglial activation (19). Importantly, fisetin prevented the activation of SAPK/JNK in the old SAMP8 mice. Although the number of microglia increased in the hippocampus of old mice compared to young mice (14), fisetin did not significantly alter their number (Figure 3D).
A detailed analysis of eicosanoids in the brain cortex was also conducted. Eicosanoids are a class of bioactive lipid mediators that are potent regulators of inflammatory responses (20). In the SAMP8 mice, fisetin significantly increased the levels of docosahexaenoic acid (DHA) (Figure 4) and significantly reduced the levels of many of its products of nonenzymatic oxidation (8-HDoHE, 10-HDoHE, 11-HDoHE, 13-HDoHE, 14-HDoHE, 16-HDoHE, 17-HdoHE, and 20-HDoHE) as well as the levels of nonenzymatically oxidized metabolites of arachidonic acid (8-HETE and 9-HETE) and linoleic acid (9-HODE) (Figure 4). These findings suggest that fisetin may reduce the pro-oxidant status in the brains of old animals.
Figure 4.

Analysis of eicosanoid metabolism in the cortex of young SAMP8, old SAMP8, and old SAMP8 mice fed with fisetin. Significant changes in the metabolites of arachidonic acid, docosahexaenoic acid, and linoleic acid derived from the actions of COX, LOXs, cytochrome P450, and nonenzymatic oxidation, with aging and fisetin treatment. With the exception of eicosanoids derived from linoleic acid, whose levels were not quantified in this study, levels of all other eicosanoids were normalized to their respective fatty acid precursor. Boxes are: white = young; black = old; gray = old + fisetin. One-way ANOVA followed by Tukey–Kramer post-hoc test (n = 5/group). *p < .05, **p < .01, ***p < .001. ANOVA = analysis of variance; SAMP8 = senescence-accelerated prone 8.
The levels of the epoxides 9,10-DiHOME and 12,13-DiHOME, which are derived enzymatically from linoleic acid and associated with an inflammatory response (21), were slightly increased in old animals and were reduced with fisetin treatment (9,10-DiHOME was significantly reduced). In addition, several COX (PGD2, TXB2, and PGF2α), 5-LOX (5-HETE and 9-HOTre), and 15-LOX (15-HETE) metabolites, all of which are associated with inflammation (22–24), were significantly lowered by fisetin (Figure 4).
The Beneficial Effects of Fisetin on the SAMP8 Phenotype Are Associated With Alterations in Specific Brain and Plasma Metabolites
Because major metabolic alterations take place with aging, a global metabolic profiling study was carried out with blood plasma and brain cortical tissue in order to understand the possible therapeutic effects of fisetin on brain and whole body health. As described previously (14), 166 of 593 (28.0%) and 82 of 493 (16.6%) assayed biochemicals differed significantly in the plasma and cortex, respectively, between young and old SAMP8 mice. Although treatment with fisetin only resulted in changes in 11 metabolites in the plasma relative to untreated old SAMP8 mice, most of these changes resulted in a prevention of the effects of aging mainly associated with metabolites involved in amino acid (4-hydroxyphenylacetate, N-acetyl tyrosine, indoleacetate, and homocitruline) and lipid (caprylate, sphingosine, and taurourso deoxycholate) metabolism (Figure 5). Interestingly, several of these metabolites (4-hydroxyphenylacetate, indoleacetate, and taurourso deoxycholate) are mainly derived from the gut microbiome (25,26). Similarly, in the brain, although fisetin only altered 12 metabolites relative to the untreated, old SAMP8 mice, most of these changes also resulted in the maintenance of the young phenotype including metabolites that play a role in amino acid (glycine, 2-hydroxybutyrate, trans-4-hydroxyproline), nucleotide (adenylosuccinate, uridine monophosphate, N-acetyl-beta-alanine, and 2ʹ-deoxycytidine), and lipid (heptanoate, 2-stearoyl-glyceroPEE, and stearoyl-arachidonoyl-glyceroPC) metabolism (Figure 5).
Figure 5.

Global metabolomic profiling of plasma and cortex demonstrate that alterations in biological pathways between young SAMP8 and old SAMP8 mice are partially rescued by fisetin. Plasma and cortex biochemicals found significantly modified. Boxes are: white = young; black = old; gray = old + fisetin. Welch’s two-sample t test was used to identify biochemicals that differed significantly between experimental groups (n = 5/group). *p < .05, **p < .01, ***p < .001. SAMP8 = senescence-accelerated prone 8.
Discussion
We used an integrated experimental approach to investigate the potential therapeutic properties of the flavonol fisetin in the SAMP8 model of aging and early sporadic AD. SAMP8 mice develop a progressive, age-associated decline in brain function, as well as pathophysiological features similar to those found in the brains of sporadic AD patients. Therefore, they may represent an excellent model for studying the relationship between aging and sporadic AD (12–14). Our data with fisetin suggest that modulation of a small subset of the physiological changes associated with aging can have a major influence on memory impairment, an alteration directly related to the clinical hallmarks of AD. Thus, these data support and extend our results on the beneficial effects of fisetin in a transgenic model of AD (6).
Given the positive action of fisetin on cognitive function in the old SAMP8 mice, we first examined its effects on several proteins involved in synaptic function whose expression was previously found to be decreased in old SAMP8 mice (14). Importantly, it has been suggested that AD is primarily a synaptic disorder with the number of neocortical synapses being a better correlate of cognition than either beta amyloid plaques or neurofibrillary tangles (27). The levels of the postsynaptic density scaffolding protein Homer 1a were recently shown to be positively correlated with cognitive function in aged mice (28), consistent with our observation that the levels were increased by fisetin treatment. Arc is necessary for durable LTP and lasting memories. Several lines of evidence (29) suggest that dysregulation of Arc expression in the hippocampus could contribute to cognitive impairment in AD and aging. Fisetin also maintained Arc levels in the old SAMP8 mouse brains. The postsynaptic scaffold protein SAP102 has been shown to be decreased in human AD brains with the level of reduction directly correlating with disease severity (30). Fisetin prevented the loss of SAP102 in the old SAMP8 mouse brains. Together, the maintenance of the levels of these proteins in the aging brain may contribute to the positive effects of fisetin on cognitive function in the old SAMP8 mice. We then asked whether fisetin might also modulate age-associated stress pathways whose inappropriate activation could contribute to cognitive dysfunction.
HSPs represent a major cellular defense against the proteotoxic stress that is characteristic of age-related neurodegenerative disorders. The consequences of HSP expression depend on the type of HSP, the disease, cell type, and brain region (31). The changes in HSP40, 60, and 90 observed in the hippocampus of old SAMP8 mice are indicative of stress, and fisetin maintained the levels of HSP60 and 90 at those seen in young mice. Consistent with our results, HSP60 has been reported to increase in senescent cells both in vitro and in vivo and may contribute to the age-dependent increase in inflammation (32).
Indeed, one of the most prominent manifestations of stress during aging is the production of inflammatory mediators accompanied by metabolic alterations. Although clinical trials with a few antiinflammatory drugs failed to prevent AD disease progression, epidemiological studies suggest that long-term use of antiinflammatory drugs may reduce the risk (33). Our data (14) showed an increase in inflammatory parameters in old SAMP8 mice. In the CNS, inflammation is often characterized by the activation of glial cells, mainly astrocytes and microglia. This age-associated phenotype is characteristic of the AD brain (34). Old SAMP8 mice also show astrocyte activation as determined by both Western blotting and immunohistochemistry for GFAP (14,15), and we demonstrate here that fisetin reduces the age-dependent increase in GFAP expression in the hippocampus as determined by Western blotting.
Recently, using mice overexpressing p25 specifically in neurons, it was shown that p25 induces neuroinflammation that is associated with astrocyte activation and synaptic damage (35,36). Previously, we found that in transgenic AD mice, fisetin prevented the highly significant increase in p25 levels in the AD mice and greatly reduced the p25/p35 ratio in both wild-type and AD mice as well as reducing astrocyte activation (6). However, in the SAMP8 mice, we found a decrease in p25 levels with aging despite an increase in astrocyte activation. Although several treatments have been shown to reduce p25 levels in SAMP8 mice (e.g., (37, 38)), the levels of p25 in old and young SAMP8 mice have not been previously compared. Moreover, recently, it was suggested (39) that the increase in p25 levels in transgenic AD mice is an artifact due to the overexpression of the amyloid precursor protein and presenilin. Importantly, the results presented here showing that fisetin can reduce astrocyte activation in the absence of increases in p25 indicate that fisetin’s beneficial effects are not specific to transgenic AD mice.
Microglia are the resident macrophages of the brain and play a central role during inflammation in the aging and AD brains (40). Although fisetin did not reduce the increased number of microglia cells found in the hippocampus of old SAMP8 mice, it reduced activation of the stress-induced kinase SAPK/JNK, which we had previously shown to be a good marker of microglial activation (19). Moreover, SAPK/JNK is activated in AD brains (41). An antiinflammatory action of fisetin in the brain is further supported by consistent decreases in a number of COX, 5-LOX, 15-LOX, and P450 epoxide metabolites with fisetin treatment. In transgenic AD mice, we also found that fisetin modulated the levels of several products of both COX and LOX metabolism (6).
Among the most interesting observations with fisetin is that it significantly increased the level of DHA in the brain. DHA is the primary structural fatty acid in the human brain and has been linked to cognitive performance. Although low plasma levels of DHA are associated with cognitive decline in elderly and AD patients, higher DHA intake and plasma levels inversely correlate with AD risk (42). DHA supplementation in aged animals enhances learning and memory and protects against Aβ and tau pathology in AD mouse models (42–44). The increase in DHA with fisetin could be a consequence of its reduced nonenzymatic oxidation, as suggested by lower levels of its oxidized metabolites, HDoHes. Importantly, similar effects of fisetin on DHA oxidation were seen in transgenic AD mice (6).
Aging is accompanied by strong metabolic alterations at the organismal level that are often associated with mitochondrial dysfunction (45,46). Our analysis of the metabolomic profile of the plasma of old SAMP8 mice revealed global pathway changes regarding the metabolism of amino acids, peptides, and lipids in comparison to the young SAMP mice (14). However, in contrast, to the results with J147 (14), fisetin only altered a few of these metabolites. Nevertheless, its effects on behavior were very similar to those of J147 (14). Moreover, it modulated the levels of proteins involved in synaptic function and inflammation in a manner similar to J147 (14). Thus, these results suggest that at least with respect to cognitive function, there may be a core set of pathways that contribute to dysfunction with aging and it is sufficient to modulate just these pathways to have a beneficial effect. However, the broader effects of J147 on the aging phenotype (14) may be important for protecting other functions that were not examined.
In summary, the purpose of this study was to determine if, similar to its effects in transgenic AD mice, fisetin could also prove beneficial in old SAMP8 mice, a potential model of sporadic AD. By assessing the relationship between aging and the therapeutic effects of fisetin in the SAMP8 mouse model of aging and sporadic AD, we identified several key processes that correlated well with its beneficial effects on cognitive function. These include the modulation of proteins involved in synaptic function and inflammation. Changes in both of these are implicated in the cognitive decline seen in normal aging as well as the more severe decline associated with AD. In addition, we provide a set of specific metabolic alterations that may underlie these processes. Thus, these results provide further support that fisetin or a derivative (47) might be beneficial for the treatment of age-associated neurodegenerative diseases.
Supplementary Material
Supplementary data is available at The Journals of Gerontology, Series A: Biological Sciences and Medical Sciences online.
Funding
This work was supported by the Salk Institute Pioneer Fund Postdoctoral Scholar Award and the Salk Nomis Fellowship Award to AC and grants from the Alzheimer’s Association, Burns Foundation, and National Institutes of Health (grant numbers RO1AG046153, RO1AG035055, and R42AI104034) to PM and DS. This work was also supported by a NINDS Neuroscience Core Grant for the Behavioral Testing Core Facility.
Conflict of Interest
The authors have no conflicts of interest.
Supplementary Material
Acknowledgments
We thank Joseph Chambers, Maria Encizo and Karen Suter for valuable help with the breeding and husbandry of the mice.
References
- 1. Schubert D, Maher P. An alternative approach to drug discovery for Alzheimer’s disease dementia. Future Med Chem. 2012;4:1681–1688. doi:10.4155/fmc.12.109 [DOI] [PubMed] [Google Scholar]
- 2. Frautschy SA, Cole GM. Why pleiotropic interventions are needed for Alzheimer’s disease. Mol Neurobiol. 2010;41:392–409. doi:10.1007/s12035-010-8137-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Golde TE, Schneider LS, Koo EH. Anti-aβ therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron. 2011;69:203–213.doi:10.1016/j.neuron.2011.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prior M, Chiruta C, Currais A, et al. Back to the future with phenotypic screening. ACS Chem Neurosci. 2014;5:503–513. doi:10.1021/cn500051h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maher P, Akaishi T, Abe K. Flavonoid fisetin promotes ERK-dependent long-term potentiation and enhances memory. Proc Natl Acad Sci USA. 2006;103:16568–16573. doi:10.1073/pnas.0607822103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Currais A, Prior M, Dargusch R, et al. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell. 2014;13:379–390. doi:10.1111/acel.12185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maher P, Dargusch R, Bodai L, Gerard PE, Purcell JM, Marsh JL. ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum Mol Genet. 2011;20:261–270. doi:10.1093/hmg/ddq460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maher P, Salgado KF, Zivin JA, Lapchak PA. A novel approach to screening for new neuroprotective compounds for the treatment of stroke. Brain Res. 2007;1173:117–125. doi:10.1016/j.brainres.2007.07.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gelderblom M, Leypoldt F, Lewerenz J, et al. The flavonoid fisetin attenuates postischemic immune cell infiltration, activation and infarct size after transient cerebral middle artery occlusion in mice. J Cereb Blood Flow Metab. 2012;32:835–843. doi:10.1038/jcbfm.2011.189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Swerdlow RH. Is aging part of Alzheimer’s disease, or is Alzheimer’s disease part of aging?Neurobiol Aging. 2007;28:1465–1480. doi:10.1016/j.neurobiolaging.2006.06.021 [DOI] [PubMed] [Google Scholar]
- 11. Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015;18:794–799. doi:10.1038/nn.4017 [DOI] [PubMed] [Google Scholar]
- 12. Morley JE, Armbrecht HJ, Farr SA, Kumar VB. The senescence accelerated mouse (SAMP8) as a model for oxidative stress and Alzheimer’s disease. Biochim Biophys Acta. 2012;1822:650–656. doi:10.1016/j.bbadis.2011.11.015 [DOI] [PubMed] [Google Scholar]
- 13. Cheng XR, Zhou WX, Zhang YX. The behavioral, pathological and therapeutic features of the senescence-accelerated mouse prone 8 strain as an Alzheimer’s disease animal model. Ageing Res Rev. 2014;13:13–37. doi:10.1016/j.arr.2013.10.002 [DOI] [PubMed] [Google Scholar]
- 14. Currais A, Goldberg J, Farrokhi C, et al. A comprehensive multiomics approach toward understanding the relationship between aging and dementia. Aging (Albany NY). 2015;7:937–955. doi:10.18632/aging.100838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Currais A, Prior M, Lo D, Jolivalt C, Schubert D, Maher P. Diabetes exacerbates amyloid and neurovascular pathology in aging-accelerated mice. Aging Cell. 2012;11:1017–1026. doi:10.1111/acel.12002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Prior M, Dargusch R, Ehren JL, Chiruta C, Schubert D. The neurotrophic compound J147 reverses cognitive impairment in aged Alzheimer’s disease mice. Alzheimers Res Ther. 2013;5:25. doi:10.1186/alzrt179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi:10.1007/s00401-009-0619-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lopes JP, Oliveira CR, Agostinho P. Neurodegeneration in an Abeta-induced model of Alzheimer’s disease: the role of Cdk5. Aging Cell. 2010;9:64–77. doi:10.1111/j.1474-9726.2009.00536.x [DOI] [PubMed] [Google Scholar]
- 19. Currais A, Farrokhi C, Dargusch R, Goujon-Svrzic M, Maher P. Dietary glycemic index modulates the behavioral and biochemical abnormalities associated with autism spectrum disorder. Mol Psychiatry. 2016;21:426–436. doi:10.1038/mp.2015.64 [DOI] [PubMed] [Google Scholar]
- 20. Buczynski MW, Dumlao DS, Dennis EA. An integrated omics analysis of eicosanoid biology. J Lipid Res. 2009;50:1015–1038. doi:10.1194/jlr.R900004-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thompson DA, Hammock BD. Dihydroxyoctadecamonoenoate esters inhibit the neutrophil respiratory burst. J Biosci. 2007;32:279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dobrian AD, Lieb DC, Cole BK, Taylor-Fishwick DA, Chakrabarti SK, Nadler JL. Functional and pathological roles of the 12- and 15-lipoxygenases. Prog Lipid Res. 2011;50:115–131. doi:10.1016/j.plipres.2010.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi:10.1161/ATVBAHA.110.207449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Joshi YB, Praticò D. The 5-lipoxygenase pathway: oxidative and inflammatory contributions to the Alzheimer’s disease phenotype. Front Cell Neurosci. 2014;8:436. doi:10.3389/fncel.2014.00436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nicholson JK, Holmes E, Kinross J, et al. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–1267. doi:10.1126/science.1223813 [DOI] [PubMed] [Google Scholar]
- 26. Guo L, Milburn MV, Ryals JA, et al. Plasma metabolomic profiles enhance precision medicine for volunteers of normal health. Proc Natl Acad Sci USA. 2015;112:E4901–E4910. doi:10.1073/pnas.1508425112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun. 2014;2:135. doi:10.1186/s40478-014-0135-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaja S, Sumien N, Borden PK, et al. Homer-1a immediate early gene expression correlates with better cognitive performance in aging. Age (Dordr). 2013;35:1799–1808. doi:10.1007/s11357-012-9479-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bramham CR, Alme MN, Bittins M, et al. The Arc of synaptic memory. Exp Brain Res. 2010;200:125–140. doi:10.1007/s00221-009-1959-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Proctor DT, Coulson EJ, Dodd PR. Reduction in post-synaptic scaffolding PSD-95 and SAP-102 protein levels in the Alzheimer inferior temporal cortex is correlated with disease pathology. J Alzheimers Dis. 2010;21:795–811. doi:10.3233/JAD-2010-100090 [DOI] [PubMed] [Google Scholar]
- 31. Leak RK. Heat shock proteins in neurodegenerative disorders and aging. J Cell Commun Signal. 2014;8:293–310. doi:10.1007/s12079-014-0243-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cappello F, Conway de Marcario E, Marino Gammaza A, et al. Hsp60 and human aging: Les liasons dangereuses. Front Biosci. 2013;18:626–637. doi:10.2741/4126 [DOI] [PubMed] [Google Scholar]
- 33. Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2:a006346. doi:10.1101/cshperspect.a006346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li C, Zhao R, Gao K, et al. Astrocytes: implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr Alzheimer Res. 2011;8:67–80. doi:10.2174/156720511794604543 [DOI] [PubMed] [Google Scholar]
- 35. Muyllaert D, Terwel D, Kremer A, et al. Neurodegeneration and neuroinflammation in cdk5/p25-inducible mice. Am J Pathol. 2008;172:470–485. doi:10.2353/ajpath.2008.070693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sundaram JR, Chan ES, Poore CP, et al. Cdk5/p25-induced cytosolic PLA2-mediated lysophosphatidylcholine production regulates neuroinflammation and triggers neurodegeneration. J Neurosci. 2012;32:1020–1034. doi:10.1523/JNEUROSCI.5177-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gutierrez-Cuesta J, Sureda FX, Romeu M, et al. Chronic administration of melatonin reduces cerebral injury biomarkers in SAMP8. J Pineal Res. 2007;42:394–402. doi:10.1111/j.1600-079X.2007.00433.x [DOI] [PubMed] [Google Scholar]
- 38. Zhang ZX, Zhao RP, Wang DS, Li YB. Fuzhisan ameliorates the memory deficits in aged SAMP8 mice via decreasing Aβ production and tau hyperphosphorylation of the hippocampus. Neurochem Res. 2016;41:3074–3082. doi:10.1007/s11064-016-2028-4 [DOI] [PubMed] [Google Scholar]
- 39. Saito T, Matsuba Y, Yamazaki N, Hashimoto S, Saido TC. Calpain activation in Alzheimer’s model mice is an artifact of APP and presenilin overexpression. J Neurosci. 2016;36:9933–9936. doi:10.1523/JNEUROSCI.1907-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem Pharmacol. 2014;88:594–604. doi:10.1016/j.bcp.2014.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ploia C, Antoniou X, Sclip A, et al. JNK plays a key role in tau hyperphosphorylation in Alzheimer’s disease models. J Alzheimers Dis. 2011;26:315–329. doi:10.3233/JAD-2011-110320 [DOI] [PubMed] [Google Scholar]
- 42. Yurko-Mauro K. Cognitive and cardiovascular benefits of docosahexaenoic acid in aging and cognitive decline. Curr Alzheimer Res. 2010;7:190–196. doi:10.2174/156720510791050911 [DOI] [PubMed] [Google Scholar]
- 43. Lim GP, Calon F, Morihara T, et al. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J Neurosci. 2005;25:3032–3040. doi:10.1523/JNEUROSCI.4225-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Green KN, Martinez-Coria H, Khashwji H, et al. Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid-beta and tau pathology via a mechanism involving presenilin 1 levels. J Neurosci. 2007;27:4385–4395. doi:10.1523/JNEUROSCI.0055-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi:10.1038/nature05292 [DOI] [PubMed] [Google Scholar]
- 46. Navarro A, Boveris A. Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s disease. Front Aging Neurosci. 2010;2012:2. doi:10.3389/fnagi.2010.00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chiruta C, Schubert D, Dargusch R, Maher P. Chemical modification of the multitarget neuroprotective compound fisetin. J Med Chem. 2012;55:378–389. doi:10.1021/jm2012563 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.