

Abstract
Background
We investigated the independent effects of Alzheimer’s disease (AD) and cerebrovascular disease (CVD) pathologies on brain structural changes and cognition.
Methods
Amyloid burden (Pittsburgh compound B [PiB] retention ratio), CVD markers (volume of white matter hyperintensities [WMH] and number of lacunae), and structural changes (cortical thickness and hippocampal shape) were measured in 251 cognitively impaired patients. Path analyses were utilized to assess the effects of these markers on cognition.
Results
PiB retention ratio was associated with hippocampal atrophy, which was associated with memory impairment. WMH were associated with frontal thinning, which was associated with executive and memory dysfunctions. PiB retention ratio and lacunae were also associated with memory and executive dysfunction without the mediation of hippocampal or frontal atrophy.
Conclusions
Our results suggest that the impacts of AD and CVD pathologies on cognition are mediated by specific brain regions.
Keywords: Cognition, Amyloid, Pittsburgh compound B, Atrophy, Cortical thickness, Hippocampus, Path analysis, Cerebrovascular disease
1. Introduction
Alzheimer’s disease (AD) and subcortical vascular dementia (SVaD) are common types of dementia in the elderly [1,2]. AD results from an imbalance in amyloid beta (Aβ) production and clearance, which lead to amyloid plaque accumulation. SVaD is characterized by extensive cerebrovascular disease (CVD) magnetic resonance imaging (MRI) markers, including white matter hyperintensities (WMH) and lacunae [3]. Postmortem studies have shown that AD and SVaD pathologies frequently co-occur [4,5]. There is also increasing evidence that AD and CVD pathologies contribute independently to the impairment of specific cognitive domains [6,7]. Pathological and Pittsburgh compound B (PiB) positron emission tomography (PET) studies have shown that AD pathology is associated with memory dysfunction, whereas CVD pathology affects multiple cognitive domains, including memory and executive function [7–9]. However, controversy still remains regarding these conclusions [10].
Previous studies of AD patients have shown that amyloid burden is associated with medial temporal cortical atrophy, including that of the hippocampus [11]. Also, recent studies have shown that WMH might affect brain atrophy, although those WMH were located in subcortical regions [12,13]. However, these studies did not evaluate the independent effects of amyloid burden and CVD on brain atrophy. Furthermore, no studies have evaluated the relationship among amyloid, CVD, brain atrophy, and cognitive impairments. Brain atrophy associated with amyloid burden [14] or WMH [12,13,15] was reported to correlate strongly with cognitive impairment [11,12,15]. Some researchers have also proposed that cognitive impairment may be more highly correlated with cortical atrophy than with amyloid burden [11] or WMH [16], implying that cortical atrophy may be the final common pathway of AD and SVaD. Therefore, it is possible that amyloid burden and CVD markers might drive cognitive impairment via the mediation of cortical or hippocampal atrophy. Alternatively, amyloid burden and CVD markers might affect cognitive impairment without the mediation of cortical or hippocampal atrophy. Actually, some studies have shown that the significant relationship between amyloid burden or CVD markers and cognitive impairment remains after controlling for cortical or hippocampal atrophy [17,18].
In this study, we investigated a large sample of patients with cognitive impairments who underwent PiB-PET, MRI, and detailed neuropsychological testing. The first goal was to evaluate the independent effects of amyloid burden evaluated through PiB-PET, and CVD, measured by the volume of WMH and number of lacunae, on the topography of brain atrophy, including hippocampal deformities or cortical thinning. The following hypotheses concerning the relationships among amyloid burden, CVD, brain atrophy, and cognitive impairment using path analyses were also tested. First, amyloid burden or CVD may drive memory dysfunction via hippocampal atrophy or frontal thinning. Second, amyloid burden or CVD might drive executive dysfunction via frontal thinning.
2. Methods
2.1. Participants
We prospectively recruited 251 patients with cognitive impairment who had undergone PiB-PET and structural brain MRI during the period from July 2007 to July 2011. We included 45 patients with amnestic mild cognitive impairment (aMCI), 69 with probable AD dementia, 67 with subcortical vascular MCI (svMCI), and 70 with SVaD who had all been clinically diagnosed at Samsung Medical Center. Probable AD dementia patients met the criteria proposed by the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association [19]. Patients with SVaD met the diagnostic criteria for vascular dementia as determined by the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, and also fulfilled the imaging criteria for SVaD as proposed by Erkinjuntti et al. [20]. The aMCI and svMCI patients met Petersen’s criteria for MCI with modifications, as previously described [21]. All svMCI and SVaD patients had severe WMH defined as a cap or band (periventrivular WMH or PWMH) ≥ 10 mm and deep white-matter lesions (deep WMH or DWMH) ≥ 25 mm, as modified from Fazekas’ MRI ischemia criteria [22]. All aMCI and AD were classified as having minimal (PWMH <5 mm and DWMH <10 mm) or moderate (between minimal and severe grades) WMH. Detailed criteria for aMCI and svMCI are described in online data Supplement 1. Patients with aMCI or AD were considered to have AD-related cognitive impairment (ADCI), whereas those with svMCI or SVaD were considered to have subcortical vascular cognitive impairment (SVCI). Patients with territorial infarctions and those with WMH due to radiation injury, multiple sclerosis, vasculitis, or leukodystrophy were excluded.
Each patient underwent a clinical interview; neurological examinations; a battery of neuropsychological tests; laboratory tests including a complete blood count, blood chemistry, vitamin B12/folate measurement, syphilis serology, thyroid function, apolipoprotein E (APOE) genotyping, and PiB-PET scan.
Written informed consent was obtained from every patient. The institutional review board of Samsung Medical Center approved the study protocol. Participant characteristics are summarized in Table 1.
Table 1.
Demographics, clinical characteristics, and MRI markers in study participants
| Group | Total | ADCI
|
SVCI
|
||||
|---|---|---|---|---|---|---|---|
| Subtotal | aMCI | AD | Subtotal | svMCI | SVaD | ||
| Number | 251 | 114 | 45 | 69 | 137 | 67 | 70 |
| Demographics | |||||||
| Age, y | 72.1 (8.1) | 70.0 (8.9)* | 70.0 (8.0) | 70.0 (9.5) | 73.8 (6.9) | 73.7 (6.7) | 74.0 (7.1) |
| Sex, female, n (%) | 146 (58.2) | 64 (56.1) | 20 (44.4) | 44 (63.8) | 82 (59.9) | 41 (61.2) | 41 (58.6) |
| Education, y | 10.1 (5.3) | 11.3 (5.2)* | 12.5 (4.6) | 10.6 (5.5) | 9.0 (5.2) | 9.4 (5.4) | 8.7 (5.0) |
| Cardiovascular risk factors, n (%) | |||||||
| Hypertension | 157 (62.5) | 52 (45.6)* | 17 (37.8) | 35 (50.7) | 105 (76.6) | 50 (74.6) | 55 (78.6) |
| Diabetes mellitus | 51 (20.3) | 16 (14.0)* | 5 (11.1) | 11 (15.9) | 35 (25.5) | 17 (25.4) | 18 (25.7) |
| Hyperlipidemia | 73 (29.1) | 26 (22.8)* | 10 (22.2) | 16 (23.2) | 47 (34.3) | 20 (29.9) | 27 (38.6) |
| Heart disease | 37 (14.7) | 12 (10.5) | 7 (15.6) | 5 (7.2) | 25 (18.2) | 17 (25.4) | 8 (11.4) |
| APOE genotype, n (%)† | |||||||
| ε2 allele carrier | 25 (10.2) | 8 (7.1)* | 3 (6.7) | 5 (7.5) | 17 (12.7) | 10 (14.9) | 7 (10.4) |
| ε4 allele carrier | 88 (35.8) | 53 (47.3)* | 19 (42.2) | 34 (50.7) | 35 (26.1) | 15 (22.4) | 20 (29.9) |
| Small vessel MRI markers | |||||||
| WMH volume, ml | 23.5 (22.3) | 4.8 (6.7)* | 3.1 (3.1) | 5.9 (8.1) | 39.0 (18.3) | 34.9 (17.8) | 43.0 (18.1) |
| Lacunae, n | 6.8 (11.8) | 0.5 (1.6)* | 0.5 (2.1) | 0.4 (1.0) | 11.9 (13.9) | 7.5 (8.4) | 16.1 (16.6) |
| Global PiB global retention ratio | 1.8 (0.5) | 2.0 (0.5)* | 1.8 (0.5) | 2.2 (0.4) | 1.5 (0.4) | 1.5 (0.4) | 1.5 (0.5) |
| PiB-positive patients, n (%)‡ | 134 (53.4) | 90 (78.9) | 28 (62.2) | 62 (89.9) | 44 (32.1) | 21 (31.3) | 23 (32.9) |
| Intracranial volume (ml) | 1362.2 (126.3) | 1353.2 (129.4) | 1377.5 (119.8) | 1337.4 (133.7) | 1369.6 (123.7) | 1356.5 (104.7) | 1382.1 (139.2) |
| Mean frontal thickness (mm) | 2.9 (0.2) | 2.9 (0.2)* | 3.0 (0.1) | 2.9 (0.3) | 2.8 (0.2) | 2.9 (0.2) | 2.7 (0.3) |
| Mean parietal thickness (mm) | 2.7 (0.3) | 2.7 (0.3) | 2.8 (0.2) | 2.6 (0.3) | 2.7 (0.2) | 2.7 (0.2) | 2.6 (0.2) |
| Mean temporal thickness (mm) | 2.9 (0.3) | 3.0 (0.3) | 3.1 (0.2) | 2.9 (0.3) | 2.9 (0.3) | 3.0 (0.2) | 2.8 (0.3) |
| Hippocampal volume (ml) | 2.4 (0.5) | 2.3 (0.5)* | 2.6 (0.5) | 2.1 (0.4) | 2.5 (0.5) | 2.5 (0.5) | 2.4 (0.4) |
| Executive score | 24.2 (11.6) | 26.6 (11.9)* | 33.7 (9.9) | 20.5 (10.4) | 23.1 (10.6) | 29.3 (8.5) | 15.8 (8.7) |
| Memory score | 41.8 (21.6) | 36.0 (18.5)* | 49.9 (17.0) | 25.5 (11.8) | 47.0 (22.7) | 61.0 (19.5) | 33.3 (16.2) |
| MMSE | 22.7 (5.1) | 22.0 (5.2)* | 26.1 (2.5) | 19.2 (4.7) | 23.3 (4.9) | 26.2 (3.0) | 20.6 (4.8) |
Abbreviations: MRI, magnetic resonance imaging; aMCI, amnestic mild cognitive impairment; AD, Alzheimer’s disease; ADCI, Alzheimer’s disease-related cognitive impairment; SVCI, subcortical vascular cognitive impairment; svMCI, subcortical vascular mild cognitive impairment; SVaD, subcortical vascular dementia; APOE, apolipoprotein E; WMH, white matter hyperintensities; PiB, Pittsburgh compound B; MMSE, Mini-Mental Status Examination.
NOTE. Values are expressed as mean (standard deviation) or number (%).
P value <.05 in the Chi-square test or independent t-test comparing ADCI and SVCI.
APOE genotyping was performed in 246 of 251 participants.
Patients were considered PiB-positive if their global PiB retention ratio was greater than 1.5.
2.2. PiB-PET acquisition
[11C] PiB-PET scanning was performed at Samsung Medical Center or Asan Medical Center using a Discovery STe PET/CT (computed tomography) scanner (GE Medical Systems, Milwaukee, WI) in three-dimensional (3D) scanning mode with 35 slices at 4.25-mm thickness spanning the entire brain. [11C] PiB was injected into the antecubital vein as a bolus at a mean dose of 420 MBq (i.e., range 259–550 MBq). A CT scan was performed for attenuation correction at 60 min after injection. A 30-min emission static PET scan was initiated. The specific radioactivity of [11C]-PiB at the time of administration was greater than 1500 Ci/mmol for patients, and the radiochemical yield was greater than 35%. The radiochemical purity of the tracer was greater than 95% in all PET studies.
2.3. PiB-PET data analysis
PiB-PET images were co-registered to individual MRIs, normalized to a T1-weighted MRI template. The quantitative regional values of PiB retention on the spatially normalized PiB images were obtained using an automated volume of interest (VOI) analysis drawn from the automated anatomical labeling atlas. Data processing was performed using SPM version 5 under Matlab 6.5 (Mathworks, Natick, MA).
To measure PiB retention, we used the cerebral cortical region to cerebellum uptake ratio (UR). The global PiB retention ratio was calculated from the volume-weighted average UR of 28 bilateral cerebral cortical VOIs. Detailed methods for the calculation of global PiB retention ratio are described in online data Supplement 2. We defined the PiB retention ratio as a continuous variable indexing amyloid burden.
2.4. MRI Acquisition
MRI was performed with a 3.0 Tesla MRI scanner (Achieva; Philips Medical Systems, Best, The Netherlands) at Samsung Medical Center. 3D T1 turbo field echo and 3D fluid-attenuated inversion recovery (FLAIR) were performed for all 251 subjects. Detailed imaging parameters are described in online data Supplement 3.
2.5. Measurement of WMH volume
We quantified WMH volume on FLAIR images using an automated method, as previously described [23]. Due to WMH segmentation errors, WMH volumes could not be calculated for six patients.
2.6. Lacunae on MRI
Lacunae were defined as small lesions (≥3 mm and ≤ 15 mm in diameter) with low signal on T1-weighted images, high signal on T2-weighted images, and perilesional halo on FLAIR images. Two experienced neurologists blinded to the clinical information reviewed the number and location of lacunae on FLAIR images. The rate of agreement between the two neurologists was 83.0%. Consensus was reached in all cases of discrepancy.
2.7. Cortical thickness data analysis
T1-weighted images were processed using the standard Montreal Neurological Institute anatomic pipeline. Further image processing for cortical thickness is described in online data Supplement 4. The individual cortical surface of each subject was registered to the precategorized template and divided into frontal, temporal, parietal, and occipital lobes via automated processes. The averaged values of the thickness of the whole vertices in each hemisphere and lobar region were used for the global analysis. In this way, we obtained mean thickness of the frontal lobe (mean frontal thickness). Intracranial volume (ICV) was defined as the sum of gray matter, white matter, and cerebrospinal fluid (CSF) volume. We included ICVas a covariate to control for the effect of brain size. Nine of 251 patients (one aMCI, two svMCI, and six SVaD) were excluded from the cortical thickness analysis due to registration errors during preprocessing. As a result, data from 242 patients were successfully measured.
2.8. Hippocampal shape and volume measurement
Hippocampal shape and volume were measured with a surface-based method, described in online data Supplement 5. Vertex-wise hippocampal deformation data and hippocampal volume were measured successfully in 238 patients. Due to extraction error, data for 13 patients (two aMCI, two AD, five svMCI, and four SVaD) could not be obtained.
2.9. Neuropsychological tests
All patients underwent a standardized neuropsychological battery called the Seoul Neuropsychological Screening Battery [24]. The battery contains tests evaluating attention, language, praxis, four elements of Gerstmann syndrome, visuospatial processing, verbal and visual memory, and executive function. Based on these results, we calculated memory and executive subdomain scores as described in a previous study [25] and online data Supplement 6. The memory scores ranged from 0 to 150. The executive scores ranged from 0 to 55.
2.10. Statistical analysis
To investigate independent effects of global PiB retention ratio, WMH volume, and the number of lacunae on mean cortical thickness and hippocampal volume, we entered them as predictors in general linear models controlling for age, gender, education, ICV, and clinical group (ADCI vs. SVCI).
To investigate the topography of cortical thinning and hippocampal deformity associated with global PiB retention ratio, WMH volume, and number of lacunae, we entered the three markers simultaneously in a general linear model as predictors for regional cortical thickness and hippocampal deformity on a vertex-by-vertex basis. Covariates included age, gender, education, ICV, and clinical group. The resulting statistical maps were thresholded after pooling the P-values from the regression analyses using the false discovery rate theory at a Q value of 0.05. Among the 245 patients with successful WMH volume measurement, eight patients with cortical thickness registration errors and 11 patients with hippocampal extraction errors were further excluded from cortical thickness and hippocampal shape analyses. As a result, data from 237 patients were used for cortical thickness analysis, and data from 234 patients were used for hippocampal shape analysis.
To evaluate the relative effects of amyloid burden and CVD markers on cognition with or without the mediation of brain atrophy, path analysis was performed after controlling for age, gender, education, ICV, and clinical group. Path analysis was used to simultaneously consider the direct, indirect, and total effects of predictors on outcomes through mediators. Path analyses using memory score and executive score as the outcome variables were performed using hippocampal volume and mean frontal thickness as mediators because the hippocampus is known to be associated with memory function, while the frontal cortex is associated with executive function. PiB retention ratio and WMH are also predominantly associated with hippocampal shape deformities and frontal cortical thinning, respectively. We also entered mean frontal thickness as a mediator for memory scores because frontal impairment is associated with memory dysfunction [26]. Path analysis for memory score was performed using data from 216 patients after excluding 35 patients (6 with WMH segmentation errors, 8 with registration errors for cortical thickness, 11 with hippocampal extraction errors, and 10 with missing memory score data). Path analysis for executive score was performed using data from 214 patients after excluding 37 patients (6 with WMH segmentation errors, 8 with registration errors for cortical thickness, and 23 with missing executive score data). These models showed good fit to the memory score (χ2 = 13.68, df = 16, P =.855, CFI > 0.999, root mean square error of approximation [RMSEA] <0.001) and executive score (χ2 = 14.96, df = 16, P = .52, CFI >0.999, RMSEA <0.001) data.
Amos Version 18.0 software (SPSS, Chicago, IL) was used for all path analyses using maximum likelihood estimation.
3. Results
3.1. Effects of PiB retention ratio, WMH volume, and the number of lacunae on cortical thickness
Global PiB retention ratio was not associated with mean cortical thickness (β = −0.05, SE = 0.03, P = .067). Higher WMH volume was associated with lower mean cortical thickness (β = −0.002, SE = 0.001, P =.033), whereas the number of lacunae was not (β = −0.001, SE = 0.001, P = .331).
Global PiB retention ratio was associated with cortical thinning in bilateral medial and lateral temporal, lateral parietal, and precuneus regions (Fig. 1). Higher WMH volume was associated with cortical thinning in bilateral medial and lateral frontal, lateral temporal, insula, cingulate, and lingual regions. The number of lacunae was not associated with regional cortical thinning.
Fig. 1.
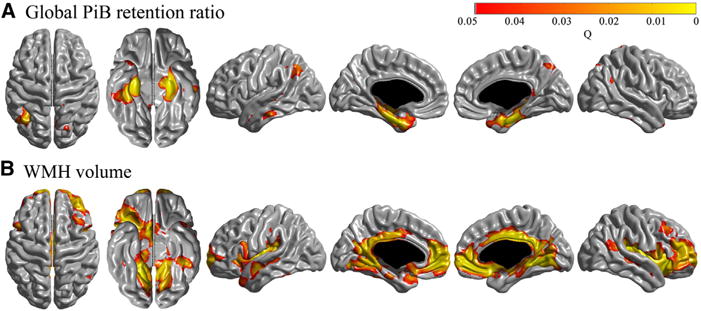
Region of cortical thinning associated with Pittsburgh compound B (PiB) (A) and white matter hyperintensities (WMH) volume (B). Global PiB retention ratio, WMH volume, and number of lacunae were entered in a general linear model. Age, gender, education, clinical group, and intracranial volume were entered as covariates. Global PiB retention ratio was associated with cortical thinning in bilateral medial, lateral temporal, lateral parietal, and precuneus regions. Higher WMH volume was associated with cortical thinning in bilateral medial and lateral frontal, lateral temporal, insula, cingulate, and lingual regions. The number of lacunae was not associated with regional cortical thinning.
3.2. Effects of PiB retention ratio, WMH volume, and the number of lacunae on hippocampal shape
Global PiB retention ratio was associated with hippocampal volume (β = −0.21, SE = 0.06, P < .001). WMH volume (β = −0.004, SE = 0.002, P =.085) and the number of lacunae (β = 0.001, SE = 0.003, P =.838) were not associated with hippocampal volume.
PiB retention ratio was associated with shape deformities in the superior-lateral heads, bilaterally in the medial and lateral bodies of the hippocampus, in the inferior body of the right hippocampus, and the inferior-medial head of the left side of the hippocampus. WMH volume was associated with shape deformities in the bilateral inferior bodies of the hippocampus and the lateral head and body of the right side of the hippocampus. The number of lacunae was associated with deformity in the lateral body and tail of the right side of the hippocampus (Fig. 2).
Fig. 2.
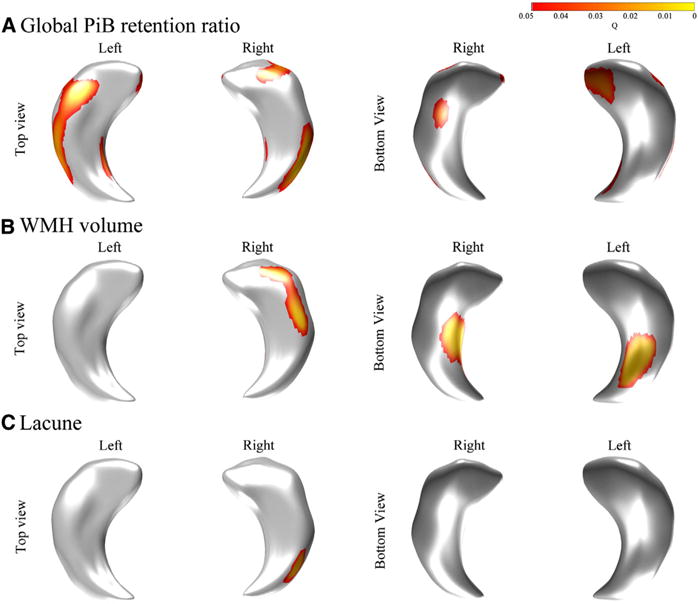
Region of hippocampal deformity associated with global Pittsburgh compound B (PiB) retention ratio (A), white matter hyperintensities (WMH) volume (B), and the number of lacunae (C). Global PiB retention ratio was related to deformity in the superior-lateral heads, bilaterally in the medial and lateral bodies of the hippocampus, in the inferior body of the right side of the hippocampus, and the inferior-medial head of the left side of the hippocampus. WMH volume was associated with deformity in the bilateral inferior bodies of the hippocampus and lateral head and body of the right side of the hippocampus. The number of lacunae was associated with deformity in the lateral body and tail of the right side of the hippocampus. Global PiB retention ratio, WMH volume, and the number of lacunae were entered in a general linear model. Age, gender, education, clinical group, and intracranial volume were entered as covariates.
3.3. Relationships among PiB retention ratio, CVD markers, brain atrophy, and memory scores
In the path analysis for memory score (Table 2), global PiB retention ratio was associated with mean hippocampal volume (P =.010), which was further associated with memory function score (P =.010). Global PiB retention ratio was associated with memory score without mediation of hippocampus volume or mean frontal thickness (P = .010). WMH were associated with mean frontal thickness (P = .010), which was further associated with memory function score (P = .010). Lacunae were associated with memory score without the mediation of hippocampus volume or mean frontal thickness (P = .029) (Fig. 3).
Table 2.
Effects of predictors (Pittsburgh compound [PiB] retention ratio, WMH volumes, and the number of lacunae) on cognition (memory and executive scores) through mediators (hippocampal volume and/or mean frontal thickness)
| Path analysis 1
|
Path analysis 2
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hippocampal volume
|
Mean frontal thickness
|
Memory score
|
Mean frontal thickness
|
Executive score
|
|||||||||||
| β | SE | P value | β | SE | P value | β | SE | P value | β | SE | P value | β | SE | P value | |
| Global PiB retention ratio | −0.20 | 0.05 | .010 | 0.002 | 0.02 | .964 | −13.77 | 2.43 | .010 | 0.002 | 0.03 | .908 | −4.44 | 1.41 | .010 |
| WMH volume | −0.004 | 0.003 | .105 | −0.003 | 0.001 | .010 | −0.05 | 0.09 | .588 | −0.003 | 0.001 | .022 | −0.06 | 0.05 | .309 |
| Lacunae | 0.0003 | 0.002 | .894 | −0.001 | 0.001 | .327 | −0.36 | 0.14 | .029 | −0.001 | 0.002 | .454 | −0.22 | 0.07 | .010 |
| Hippocampal volume | 13.60 | 3.44 | .010 | ||||||||||||
| Mean frontal thickness | 24.06 | 7.31 | .010 | 21.72 | 3.27 | .010 | |||||||||
Abbreviations: β, unstandardized beta coefficient; PiB, Pittsburgh compound B; SE, standard error; WMH, white matter hyperintensities.
NOTE. Values shown are the results of path analyses for memory score (path analysis 1) and executive score (path analysis 2).
Fig. 3.
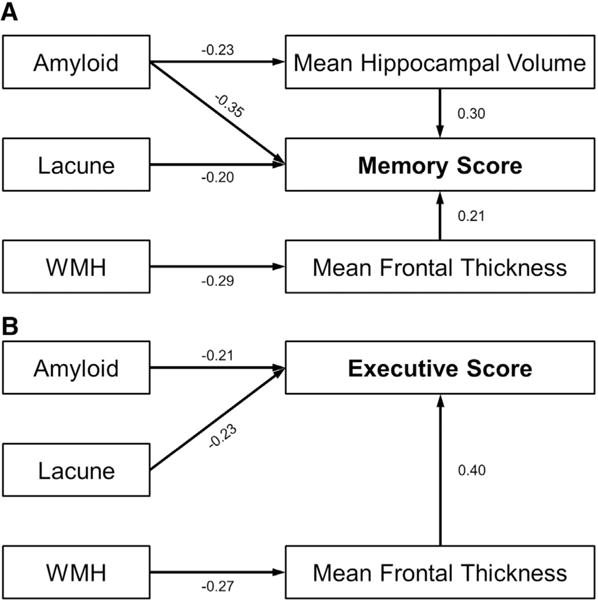
Schematic diagram of the path analyses. Path analysis for memory function score (A) and executive function score (B). Mean hippocampal volume and mean frontal thickness were entered as mediator variables for memory function score. Mean frontal thickness was entered as a mediator variable for executive function score. Amyloid burden, white matter hyper-intensities (WMH), and the number of lacunae were entered as predictors. Age, gender, education, clinical group, and intracranial volume were entered as covariates. The numbers on the paths are standardized coefficients. Direct paths that were statistically significant were reported.
Table 3 shows the total effects of PiB retention ratio, lacunae, and WMH on memory score; PiB retention ratio and lacunae account for 0.42 and 0.22 of the memory score, respectively. The total effect of WMH on memory score was 0.17, but it was not statistically significant (P = .091).
Table 3.
Total effects of imaging markers on cognition
| Domain | Predictors | Total effect
|
||
|---|---|---|---|---|
| Standardized β | SE | P value | ||
| Memory score | Global PiB retention ratio | −0.42 | 0.06 | .010 |
| WMH volume | −0.17 | 0.10 | .091 | |
| Lacunae | −0.22 | 0.08 | .026 | |
| Executive score | Global PiB retention ratio | −0.20 | 0.07 | .010 |
| WMH volume | −0.21 | 0.10 | .039 | |
| Lacunae | −0.26 | 0.07 | .010 | |
Abbreviations: PiB, Pittsburgh compound B; SE, standard error; WMH, white matter hyperintensities.
NOTE. Values shown are the results of path analyses for memory score (path analysis 1) and executive score (path analysis 2).
Because amyloid burden was associated with brain atrophy in the temporal-parietal region in the cortical thickness analysis, we performed further path analysis on memory score using mean temporal-parietal thickness as a mediator variable (Supplementary Table 1 and Supplementary Fig. 1). PiB retention ratio was associated with memory score with and without the mediation of mean temporal-parietal thickness.
3.4. Relationships among PiB retention ratio, CVD markers, brain atrophy, and executive scores
In the path analysis for executive score (Table 2), global PiB retention ratio was associated with executive score without the mediation of mean frontal thickness (P = .010). WMH were associated with mean frontal thickness (P =.022), which was further associated with executive score (P =.010). The number of lacunae was associated with executive score without the mediation of mean frontal thickness (P = .010) (Fig. 3).
Table 3 shows the total effects of PiB retention ratio, lacunae, and WMH on executive score; PiB retention ratio, lacunae, and WMH account for 0.20, 0.26, and 0.21 of the executive score, respectively.
4. Discussion
This study reports new data for potential mechanisms underlying the relationships among amyloid burden, CVD, brain atrophy, and cognitive impairment in a large cohort of carefully phenotyped, cognitively impaired patients using noninvasive amyloid imaging and structural MRI for markers of CVD, cortical thickness, and hippocampal atrophy. The major findings of this study are as follows. First, amyloid burden was found to be associated with the hippocampal atrophy or temporal-parietal thinning, which was further associated with memory impairment. Second, WMH were associated with frontal thinning, which was further associated with executive and memory dysfunctions. Finally, amyloid burden and lacunae were also associated with memory and executive dysfunction without the mediation of hippocampal or frontal atrophy. Taken together, these findings suggest that brain atrophy is the common pathway between amyloid burden or CVD and cognitive impairments. Further, these findings indicate that amyloid burden and CVD affect cognitive impairment without the mediation of brain atrophy.
We found that amyloid burden and CVD independently affect region-specific brain atrophy. That is, PiB retention ratio was associated with cortical thinning in the medial and lateral temporal, lateral parietal, and precuneus regions. Furthermore, the PiB retention ratio was associated with hippocampal atrophy, especially in the superior-lateral and inferior-medial portions of the head and body, which corresponded to the cornu ammonis (CA) 1 and subiculum [27]. The topography of hippocampal shape deformities and cortical thinning are consistent with previous studies of AD patients [28–30].
WMH were associated with cortical thinning in the frontal, perisylvian, and lingual regions. Previous studies showed that WMH were associated with brain atrophy [13,15], and a more recent study evaluated the topography of cortical thinning related to WMH volume [12]. However, unlike previous studies, our findings suggest that WMH are responsible for cortical thinning in these regions regardless of the effects of amyloid burden and lacunae. In this study, there was no association between lacunae and cortical atrophy. Previous studies have shown inconsistent results on the relationship between the number of lacunae and cortical atrophy [31,32]. One study on patients with cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalo pathy (CADASIL) showed that lacuna burden was associated with cortical changes [31]. However, these findings are inconsistent with a previous study of cognitively impaired patients with varying degrees of CVD, which is similar to the subjects in this study, suggesting that WMH had a significant effect on cortical gray matter volume, but lacunae did not [13]. We also found that WMH and lacunae were associated with shape deformities in the superior-lateral head and body (CA1) and inferior body (CA 1 and subiculum) and superior-lateral body (CA1) of the hippocampus, respectively. These subregions are known to be susceptible to ischemia [33].
Our first conclusion that brain atrophy is the common pathway between amyloid or CVD and cognitive impairments is supported by the following observations: (1) amyloid burden was associated with the temporal-parietal thinning including hippocampal atrophy, which was further associated with memory impairment; (2) WMH was associated with frontal thinning, which was further associated with executive and memory dysfunctions. Although several studies have shown that amyloid burden and CVD were independently associated with cognitive impairment [11,18], most studies did not evaluate the mediative effects of brain atrophy on the relationships between amyloid burden or CVD and cognitive impairments. A recent study showed that amyloid burden affected hippocampal atrophy, which in turn led to memory dysfunction [11]. One previous study from our group revealed that WMH were associated with frontal thinning, which is further associated with executive dysfunction [12]. Therefore, our findings suggest that amyloid burden and CVD might affect memory and executive dysfunction through hippocampal and frontal atrophy, respectively.
We found that WMH affected memory dysfunction with the mediation of frontal thinning. WMH are known to involve frontal-subcortical circuits responsible for not only executive but also memory dysfunction [34]. Indeed, previous studies have revealed that WMH [35] and frontal lesions [36] were associated with memory dysfunction.
Our study provides new evidence that amyloid burden is also associated with memory and executive dysfunction without the mediation of hippocampal or frontal atrophy. There have been only a few studies evaluating the relationships among amyloid burden, the hippocampus, and memory function [11]. Unlike the present findings, these previous studies suggested that amyloid burden affected memory dysfunction through the mediation of hippocampal atrophy. This discrepancy might be due to differences in study population (MCI or dementia patients with varying degrees of ischemia in our sample compared with nondemented subjects with mild degrees of ischemia in the previous study).
Amyloid burden had a significant effect on cognition without the mediation of hippocampal atrophy or frontal thinning (Fig. 3). Also, amyloid burden had significant effect on memory function without the mediation of temporal-parietal thinning. The direct effects of amyloid burden on cognition without the mediation of brain atrophy may be due to the fact that amyloid burden affects cortical synaptic/network dysfunction. Indeed, previous studies have shown that amyloid burden affects the default mode network [37,38], which is associated with memory dysfunction [37]. Additionally, amyloid burden may affect functional networks involving the frontal lobes because amyloid burden is distributed predominantly in the frontal regions [37]. Our suggestion seems to be supported by previous studies that have found that the PiB retention ratio is associated with executive dysfunction and memory dysfunction in nondemented subjects [39]. Also, previous studies have shown that synaptic/network dysfunction precedes cortical thinning [40]. Therefore, it is possible that amyloid burden might affect synaptic/network dysfunction without brain atrophy. Future studies are needed to investigate this hypothesis. Alternatively, amyloid burden might have affected memory or executive function through atrophy in brain regions that were not considered in the current path analyses.
We also found that lacunae, like WMH, affected executive and memory dysfunctions. However, in contrast to WMH, the effects of lacunae on cognition occurred without the mediation of frontal thinning. Furthermore, the relative effects of lacunae on cognition were greater than those of WMH. For example, the relative effect of lacunae on executive dysfunctions was 0.26, whereas that of WMH was 0.21. The reason that the effects of WMH on cortical thinning were greater than those of lacunae, whereas the effects of lacunae on cognition were greater than those of WMH remains unknown. However, this might be related to differences in the topography and the severity of involvement between WMH and lacuna. Specifically, WMH usually involves widespread white matter regions that contain long association fasciculi that connect various cortical regions. However, most lacunae occur focally in subcortical gray matter [13] that does not traverse via a long association fasciculus but that are major structures in frontal-subcortical circuits, associated with cognitive impairments [34]. Also, lacunae reflect a complete infarction, whereas WMH reflect incomplete ischemic injury [3]. Therefore, WMH affect long association fasciculi, which in turn leads to the secondary degeneration of cortical thinning, whereas lacunae cause a complete obstruction in major structures in frontal subcortical circuits, which leads to cognitive impairment.
When the relative amount of amyloid influence and CVD markers on cognition were compared after controlling for each other, amyloid burden (0.42) had a greater impact on memory dysfunction than CVD (lacunae, 0.22). In contrast, the total effect of CVD (0.47) on executive dysfunction (lacunae, 0.26 and WMH, 0.21) was greater than that of amyloid burden (0.20). Although these findings are consistent with general concepts, interestingly, the effects of amyloid burden on executive dysfunction and the effects of CVD on memory dysfunction were greater than expected. This finding might be explained by the fact that AD and SVaD patients were included as subjects. In AD patients, executive dysfunction is the domain generally affected subsequent to the memory domain [9]. SVaD patients also commonly show memory dysfunction [3]. These findings, however, support previous studies showing that AD and CVD pathologies independently affected cognitive decline and the risk of dementia [6,8,41,42].
This study had several limitations. First, the patients in this study all had cognitive impairment, which may limit the generalizability of these results to other populations. Second, it would be more relevant for predicting the clinical impact of CVD to use the location and volume of lacunae and voxel symptom lesion mapping of WMH rather than just volume or number alone [43]. Third, other pathologies such as tau, cortical microinfarct, or hippocampal sclerosis were not taken into consideration because a pathological study was not performed. Fourth, there is a possibility that the effects of WMH volume and lacunae were not completely disentangled. Although the effects of WMH volume and lacunae were adjusted for each other, the independent effects of WMH volume and lacunae should be cautiously interpreted. Fifth, neuropsychological composite scores used in this study might not purely reflect memory or executive functions because the assessment of neuropsychological performance usually requires the integration of multiple cognitive domains [44,45]. Finally, we cannot exclude the possibility that confounding factors could have been introduced by the criteria used to define the clinical groups. These patients, however, were diagnosed using clinical criteria and not PiB status. Also, after including clinical criteria as covariates, these findings were unchanged. Therefore, we believe these findings were unlikely to be affected by the criteria used to define each clinical group. Despite these limitations, results indicate that amyloid burden and CVD have independent effects on cognitive dysfunction with or without the mediation of specific regional brain atrophy.
Supplementary Material
RESEARCH IN CONTEXT.
Systematic review: The authors reviewed the literature using traditional (e.g., PubMed) sources and meeting abstracts and presentations. There have been several articles investigating the effect of Alzheimer’s disease (AD) or subcortical cerebrovascular disease (CVD) pathology on brain atrophy and cognition. Relevant citations are appropriately cited. However, independent effects of AD and CVD pathologies on cognition, with or without the mediation of brain atrophy, have not been fully elucidated.
Interpretation: Our findings provided an integrated mechanism of AD and CVD pathologies influencing cognition with or without the mediation of brain atrophy in cognitively impaired patients. This mechanism is consistent with previous clinical findings currently in the public domain.
Future directions: The manuscript proposes new hypotheses about the relationships among amyloid, CVD, brain atrophy, and cognition. These issues might extend to incorporating the information about functional or structural connectivity.
Acknowledgments
The authors would like to thank Victoria Barney for English correction. The authors also want to give special thanks to Changsoo Kim for his statistical advice. This study was supported by Basic Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2013R1A1A2065365), the Korean Healthcare Technology R&D Project Ministry for Health & Welfare Affairs (HI10C2020 & HIC120713), the KOSEF NRL program grant (MEST; 2011-0028333), Samsung Medical Center (CRL-108011&CRS110-14-1), and the Converging Research Center Program through the Ministry of Science, ICT and Future Planning, Korea (2013K000338).
Footnotes
Supplementary Material: Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.jalz.2014.04.521.
References
- 1.Ott A, Breteler MM, van Harskamp F, Claus JJ, van der Cammen TJ, Grobbee DE, et al. Prevalence of Alzheimer’s disease and vascular dementia: association with education. The Rotterdam study. BMJ. 1995;310:970–3. doi: 10.1136/bmj.310.6985.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorelick PB. Risk factors for vascular dementia and Alzheimer disease. Stroke. 2004;35:2620–2. doi: 10.1161/01.STR.0000143318.70292.47. [DOI] [PubMed] [Google Scholar]
- 3.Roman GC, Erkinjuntti T, Wallin A, Pantoni L, Chui HC. Subcortical ischaemic vascular dementia. Lancet Neurol. 2002;1:426–36. doi: 10.1016/s1474-4422(02)00190-4. [DOI] [PubMed] [Google Scholar]
- 4.Lim A, Tsuang D, Kukull W, Nochlin D, Leverenz J, McCormick W, et al. Clinico-neuropathological correlation of Alzheimer’s disease in a community-based case series. J Am Geriatr Soc. 1999;47:564–9. doi: 10.1111/j.1532-5415.1999.tb02571.x. [DOI] [PubMed] [Google Scholar]
- 5.Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS) Lancet. 2001;357:169–75. doi: 10.1016/s0140-6736(00)03589-3. [DOI] [PubMed] [Google Scholar]
- 6.Schneider JA, Wilson RS, Bienias JL, Evans DA, Bennett DA. Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology. 2004;62:1148–55. doi: 10.1212/01.wnl.0000118211.78503.f5. [DOI] [PubMed] [Google Scholar]
- 7.Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66:1837–44. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 8.Chui HC, Zarow C, Mack WJ, Ellis WG, Zheng L, Jagust WJ, et al. Cognitive impact of subcortical vascular and Alzheimer’s disease pathology. Ann Neurol. 2006;60:677–87. doi: 10.1002/ana.21009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reed BR, Mungas DM, Kramer JH, Ellis W, Vinters HV, Zarow C, et al. Profiles of neuropsychological impairment in autopsy-defined Alzheimer’s disease and cerebrovascular disease. Brain. 2007;130:731–9. doi: 10.1093/brain/awl385. [DOI] [PubMed] [Google Scholar]
- 10.Marchant NL, Reed BR, Sanossian N, Madison CM, Kriger S, Dhada R, et al. The aging brain and cognition: contribution of vascular injury and abeta to mild cognitive dysfunction. JAMA Neurol. 2013;70:488–95. doi: 10.1001/2013.jamaneurol.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mormino EC, Kluth JT, Madison CM, Rabinovici GD, Baker SL, Miller BL, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009;132:1310–23. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seo SW, Lee JM, Im K, Park JS, Kim SH, Kim ST, et al. Cortical thinning related to periventricular and deep white matter hyperintensities. Neurobiol Aging. 2012;33:1156–67. doi: 10.1016/j.neurobiolaging.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Du AT, Schuff N, Chao LL, Kornak J, Ezekiel F, Jagust WJ, et al. White matter lesions are associated with cortical atrophy more than entorhinal and hippocampal atrophy. Neurobiol Aging. 2005;26:553–9. doi: 10.1016/j.neurobiolaging.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 14.Becker JA, Hedden T, Carmasin J, Maye J, Rentz DM, Putcha D, et al. Amyloid-beta associated cortical thinning in clinically normal elderly. Ann Neurol. 2011;69:1032–42. doi: 10.1002/ana.22333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt R, Ropele S, Enzinger C, Petrovic K, Smith S, Schmidt H, et al. White matter lesion progression, brain atrophy, and cognitive decline: the Austrian stroke prevention study. Ann Neurol. 2005;58:610–6. doi: 10.1002/ana.20630. [DOI] [PubMed] [Google Scholar]
- 16.Mungas D, Jagust WJ, Reed BR, Kramer JH, Weiner MW, Schuff N, et al. MRI predictors of cognition in subcortical ischemic vascular disease and Alzheimer’s disease. Neurology. 2001;57:2229–35. doi: 10.1212/wnl.57.12.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jokinen H, Lipsanen J, Schmidt R, Fazekas F, Gouw AA, van der Flier WM, et al. Brain atrophy accelerates cognitive decline in cerebral small vessel disease: the LADIS study. Neurology. 2012;78:1785–92. doi: 10.1212/WNL.0b013e3182583070. [DOI] [PubMed] [Google Scholar]
- 18.Whitwell JL, Josephs KA, Murray ME, Kantarci K, Przybelski SA, Weigand SD, et al. MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology. 2008;71:743–9. doi: 10.1212/01.wnl.0000324924.91351.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 20.Erkinjuntti T, Inzitari D, Pantoni L, Wallin A, Scheltens P, Rockwood K, et al. Research criteria for subcortical vascular dementia in clinical trials. J Neural Transm Suppl. 2000;59:23–30. doi: 10.1007/978-3-7091-6781-6_4. [DOI] [PubMed] [Google Scholar]
- 21.Seo SW, Cho SS, Park A, Chin J, Na DL. Subcortical vascular versus amnestic mild cognitive impairment: comparison of cerebral glucose metabolism. J Neuroimaging. 2009;19:213–9. doi: 10.1111/j.1552-6569.2008.00292.x. [DOI] [PubMed] [Google Scholar]
- 22.Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. AJR Am J Roentgenol. 1987;149:351–6. doi: 10.2214/ajr.149.2.351. [DOI] [PubMed] [Google Scholar]
- 23.Jeon S, Yoon U, Park J-S, Seo SW, Kim J-H, Kim ST, et al. Fully automated pipeline for quantification and localization of white matter hyperintensity in brain magnetic resonance image. Int J Imaging Syst Technol. 2011;21:193–200. [Google Scholar]
- 24.Kang Y, Na DL. Seoul Neuropsychological Screening Battery (SNSB) Incheon: Human Brain Research & Consulting Co.; 2003. [Google Scholar]
- 25.Ahn HJ, Chin J, Park A, Lee BH, Suh MK, Seo SW, et al. Seoul Neuropsychological Screening Battery-dementia version (SNSB-D): a useful tool for assessing and monitoring cognitive impairments in dementia patients. J Korean Med Sci. 2010;25:1071–6. doi: 10.3346/jkms.2010.25.7.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reed BR, Eberling JL, Mungas D, Weiner M, Kramer JH, Jagust WJ. Effects of white matter lesions and lacunes on cortical function. Arch Neurol. 2004;61:1545–50. doi: 10.1001/archneur.61.10.1545. [DOI] [PubMed] [Google Scholar]
- 27.Apostolova LG, Dutton RA, Dinov ID, Hayashi KM, Toga AW, Cummings JL, et al. Conversion of mild cognitive impairment to Alzheimer disease predicted by hippocampal atrophy maps. Arch Neurol. 2006;63:693–9. doi: 10.1001/archneur.63.5.693. [DOI] [PubMed] [Google Scholar]
- 28.Wang L, Miller JP, Gado MH, McKeel DW, Rothermich M, Miller MI, et al. Abnormalities of hippocampal surface structure in very mild dementia of the Alzheimer type. Neuroimage. 2006;30:52–60. doi: 10.1016/j.neuroimage.2005.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerardin E, Chetelat G, Chupin M, Cuingnet R, Desgranges B, Kim HS, et al. Multidimensional classification of hippocampal shape features discriminates Alzheimer’s disease and mild cognitive impairment from normal aging. Neuroimage. 2009;47:1476–86. doi: 10.1016/j.neuroimage.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bakkour A, Morris JC, Dickerson BC. The cortical signature of prodromal AD: regional thinning predicts mild AD dementia. Neurology. 2009;72:1048–55. doi: 10.1212/01.wnl.0000340981.97664.2f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jouvent E, Mangin JF, Porcher R, Viswanathan A, O’Sullivan M, Guichard JP, et al. Cortical changes in cerebral small vessel diseases: a 3D MRI study of cortical morphology in CADASIL. Brain. 2008;131:2201–8. doi: 10.1093/brain/awn129. [DOI] [PubMed] [Google Scholar]
- 32.Kloppenborg RP, Nederkoorn PJ, Grool AM, Vincken KL, Mali WP, Vermeulen M, et al. Cerebral small-vessel disease and progression of brain atrophy: the SMART-MR study. Neurology. 2012;79:2029–36. doi: 10.1212/WNL.0b013e3182749f02. [DOI] [PubMed] [Google Scholar]
- 33.Kirino T, Sano K. Selective vulnerability in the gerbil hippocampus following transient ischemia. Acta Neuropathol. 1984;62:201–8. doi: 10.1007/BF00691853. [DOI] [PubMed] [Google Scholar]
- 34.Cummings JL. Frontal-subcortical circuits and human behavior. Arch Neurol. 1993;50:873–80. doi: 10.1001/archneur.1993.00540080076020. [DOI] [PubMed] [Google Scholar]
- 35.Au R, Massaro JM, Wolf PA, Young ME, Beiser A, Seshadri S, et al. Association of white matter hyperintensity volume with decreased cognitive functioning: the Framingham Heart Study. Arch Neurol. 2006;63:246–50. doi: 10.1001/archneur.63.2.246. [DOI] [PubMed] [Google Scholar]
- 36.Wheeler MA, Stuss DT, Tulving E. Frontal lobe damage produces episodic memory impairment. J Int Neuropsychol Soc. 1995;1:525–36. doi: 10.1017/s1355617700000655. [DOI] [PubMed] [Google Scholar]
- 37.Sperling RA, Laviolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–88. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheline YI, Raichle ME, Snyder AZ, Morris JC, Head D, Wang S, et al. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry. 2010;67:584–7. doi: 10.1016/j.biopsych.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Resnick SM, Sojkova J, Zhou Y, An Y, Ye W, Holt DP, et al. Longitudinal cognitive decline is associated with fibrillar amyloid-beta measured by [11C]PiB. Neurology. 2010;74:807–15. doi: 10.1212/WNL.0b013e3181d3e3e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–92. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- 42.Schneider JA, Boyle PA, Arvanitakis Z, Bienias JL, Bennett DA. Subcortical infarcts, Alzheimer’s disease pathology, and memory function in older persons. Ann Neurol. 2007;62:59–66. doi: 10.1002/ana.21142. [DOI] [PubMed] [Google Scholar]
- 43.Duering M, Zieren N, Herve D, Jouvent E, Reyes S, Peters N, et al. Strategic role of frontal white matter tracts in vascular cognitive impairment: a voxel-based lesion-symptom mapping study in CADA-SIL. Brain. 2011;134:2366–75. doi: 10.1093/brain/awr169. [DOI] [PubMed] [Google Scholar]
- 44.Sweet JJ, Suchy Y, Leahy B, Abramowitz C, Nowinski CJ. Normative clinical relationships between orientation and memory: age as an important moderator variable. Clin Neuropsychol. 1999;13:495–508. doi: 10.1076/1385-4046(199911)13:04;1-Y;FT495. [DOI] [PubMed] [Google Scholar]
- 45.Kipps CM, Hodges JR. Cognitive assessment for clinicians. J Neurol Neurosurg Psychiatry. 2005;76(Suppl 1):i22–30. doi: 10.1136/jnnp.2004.059758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.