

Abstract
Introduction
The aetiology of congenital hearing loss is heterogeneous, and in many infants a genetic cause is suspected. Parents face a diagnostic odyssey when searching for a cause of their infant’s hearing loss. Through the Melbourne Genomics Health Alliance, a prospective cohort of infants will be offered whole-exome sequencing (WES) with targeted analysis in conjunction with chromosome microarray to determine the genetic causes of congenital hearing loss. Parents will also be offered the opportunity to receive additional results from their infant’s WES.
Methods
Eligible infants will be identified through the Victorian Infant Hearing Screening Program and offered an appointment in a paediatrician-run clinic, a genetics assessment and enrolment in the Victorian Childhood Hearing Impairment Longitudinal Databank. If parents consent to WES, genes causing deafness will be analysed and they can choose to obtain additional findings. For the additional results component, a modified laboratory protocol has been designed for reporting of results in the absence of a relevant phenotype. Parents’ experience of being offered WES will be evaluated using surveys.
Discussion
This project will provide descriptive analysis of the genetic aetiology of congenital hearing loss in this cohort and may provide data on genotype–phenotype correlations. Additionally, choices regarding additional findings will be analysed. Participants will represent a diverse cross section of the population, increasing the ability to generalise results beyond the study group. Evaluation surveys will allow analysis of preferences around counselling, usefulness of a decision aid and adequacy of information provision.
Keywords: Deafness, Genetics, Screening
What is already known in this topic?
50% of congenital bilateral moderate or worse hearing loss is presumed to have a genetic aetiology.
Many individuals do not receive a genetic diagnosis due to the heterogeneous nature of the disease and the expense of iterative gene testing.
A majority of individuals, including parents, are interested in receiving additional genomic information when they have next-generation sequencing performed as a diagnostic test.
What this study hopes to add?
Descriptive evidence of what proportion of congenital hearing loss is due to a genetic aetiology and what the genetic causes are in this cohort.
A streamlined approach for information delivery and comprehensive care for parents who have a newborn diagnosed with hearing loss.
An opportunity to investigate if, why and how people want additional findings when they have genomic sequencing in their children.
Strengths and limitations of this study.
Strengths are that this is a population based study with the potential to provide actionable results for families, that the study is integrated into a pre-existing framework, and that it will also provide evidence around the scope of results that should be returned for genomic sequencing.
Limitations are genomic testing may not identify all genetic causes of hearing loss.
Introduction
Congenital hearing loss affects 0.1% of the Australian population. Universal newborn hearing screening has lowered the mean age of detection of moderate to profound hearing loss to around 2 months of age1 2 and early identification of hearing loss allows early amplification (hearing aids and cochlear implants) and early intervention3 to improve cognitive and communication development.
Although the aetiology of congenital hearing loss is heterogeneous, a significant proportion has a genetic basis.4 Several hundred genes have been identified as playing a role in hearing and all modes of inheritance are described: autosomal dominant, recessive, x-linked and mitochondrial.5 Congenital hearing loss is most commonly an isolated finding; however, in 30% there is an associated syndrome diagnosis.5 Syndromic associations may not be evident until later childhood or adulthood, for example in Usher syndrome, which results in retinitis pigmentosa and visual loss starting in the later childhood years.
Despite the high frequency of an underlying genetic aetiology, few families receive a genetic diagnosis because of the iterative nature of single gene testing and the significant expense and time required to engage in this process. Whole-exome sequencing (WES) allows simultaneous and rapid analysis of human genes, therefore detection of all exome tractable mutations in genes known to be associated with deafness should be possible for these patients. Benefits of receiving an early genetic diagnosis include provision of prognostic information, streamlined care, accurate recurrence risk advice, and where appropriate, screening for associated health conditions.
Through the Melbourne Genomic Health Alliance (the Alliance), WES will be offered in a clinical context to a 2-year prospective population-based cohort of patients diagnosed in the newborn period with hearing loss. Established as a collaboration in 2013, the members of the Alliance are 10 healthcare, education and medical research institutions committed to incorporating genomics into healthcare to benefit patients.6 This project is part of the Alliance’s broader programme of work to develop an evidence base to inform the cost-effective use of genomics in clinical practice and to build workforce capability in Victoria, Australia.6 Alliance processes, procedures, and systems are technically agnostic, that is, they are designed to be modifiable as utility in routine clinical practice is demonstrated for other ‘-omic’ technologies. WES with targeted analysis, the first of these technologies, is ready for clinical application. Congenital hearing loss is an ideal candidate condition to further this work because of its extensive genetic heterogeneity, and a population-based cohort is readily identifiable.
In line with international research on the implementation of genomic sequencing7 8 this cohort of infants will be used to investigate parents’ preferences around receipt of additional genomic information about their newborn. This will help inform the scope of genomic sequencing results provided to families and may provide baseline data to inform studies of genomic sequencing in healthy newborns.
Studies of the return of results from genomic sequencing have indicated that the majority of individuals express a desire for additional genetic information.8–10 Hypothetical preferences for return of additional information indicate that a large proportion (>80%) of survey respondents from the Melbourne Genomics demonstration project want this information for preventable conditions (C. Gaff, unpublished data, 2017). We will contribute to this pool of research, with this study having the benefit of recruiting a population sample with diversity of ethnicity, language, socioeconomic status and education level.
Study aims and hypothesis
The primary aims of the Melbourne Genomics Congenital Deafness project are to understand the reasons parents choose to have genetic testing for their infant and to describe the genetic aetiologies that cause congenital hearing loss in this cohort. A secondary aim is to investigate the feasibility and acceptability of genomic screening of newborns for other health conditions that occur in childhood. This component of the study is titled ‘Baby Beyond Hearing’.
The study hypotheses are that: (1) the majority of parents will choose to have WES for their child because accurate genetic diagnosis at an early age will benefit families (understanding the cause, recurrence risk and prognosis information) and health systems (limiting the need for further investigation and tailoring screening as indicated by the genetic diagnosis), (2) up to 50% of children born with moderate or worse bilateral permanent hearing loss will have a genetic cause identifiable by a combination of WES and microarray and (3) parents will want to receive additional genetic information from the WES data about their children.
Methods/design
The state of Victoria, Australia, has a unique infrastructure of three major platforms that jointly enable this study to be conducted at the population level, as follows:
Victorian Infant Hearing Screening Program (VIHSP): identification and recruitment
The VIHSP is a well-established state-wide newborn hearing screening programme delivered in all birthing hospitals via a single provider, the Royal Children’s Hospital, Melbourne. The programme commenced in 2005 and by 2012 established state-wide coverage of approximately 80 000 births annually. All newborns are offered automated auditory brainstem response (AABR) testing. Those who do not pass the first screening test are referred for a repeat AABR, with a second positive screen result leading to a referral to audiology for confirmatory diagnosis.2 Based on VIHSP data from previous years, it is estimated that 140 patients will meet the criteria for entry into the study over this 2-year period.11 There are no similar studies or empirical data on which to predicte uptake and therefore sample size.
Victorian Childhood Hearing Impairment Longitudinal Databank (VicCHILD): recruitment, programme evaluation, long-term outcomes
VicCHILD is a longitudinal databank for Victorian children diagnosed with hearing loss. This programme follows children and families over time to: (1) help researchers and health professionals gain a better understanding of the causes and outcomes of childhood hearing loss, (2) understand why some children do well while others face greater difficulties and (3) improve intervention and treatment and ultimately the lives of children with permanent hearing loss and their families. Over the 2-year period prior to this project commencing VicCHILD identified that of babies born with congenital hearing loss, 40% of their parents were born overseas with the majority of these being of Asian ethnicity and 20% of families identified a primary language other than English. Families came from a range of socioeconomic backgrounds. This provides an insight into the diversity of the cohort that will be recruited in this project.
Paediatrician clinics: recruitment, phenotyping, long-term outcomes
Two clinical services exist in Victoria, specifically catering to infants diagnosed with hearing loss through VIHSP. These are run through two of the major tertiary hospitals in the state. The Paediatric Hearing Loss Investigation Clinic (PHLIC) through Monash Children’s Hospital and the Caring for Hearing Impaired Children (CHIC) clinic through the Royal Children’s Hospital.
Eligibility criteria
Infants born between January 2016 and December 2017 (inclusive) in Victoria and diagnosed, following two positive AABR screens delivered by VIHSP, with bilateral permanent hearing loss greater than 40 dB over three frequencies (moderate, severe or profound hearing loss). These criteria include infants classified as having sensorineural, mixed conductive and sensorineural and auditory neuropathy. These infants may have isolated hearing loss or suspected syndromic hearing loss.
Exclusion criteria
Unilateral hearing loss of any degree or mild hearing loss or better in the best ear or hearing loss that is confirmed as conductive and temporary.
Recruitment procedures
Recruitment procedures are depicted in figure 1.
Figure 1.
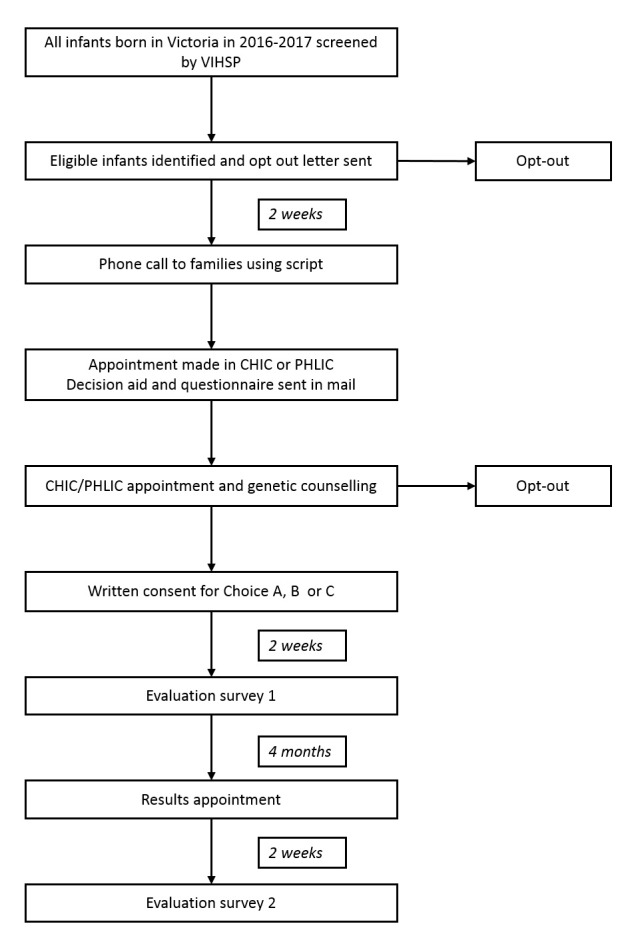
Flowchart of recruitment and testing. CHIC, Caring for Hearing Impaired Children; PHLIC, Paediatric Hearing Loss Investigation Clinic; VIHSP, Victorian Infant Hearing Screening Program.
Opt-out letter
Infants who fulfil the eligibility criteria will be identified by VIHSP and sent an ‘opt-out’ letter, which briefly outlines the paediatrician-run clinics, VicCHILD and the Melbourne Genomics Congenital Deafness project. Parents will be given the option of doing nothing and therefore being contacted over telephone, sending an ‘opt-out’ notification back to VIHSP (via prepaid post, email or fax) or deferring contact by several months.
Telephone call
A genetic counsellor or genetic doctor will telephone parents who do not opt-out and provide standardised information using a guideline. Parents will be offered an appointment in CHIC or PHLIC clinic. They will be able to book a genetic counselling appointment to discuss genetic testing on the same day or at an alternate time according to their preference.
Information about VicCHILD will also be provided and parents will be given the opportunity to meet with a VicCHILD staff member on the day of their appointment.
Interpreter services will be available for families from a non-English speaking background. If the family is not contactable over telephone, alternative attempts will be made such as via email or by review of the medical record (if one exists) for an alternative contact number.
At the phone call, parents indicate which of the three initiatives (VicCHILD, Melbourne Genomics and paediatrician clinics) they wish to pursue. For patients who book an appointment for genetic counselling, an information pack will be sent in the post, including a questionnaire and a decision aid. Both parents are encouraged to attend the appointment to facilitate joint decision-making.
Baseline questionnaire
The questionnaire was designed in collaboration with the paediatricians providing the CHIC and PHLIC clinics and the VicCHILD research team. The questionnaire enables parents to complete a single document that provides the necessary information for: (1) the clinical appointment, (2) inclusion in VicCHILD and (3) data collection for the Melbourne Genomics Congenital Deafness project. Parents can opt to share this information with all three initiatives or exclude any in which they do not wish to participate.
The questionnaire asks about pregnancy, delivery, family history, the child’s hearing loss diagnosis, investigations, treatments and service use to date, general health and development (ASQ)12 as assessed by the parent, demographic information, quality of life (PEDS QL 3.0)13 and parental mental health.14 Parents can complete the questionnaire in paper form or online.
Decision aid
A decision aid has been developed specifically for this study to provide families with information about testing prior to their appointment. The decision aid was modelled on a tool designed by the Public Health Genetics team at the Murdoch Children’s Research Institute for couples to use when deciding what investigations to have during pregnancy.15 The decision aid helps to ensure parents are giving true informed consent for genomic testing, and will also be used to evaluate what resources are required to integrate genomic sequencing into standard clinical practice.16 The decision aid is designed to help families consider if they would like their child to have WES, and also how much information they would like to receive from the test. The document contains two worksheets where parents can explore their own personal views on: (1) having genetic testing and (2) how much information they are interested in receiving from this testing. Infants whose parents consent to investigate the cause of hearing loss will be offered options regarding additional gene analysis (table 1).
Table 1.
The three choices offered to parents regarding which genes will be analysed and reported following whole-exome sequence
| Choice A | Choice B | Choice C |
| Only genes that are known to cause hearing loss will be examined. We estimate that we will identify a genetic cause for hearing loss in about half of the infants that we test. | Choice B is the same as Choice A plus We will analyse additional genes that cause diseases that:
|
Choice C is the same as Choice B plus We will analyse additional genes that cause diseases that:
|
Parents are asked to bring the decision aid to the appointment where a genetic counsellor will go through their preferences and use the document as a talking point for exploring their decisions around genetic testing.
Clinic appointment
A paediatrician with special interest in hearing loss and either a doctor with training in genetics or a genetic counsellor will conduct the clinic appointment. During the first hour of the appointment, the patient will have a full clinical and developmental assessment and non-genetic investigations and referrals will be ordered as indicated. During the second hour of the appointment, a genetic counsellor will discuss WES with the parents in more detail, using the completed decision aid as a basis for discussion.
Talking points for genetic counsellors will be created in addition to a training session facilitated by an educator and experienced genetic counsellors to upskill counsellors in obtaining consent for additional findings. This ensures that the information and counselling provided is standardised across sites and between clinicians. If the family wishes to participate in the research, clinical grade WES and chromosome microarray will be ordered and a venous blood sample will be taken from the child on the day. Families will also have the option of having more time to consider the testing with follow-up from the genetic counsellor.
Written consent will be required to participate in the study from one parent; however, counsellors will ensure that both parents are aware of the nature of the testing and in agreeance regarding the scope of results to be returned.
Appointments to disclose results will be scheduled when reports are issued for the WES. These may be with a geneticist or paediatrician and genetic counsellor.
Gene lists
Gene lists are designed to allow rapid interpretation of test data as well as filtering of unwanted information such as variants in genes known to cause adult-onset untreatable disease (online supplementary appendix 1).
bmjpo-2017-000119supp001.pdf (25.5KB, pdf)
Choice A contains all of the genes known to cause hearing loss. It was constructed by comparing and merging gene lists from two pre-existing clinical diagnostic panels: OtoSCOPE17 and OtoGenome,18 and several research gene lists sourced from Genomics England (the PanelApp Congenital Hearing Impairment (profound/severe) panel v1.8),19 the Avraham laboratory,20 the Gasparini laboratory21 and the Rehm laboratory.22 Additional genes were added to the list based on recently reported findings in the literature. The list is separated into two tiers. Tier 1 comprises 141 genes for which there is validated evidence that mutations are causative in human patients with hearing loss. Tier 2 contains a further 241 genes with good evidence from a range of sources for their involvement in hearing loss, substantively based on evidence from animal models.
Choice B was adapted from a gene list created, and generously shared, by the NC NEXUS group. This list is based on a metric they devised and used to ‘score’ gene/disease pairs.23 These conditions all have treatment or intervention available.
Choice C was devised using a set of criteria that were agreed by the investigative team. These include:
definitive gene disease association (as defined by the BabySeq group10);
onset of symptoms prior to age 16 the majority of the time;
validation method available to confirm diagnosis at the time of testing.
All of the diseases associated with genes in choice C have a validation method available such as a biochemical test, X-ray or a one-off clinical examination in order to provide additional certainty around a presymptomatic diagnosis.
Laboratory protocol
Exome sequencing, variant detection, variant filtering and interpretation will be performed in a clinically accredited laboratory using similar methods to those previously described in other patient cohorts of the Melbourne Genomics Health Alliance.24
Whole-exome sequencing
DNA will be extracted from peripheral blood, and exome sequencing will be performed using SureSelect Clinical Research Exome V.1 (Agilent) on either a HiSeq4000 or NextSeq500 (Illumina) with a targeted average sequencing depth of 100×.
Bioinformatic analysis and variant filtering
Data analysis and variant calling will be performed using Cpipe.25 Variants will be assessed using LOVDplus.26 Variants will be prioritised based on the clinician’s assessment using information such as likely mode of inheritance (assumed to be autosomal recessive unless family history indicates otherwise) and any phenotypic information available such as MRI brain findings or dysmorphic features on examination. We predict a large proportion of patients will have no family history and no additional phenotypic information apart from hearing loss and in this instance all variants detected will be curated.
Variant interpretation
The methodology around the interpretation of the additional findings gene lists focuses on the shift in principle from using WES as a diagnostic tool to use of WES as a screening tool. In a diagnostic situation, a high level of sensitivity is required from the test to capture all possible causes of a patient’s phenotype. In a ‘screening’ or ‘predictive’ analysis, a high level of specificity is required to ensure there is adequate evidence and understanding of the outcome of a gene variant to return results to patients who will often be asymptomatic at that time.
The standard diagnostic laboratory variant interpretation protocol was adapted for this cohort due to the novel situation of offering analysis of genes outside of the patient’s current phenotype.
Choice A, tier 1 genes will be analysed in accordance with the laboratory’s National Association of Testing Authorities accredited procedures which are aligned with American College of Medical Genetics guidelines,27 resulting in a clinical report. Choice A, tier 2 genes will be analysed by a research group only in patients who do not receive a clinical result from tier 1.
Choice B and C genes will be analysed by the same clinically accredited diagnostic laboratory for patients who consented to the analysis, but only variants in class 4 or 528 will be reported to families for these genes. Variants of uncertain significance, as well as carrier-only variants, will not be reported. In order for this process to be streamlined, any missense variant identified in the genes of interest will first be searched for in the literature. If the variant is not previously described in published literature or in ClinVar, no further curation will be performed. This is on the basis that missense variants cannot be classified any higher than class 3 without supporting case reports. Truncating and nonsense variants are curated in the same way as variants in choice A.
Criteria for classification will be based on the principles outlined in the American College of Medical Genetics and Genomics standards for interpretation of sequence variants.27 Variant classifications will be reviewed in a multidisciplinary team meeting attended by clinical geneticists and other medical subspecialists, genetic counsellors, molecular geneticists and bioinformaticians.
Evaluation surveys
Evaluation surveys have been designed and can be completed online or on paper. Patients will complete the first survey 2 weeks after they consent for genomic testing and a second survey 2 weeks following their results appointment (see figure 1). The surveys include data captured for Melbourne Genomics across all patient groups as well as data that is specific to children within the Melbourne Genomics Congenital Deafness project.
The first survey includes questions about the parents’ experience of the consent process including the decision aid and the genetic counselling appointment. The Decisional Conflict Scale and Decisional Regret Scales29 are included in survey 1 and 2, respectively, to evaluate parental decision-making around which gene list they selected for analysis. The first survey includes closed and open questions exploring families’ reasons for wanting to have genomic sequencing for their child. Knowledge questions and a question on the likelihood of the test finding the cause of the hearing loss are included to gauge understanding. Validated measures to assess potential predictors of uptake of the expanded analysis are included: tolerance of uncertainty,30 State-Trait Anxiety scale,31 medical literacy32 and information-seeking preferences.33 34 The Perceived Personal Control scale35 is included as a measurement of patient empowerment in both surveys.
The second survey includes study-specific questions assessing the parents’ understanding of the genomic results, impact these will have on them and their family and what they have valued about having the test done for their child.
Analysis of data
The majority of data analysis will be descriptive in nature due to the small sample size, which is estimated to be approximately 100. The primary outcome measures reported will be uptake of testing, the detection rate of causative/pathogenic variants and identification of carrier status. Detailed sociodemographic characteristics of the participants (eg, socioeconomic status, age of parents, language and cultural background, education) and variable aspects of the process and outcomes will be presented, followed by comparisons between groups of interest. Examples of these include:
information delivery methods (phone call, face-to-face consultation and written information) and parental responses to the offer of testing and their knowledge about testing in relation to these various modalities;
psychosocial measures of parents who accept versus decline testing and the scope of results chosen by those who participate;
differences in uptake between parents of children who require cochlear implant, those who are systemically unwell or have a broader phenotype (not isolated hearing loss) or whose infant was born prematurely for example.
Multivariable statistical analysis may be performed, but this will be dependent on sample size for each group, for example, accept testing versus decline testing and no additional findings versus additional findings.
Audiological and general phenotype data will be correlated with genotype and a summary of these findings will be presented.
Discussion
This project is novel in offering genomic sequencing to infants with congenital hearing loss in conjunction with comprehensive developmental and general health assessments and long-term follow-up.
The primary strengths of the study are that it is a population-based study, has the potential to provide actionable results for families at an early stage and is integrated into a pre-existing framework, making the process streamlined for patients and care providers. It will also provide new and important evidence around the scope of results that should be returned for genomic sequencing. The limitations of the study are concordant with the limitations of all WES. These relate to sequencing gaps which may result in a pathogenic variant not being detected, as well as the technology’s inability to detect some types of DNA changes such as copy number variation, and the possible detection of a variant in a gene that is not yet known to have an association with hearing loss. Evidence suggests a significant proportion of congenital hearing loss (15%–20%) is due to copy number variants36 and for this reason, all patients will simultaneously have a chromosome microarray.
Genomic results will add to the knowledge around the genetic aetiology of congenital hearing loss and will provide important data on genotype–phenotype correlations. The use of genomic sequencing means that families who do not receive a molecular diagnosis initially can participate in research around genes related to hearing loss and enable gene discovery in this area by having their child’s WES data reanalysed.
Descriptive analyses of parents’ interest in gaining additional results from genomic sequencing will provide novel information that will help to determine the clinical utility of an offer to provide sequencing results beyond those directly related to the child’s phenotype, that is, deafness in this case. The study will assess previously described concepts, for example, what has been described as ‘inflicted ought’ where parents have a desire to do what they believe is the best thing for the child despite the individuals not necessarily seeking similar predictive information for themselves.37 It will also provide important data on parents’ decisions when faced with the reality of testing. Previous studies have found that parent preferences when offered theoretical versus actual testing can produce quite disparate results. In the BabySeq study, a large proportion of parents indicated in preparatory research that they would be interested in genomic sequencing for their newborn;10 however, in reality the uptake for the study has been very low.38
Evaluation surveys will produce data for further descriptive analysis of preferences around counselling, use of a decision aid and the information provided. Together, these results will add substantially to the development of strategies and policies related to implementation of genomic sequencing in the newborn period.
Supplementary Material
Acknowledgments
The authors thank all collaborators in the Melbourne Genomics Health Alliance demonstration project.
Footnotes
Contributors: LD drafted the manuscript, DA, RB, JH, CG and EL were major contributors to the design of the study protocol, SL established the laboratory protocol, and MM designed the evaluation surveys. ZP, MW, KS, MH, VS and ER have made substantial contributions to recruitment design. HLR, JH and CG were collaborators in designing gene lists. All authors read and approved the final manuscript.
Funding: The study was funded by the founding organisations of the Melbourne Genomics Health Alliance (Royal Melbourne Hospital, Royal Children’s Hospital, University of Melbourne, Walter and Eliza Hall Institute, Murdoch Children’s Research Institute, Australian Genome Research Facility (AGRF) and CSIRO) and the State Government of Victoria (Department of Health and Human Services). The involvement of AGRF was supported by sponsorship from Bioplatforms Australia and the NCRIS program.
Competing interests: None declared.
Ethics approval: The establishment of this project was approved by the Royal Melbourne Hospital Human Research Ethics Committee as an amendment to the Melbourne Genomics protocol 2013.245.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Collaborators: Collaborators in the Melbourne Genomics Health Alliance include Libby Smith, Bibi Gerner, Nessie Mupfeki, Michelle Burns, Ivan Macciocca, Gemma Brett, Anna Jarmolowicz, Yael Prawer, Belinda Chong, Jonathan Berg, Cynthia Powell, Holly Feller and Emily Shepard.
References
- 1.Russ SA, Poulakis Z, Barker M, et al. . Epidemiology of congenital hearing loss in Victoria, Australia. Int J Audiol 2003;42:385–90. [DOI] [PubMed] [Google Scholar]
- 2.Wake M, Ching TY, Wirth K, et al. . Population outcomes of three approaches to detection of congenital hearing loss. Pediatrics 2016;137:e20151722 doi:10.1542/peds.2015-1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fitzpatrick E. Neurocognitive development in congenitally deaf children. Handb Clin Neurol 2015;129:335–56. doi:10.1016/B978-0-444-62630-1.00019-6 [DOI] [PubMed] [Google Scholar]
- 4.Shearer AE, Smith RJ. Massively parallel sequencing for genetic diagnosis of hearing loss: the new standard of care. Otolaryngol Head Neck Surg 2015;153:175–82. doi:10.1177/0194599815591156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith RJH, Bale JF, White KR. Sensorineural hearing loss in children. The Lancet 2005;365:879–90. doi:10.1016/S0140-6736(05)71047-3 [DOI] [PubMed] [Google Scholar]
- 6.Gaff CL, M. Winship I, M. Forrest S, et al. . Preparing for genomic medicine: a real world demonstration of health system change. NPJ Genom Med 2017;2 doi:10.1038/s41525-017-0017-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berg JS, Powell CM. Potential uses and inherent challenges of using genome-scale sequencing to augment current newborn screening. Cold Spring Harb Perspect Med 2015;5:a023150 doi:10.1101/cshperspect.a023150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldenberg AJ, Dodson DS, Davis MM, et al. . Parents' interest in whole-genome sequencing of newborns. Genet Med 2014;16:78–84. doi:10.1038/gim.2013.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fiallos K, Applegate C, Mathews DJ, et al. . Choices for return of primary and secondary genomic research results of 790 members of families with Mendelian disease. Eur J Hum Genet 2017;25:530–7. doi:10.1038/ejhg.2017.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green RCaB AH. The BabySeq Project. 2016. www.genomes2people.org/babyseqproject.
- 11.Program VIHS. Infant Hearing. http://infanthearing.vihsp.org.au/screening-and-diagnosis/hearing-loss-in-children2017.
- 12.Squires JB D, et al. : RN ETMS, Clifford J, Murphy K, ASQ-3. 409 Washington Ave, Suite 500, Towson, MD 21204: Brookes Publishing Co. Inc, 2009. [Google Scholar]
- 13.Varni JW, Sherman SA, Burwinkle TM, et al. . The PedsQL™ family impact module: preliminary reliability and validity. The PedsQL™ Family Impact Module: Preliminary reliability and validity. Health and Quality of Life Outcomes. Health Qual Life Outcomes 2004;2:55–6. doi:10.1186/1477-7525-2-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kessler RC, Barker PR, Colpe LJ, et al. . Screening for serious mental illness in the general population. Arch Gen Psychiatry 2003;60:184–9. doi:10.1001/archpsyc.60.2.184 [DOI] [PubMed] [Google Scholar]
- 15.Nagle C, Gunn J, Bell R, et al. . Use of a decision aid for prenatal testing of fetal abnormalities to improve women’s informed decision making: a cluster randomised controlled trial [ISRCTN22532458]. BJOG 2008;115:339–47. doi:10.1111/j.1471-0528.2007.01576.x [DOI] [PubMed] [Google Scholar]
- 16.Lewis MA, Paquin RS, Roche MI, et al. . Supporting Parental Decisions About Genomic Sequencing for Newborn Screening: The NC NEXUS Decision Aid. Pediatrics 2016;137:S16–S23. doi:10.1542/peds.2015-3731E [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shearer AE, DeLuca AP, Hildebrand MS, et al. . Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc Natl Acad Sci U S A 2010;107:21104–9. doi:10.1073/pnas.1012989107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Healthcare' P. OtoGenome Test for Hearing Loss and Related Syndromes (87 Genes) Details. 2016. http://personalizedmedicine.partners.org/Laboratory-For-Molecular-Medicine/Tests/Hearing-Loss/OtoGenome.aspx
- 19.England G. Genomics England PanelApp. 2015. https://bioinfo.extge.co.uk/crowdsourcing/PanelApp/.
- 20.Smith RS, Hildebrand A, Van Camp, G M. Deafness and hereditary hearing loss overview. www.ncbi.nlm.gov2014 (updated 9 Jan 2014).
- 21.Vozzi D, Morgan A, Vuckovic D, et al. . Hereditary hearing loss: a 96 gene targeted sequencing protocol reveals novel alleles in a series of Italian and Qatari patients. Gene 2014;542:209–16. doi:10.1016/j.gene.2014.03.033 [DOI] [PubMed] [Google Scholar]
- 22.Abou Tayoun AN, Al Turki SH, Oza AM, et al. . Improving hearing loss gene testing: a systematic review of gene evidence toward more efficient next-generation sequencing-based diagnostic testing and interpretation. Genet Med 2016;18:545–53. doi:10.1038/gim.2015.141 [DOI] [PubMed] [Google Scholar]
- 23.Berg JS, Foreman AK, O’Daniel JM, et al. . A semiquantitative metric for evaluating clinical actionability of incidental or secondary findings from genome-scale sequencing. Genet Med 2016;18:467–75. doi:10.1038/gim.2015.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stark Z, Tan TY, Chong B, et al. . A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med 2016;18:1090–6. doi:10.1038/gim.2016.1 [DOI] [PubMed] [Google Scholar]
- 25.Sadedin SP, Dashnow H, James PA, et al. . Cpipe: a shared variant detection pipeline designed for diagnostic settings. Genome Med 2015;7:68 doi:10.1186/s13073-015-0191-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leiden Open Variation Database for Diagnostics lovdclinical.mcri.edu.au: Leiden University Medical Centre;. 2014-2016 [. [Google Scholar]
- 27.Richards CS, Bale S, Bellissimo DB, et al. . ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med 2008;10:294–300. doi:10.1097/GIM.0b013e31816b5cae [DOI] [PubMed] [Google Scholar]
- 28.Richards S, Aziz N, Bale S, et al. . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–23. doi:10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Connor AM. Validation of a decisional conflict scale. Med Decis Making 1995;15:25–30. doi:10.1177/0272989X9501500105 [DOI] [PubMed] [Google Scholar]
- 30.Carleton RN, Norton MA, Asmundson GJ. Fearing the unknown: a short version of the Intolerance of Uncertainty Scale. J Anxiety Disord 2007;21:105–17. doi:10.1016/j.janxdis.2006.03.014 [DOI] [PubMed] [Google Scholar]
- 31.Marteau TM, Bekker H. The development of a six-item short-form of the state scale of the Spielberger State-Trait Anxiety Inventory (STAI). Br J Clin Psychol 1992;31:301–6. doi:10.1111/j.2044-8260.1992.tb00997.x [DOI] [PubMed] [Google Scholar]
- 32.Chew LD, Bradley KA, Boyko EJ. Brief questions to identify patients with inadequate health literacy. Fam Med 2004;36:588–94. [PubMed] [Google Scholar]
- 33.Cassileth BR, Zupkis RV, Sutton-Smith K, et al. . Information and participation preferences among cancer patients. Ann Intern Med 1980;92:832–6. doi:10.7326/0003-4819-92-6-832 [DOI] [PubMed] [Google Scholar]
- 34.Blanchard CG, Labrecque MS, Ruckdeschel JC, et al. . Information and decision-making preferences of hospitalized adult cancer patients. Soc Sci Med 1988;27:1139–45. doi:10.1016/0277-9536(88)90343-7 [DOI] [PubMed] [Google Scholar]
- 35.Berkenstadt M, Shiloh S, Barkai G, et al. . PPC): a new concept in measuring outcome of genetic counseling. American journal of medical genetics 1999;82:53–9. [DOI] [PubMed] [Google Scholar]
- 36.Shearer AE, Kolbe DL, Azaiez H, et al. . Copy number variants are a common cause of non-syndromic hearing loss. Genome Med 2014;6:37 doi:10.1186/gm554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson JA, Meyn MS, Shuman C, et al. . Parents perspectives on whole genome sequencing for their children: qualified enthusiasm? J Med Ethics 2017;43:medethics-2016-103564 doi:10.1136/medethics-2016-103564 [DOI] [PubMed] [Google Scholar]
- 38.Kaiser J. Surprisingly few new parents enlist in study to have baby’s genome sequenced. Science 2016. doi:10.1126/science.aal0279 [Google Scholar]
- 39.Howard HC, Knoppers BM, Cornel MC, et al. . Whole-genome sequencing in newborn screening? A statement on the continued importance of targeted approaches in newborn screening programmes. Eur J Hum Genet 2015;23:1593–600. doi:10.1038/ejhg.2014.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjpo-2017-000119supp001.pdf (25.5KB, pdf)