

These findings indicate the long term tolerability and chemopreventive potential of dietary celastrol in colitis-associated colorectal cancer, a disease for which current preventive therapies are accompanied by severe and unwanted side effects.
Abstract
Celastrol is an anti-inflammatory natural triterpenoid, isolated from the herb Tripterygium wilfordii or thunder god vine. Here, we define mechanisms mediating anti-inflammatory activity of celastrol and demonstrate efficacy of a dietary celastrol supplement for chemoprevention of inflammation-driven carcinogenesis in mice. Dietary celastrol (31.25 ppm in rodent diet from 8 weeks to 25 weeks of age) is well tolerated and protects against LPS-induced acute inflammation in C57BL/6 mice, potently suppressing LPS-induction of inducible nitric oxide synthase (iNOS), cyclooxygenase (COX)-2, Interleukin (IL)-6 and IL-1β. To test whether dietary celastrol suppresses inflammation-driven colorectal cancer (CRC), we employed a unique model of spontaneous, inflammation-driven CRC in mice harboring a germ line deletion of the p27Kip1 gene and a T cell-specific deletion of Smad4 gene (Smad4co/co;Lck-crep27Kip1-/-or DKO), which develop severe intestinal inflammation and carcinogenesis as early as 3 months of age. Exposure of DKO mice to daily dietary celastrol (12.5 ppm in diet) from 6 weeks of age significantly suppressed development of colitis-associated CRC (CAC). Celastrol chemoprevention of CAC in this new model of intestinal neoplasia was associated with significant suppression of iNOS at 4 months of age, and iNOS, COX-2 and NFκB at 6 months of age, with significant reduction in inflammatory cytokines, IL-6 and IL-1β. Chemoprevetion of CAC by dietary celastrol was further confirmed in the model of azoxymethane (AOM) plus dextran sodium sulfate (DSS)-induced carcinogenesis in C57BL/6 mice. These data suggest the potential for celastrol as a safe and effective dietary supplement in the chemoprevention of CAC in humans.
Introduction
Colorectal cancer (CRC) is the second most common cause of cancer-related deaths in Western countries (1). Patients with inflammatory bowel disease (IBD) are at increased risk for development of CRC, which accounts for 1 in 6 deaths in such individuals (2). Risk for CRC in patients with ulcerative colitis (a form of IBD) is 2% after 10 years active disease, 8% after 20 years active disease and 18% after 30 years active disease (2,3). Reduced incidence of colitis-associated CRC (CAC) in response to non-steroidal anti-inflammatory drugs (NSAIDs) in colitis patients illustrates the inflammatory component of carcinogenesis leading to CAC, as well as the potential for prevention or delay of carcinoma through pharmacologic suppression of the inflammatory response. Adverse side effects to current anti-inflammatory compounds and lack of response to available treatments in selected patient populations necessitate the exploration of additional compounds and classes of compounds for use in the pre-emption of CAC (1,4–7).
We previously reported the potential of the semi-synthetic agent, methyl 2-cyano-3,12-dioxoleana-1,9(11)dien-28-oate (CDDO-Me) in the chemoprevention of CAC using the genetic spontaneous CAC model harboring conditional deletion of the Smad4 gene in T cells in mice (Smad4TKO) (8). CDDO-Me is derived synthetically from the natural product oleanolic acid, one of a family of phytochemicals known as the triterpenoids which are ubiquitous in nature (9). Celastrol (Figure 1A) is a quinone methide natural triterpenoid derived from Tripterygium wilfordii, a vine indigenous to Eastern Asia, also known as “Lei gong teng” or thunder god vine. T. wilfordii has been used for centuries in traditional medicine for the treatment of arthritis and other diseases associated with inflammation and celastrol has been demonstrated to have potent anti-inflammatory and anti-cancer activities, both in vitro and in animal models (10,11). There is increased public awareness and scientific interest in plant-based and dietary supplement therapy in disease treatment and prevention, particularly in prevention of CRC (12,13). Thus, we have investigated the potential for incorporation of the natural triterpenoid celastrol as a dietary supplement in mouse models for acute inflammation and chronic inflammation-associated colon carcinogenesis.
Figure 1.

Celastrol administration in diet suppresses acute LPS-induced inflammation. (A) Chemical structure of celastrol. (B) Experimental scheme: C57BL/6 mice were maintained on control diet or 31.25 ppm celastrol-supplemented diet beginning at 8 weeks of age. At 25 weeks of age, mice received 1 mg/kg LPS or vehicle IP and were euthanized 6 hours later. Serum and livers were collected for analysis. (C) Serum nitric oxide concentration (μM) in mice receiving vehicle or LPS and consuming control or celastrol diet. (D) Expression of iNOS, COX-2 and cytokines in liver lysate from mice treated with vehicle or LPS and consuming control or celastrol diet, relative to liver expression in vehicle-treated mice on control diet, as measured by RT-PCR. Results are mean ± SEM. Statistical analyses include n = 3–5 mice. *P < 0.05.
We determined that long-term administration of celastrol in rodent diet is well tolerated and suppresses acute inflammation induced by systemic lipopolysaccharide (LPS) administration in mice. More importantly, we demonstrate the potential of daily dietary celastrol in the chemoprevention of chronic inflammation-driven colon carcinogenesis in a model for spontaneous CAC in mice harboring a T cell-conditional deletion of the Smad4 gene and germ line deletion of the p27Kip1 gene (Smad4co/co;Lck-crep27Kip1-/-, double knockout (DKO) mice). DKO mice display even more rigorous disease presentation than the Smad4TKO model (a full characterization of this novel mouse model is described in a separate report). The chemopreventive activity of celastrol was confirmed further in the traditional, carcinogen-induced CAC model utilizing azoxymethane (AOM) and dextran sodium sulfate (DSS) in C57BL/6 mice. Dietary celastrol markedly allayed disease presentation of CAC in these models and substantially suppressed tumorigenesis and mediators of inflammation, most principally inducible nitric oxide synthase (iNOS) and downstream carcinogenic DNA damage. Our results support the pursuit of celastrol as a safe and effective dietary supplement in the chemoprevention of CAC.
Methods
Materials
Purified celastrol was purchased from Ontario Chemical Inc. (C1307, 98.5% purity). LPS (L2630), anti-β-actin (A5441) and azoxymethane (A5486) were purchased from Sigma Aldrich. Gene expression assays for iNOS (Mm00440502_m1), cyclooxygenase (COX)-2 (Mm00478374_m1), IFNγ (Mm0168134_m1), IL-1β (Mm00434228_m1), IL-6 (MM0046190), TNFα (Mm0043258_m1) and β-actin (Mm0060939_s1) were purchased from Applied Biosystems. Antibodies for iNOS (sc-650), COX-2 (sc-1747) and phospho-IκB (sc-8404) were purchased from Santa Cruz Biotechnology. Phospho-H2AX (γH2AX) antibody was purchased from Cell Signaling Technology (9718). DSS was purchased from MP Biomedicals (2160110). Custom rodent diets were mixed by the LabDiet/TestDiet division of Land O’Lakes Purina Feed. The base diet for control diet and for celastrol supplementation was from LabDiet (ProLab IsoPro RMH 3000 5P76).
Animals and treatments
For acute exposure to LPS in C57BL/6 mice (Jackson Labs), all male animals were maintained on non-supplemented diet (control, LabDiet 5P76) or 31.25 ppm celastrol-supplemented diet from 8 weeks of age until 25 weeks of age. For a mouse weighing 30 g and consuming 4.8 g of diet per day, 31.25 ppm correlates to oral administration of 5 mg/kg celastrol per day. The doses of celastrol selected for our in vivo experiments are within range of those used in prior studies in other models (14,15). Following administration of celastrol in diet until 25 weeks of age, mice received 1 mg/kg LPS, intraperitoneally (IP). After 6 hours, mice were euthanized by CO2. Serum and liver were collected for analysis.
Mice with conditional deletion of the Smad4 gene in T cells have been described (16). Mice deficient for the Cdk inhibitor p27Kip1 were provided by A. Koff (Memorial Sloan-Kettering, New York, NY) (17). To generate mice deficient for both p27Kip1 in germ line and Smad4 in T cells, we bred Smad4co/co;Lck-cre females with p27Kip1-/- males (p27Kip1-/- females are infertile). The resulting F1 heterozygotes were then bred to generate Smad4co/co;Lck-crep27Kip1-/- (double knockout or DKO). Mice were housed in a pathogen-free facility. For studies in DKO mice, animals received either a non-supplemented (control, LabDiet 5P76) diet or diet containing 12.5 ppm celastrol beginning at 6 weeks of age. For a mouse weighing 30 g and consuming 4.8 g of diet per day, 12.5 ppm correlates to oral administration of 2 mg/kg celastrol per day. Mice were sacrificed at 16–17 weeks of age (4 months) or 21–25 weeks of age (6 months). Each treatment group contained a mix of male and female animals and no difference in disease development or response to treatment was observed between genders.
The AOM/DSS study is described with results (see also schematic in Figure 5A). For this study, mice (all male C57BL/6, Jackson Labs) treated with AOM/DSS were divided into two groups receiving either control diet (LabDiets, 5P76) or 12.5 ppm celastrol-supplemented diet beginning with the first administration of 2% DSS and continuing until sacrifice.
Figure 5.

Celastrol suppresses colitis-associated colon cancer in AOM/DSS mouse model. (A) Experimental procedure used to induce and treat colon cancer in C57BL/6 mice. After initial AOM injection (10 mg/kg IP), DSS (2%) was given in drinking water for 5 days, followed by normal drinking water for 9 days in 3 cycles. Mice were either maintained on control diet or 12.5 ppm celastrol diet beginning with first administration of DSS and continuing until termination of experiment. (B) Mortality, n = 15 (control) and n = 10 (celastrol). (C) Percent weight defined as follows: (final weight-initial weight)/initial weight x 100. Arrows indicate start days for DSS administration, n = 13 (control) and n = 10 (celastrol). (D) Colon length, weight and weight per length of AOM/DSS-treated mice receiving control diet or celastrol diet. (E) Representative photographs of excised colons from AOM/DSS-treated mice on control diet or celastrol diet. (F) Hematoxylin and eosin staining for longitudinal colon sections. Scale bar = 200 μm. (G) Tumor incidence. (H) Number of tumors per mouse. Analyses for D, G and H include n = 11 (control) and n = 10 (celastrol). Results are mean ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001.
In the CAC studies in mice, weights and survival were monitored throughout the experiments. At the time of sacrifice, mice were euthanized by CO2 and colons were excised, washed with cold PBS, measured and weighed. Epithelial surface of the colon was photographed for tumor counting and measuring. Sections were taken from the distal portion of the large bowel for histological assessment. Serum was collected and colon epithelial scrapings were taken for protein and RNA experiments.
Study approval
All animal experiments were performed in accordance with institutional guidelines and with approval of Institutional Animal Care and Use Committees at Case Western Reserve University.
Histology
A distal portion of each excised colon was fixed in 10% formalin. Samples were embedded in paraffin wax and sections were stained with H&E at Histoserv, Inc. (Germantown, MD).
Immunohistochemistry
Slides were deparaffinized and rehydrated and heat-induced epitope retrieval was performed. Slides were incubated with primary antibodies, iNOS (1:200) and γH2AX (1:400) for 1 h at room temperature. Immunohistochemistry was performed in the Tissue Resources Core Facility in the Comprehensive Cancer Center at Case Western Reserve University.
Cell culture
Peritoneal macrophages were collected by peritoneal lavage 7 days after IP administration of 2 ml 3% thioglycollate. Peritoneal macrophages were cultured in DMEM with 10% low endotoxin FBS. Cells were plated overnight at 9.0 × 106 in 60 mm plates. Following washing with 1× PBS, cells were treated with media containing celastrol and/or LPS as indicated. Cells were harvested by scraping at indicated time points and pelleted and frozen prior to processing for Western blot.
Nitric oxide assays in serum
For nitric oxide detection in serum, Nitrate/Nitrite colorimetric assay kit was used (Cayman Chemical, 780001). Serum was filtered through a centrifugal filter device and 40 μl were evaluated and quantified according to kit directions for combined concentrations of both nitrate and nitrite ions.
Quantitative RT-PCR analysis
Total RNA was isolated from tissues using TRIzol reagent (Invitrogen, 15596018). cDNA was synthesized using High Capacity cDNA synthesis kit (Applied Biosystems, 4368814). Quantitative RT-PCR was performed using BioRad CFX96 Real-Time System C1000 Thermal Cycler. ΔΔCT was calculated using β-actin as control gene with samples normalized to expression in wild type tissue. Quantification was performed using instrument software.
Western blotting
Frozen cells or epithelial scrapings from colons of mice were lysed by intermittent agitation and incubation in radioimmunoprecipitation assay (RIPA) buffer (Thermoscientific 89901), supplemented with protease and phosphatase inhibitors. 10–50 μg of denatured proteins were separated on tris-glycine gels and transferred to nitrocellulose membranes. Antibodies were used at a 1:1000 dilution except for β-actin, which was 1:25000.
Statistics
Data are expressed as mean ± SEM. Statistics were performed in Prism GraphPad and statistical differences were calculated using un-paired two-tailed Student’s t-test. Kaplan-Meier method was used to evaluate survival with Log-rank (Mantel-Cox) test to determine statistical difference between groups. Statistical significance was accepted to be a P value less than or equal to 0.05.
Results
Dietary celastrol administration suppresses LPS-induced acute inflammation
To evaluate the protective effect of chronic dietary celastrol administration against acute inflammation, we examined the response of mice to systemic LPS following long-term exposure to celastrol, as incorporated in the diet. Mice (C57BL/6) were maintained on either control diet or celastrol-supplemented diet with a concentration of 31.25 ppm celastrol beginning at 8 weeks of age and continuing until 25 weeks of age. At 25 weeks of age, all mice received an IP injection of either 1 mg/kg LPS or vehicle (PBS). Six hours post-injection, mice were euthanized and liver and serum were evaluated for expression of inflammatory mediators (see schematic Figure 1B). Mice receiving control diet showed robust induction of nitric oxide (NO) in serum as well as expression of inflammatory mediators in liver 6 h post-LPS exposure (Figure 1C and Figure 1D). However, mice chronically exposed to a celastrol-supplemented diet showed a significantly lower level of NO in serum following LPS exposure (Figure 1C) as well as decreased iNOS expression in liver, compared to mice on control diet (Figure 1D). Chronic dietary celastrol exposure significantly inhibited the induction of other inflammatory mediators by LPS, including COX-2, IL-1β and IL-6 (Figure 1D).
To evaluate whether diet intake was altered by supplementation of celastrol, diet consumption was monitored on a weekly basis for the first 13 weeks of chronic exposure (from age 9 weeks to 21 weeks). Over this time, average diet intake as grams per mouse per day was calculated and normalized to average body weight for mice receiving control diet or celastrol-supplemented diet. Diet intake was shown to be similar between groups (Supplementary Figure S1). When the actual dose of celastrol intake was calculated from these averages, it was determined that mice received a range of 4.0 to 5.3 mg/kg celastrol per day (data not shown). In total the results from these experiments indicate that celastrol incorporation in the diet at 31.25 ppm is palatable and well tolerated in mice, showing no signs of toxicity and also affording protection against acute, systemic, LPS-induced inflammation.
Dietary celastrol administration improves survival and suppresses spontaneous inflammation-driven carcinogenesis
We next explored the potential for cancer chemoprevention by celastrol in the context of chronic inflammation in a genetic model for spontaneous inflammation-associated carcinogenesis. Mice harboring a T cell restricted deletion of tumor suppressor gene Smad4 and a germ line deletion of cell cycle regulator p27Kip1 (Smad4co/co;Lck-crep27Kip1-/-, double knockout or DKO) spontaneously develop CAC at 3 to 4 months of age and the model serves as an effective platform for the investigation of potential cancer chemopreventive compounds. To test whether the natural triterpenoid celastrol could serve as an effective chemopreventive agent in the context of CAC, celastrol was administered as a dietary supplement to DKO mice at 12.5 ppm in the diet beginning at 6 weeks of age and continuing until 4 months (16–17 weeks) or 6 months (21–26 weeks) of age (Figure 2A). DKO mice receiving non-supplemented diet for the duration of the experiment were used as a control group. The survival of DKO mice on control diet was compromised, showing reduction to 80% by 20 weeks of age, 60% by 21 weeks of age, and 33% at 24 weeks of age. Impressively, 100% survival persisted at 20 weeks of age in mice on celastrol-supplemented diet and was 77% after 25 weeks (6 months) (P = 0.0157) (Figure 2B). Moreover, dietary celastrol led to a decrease in DKO mouse morbidity by preventing chronic diarrhea typically associated with development of CAC. DKO mice receiving celastrol-supplemented diet demonstrated improved ability to maintain and gain weight compared to DKO mice receiving control diet. After only 3 weeks on celastrol-supplemented diet (9 weeks of age), DKO experimental mice showed a statistically significant difference (5%) in weight gain over the control group (P = 0.0070). A significant margin over control group persisted for weight gain in mice receiving celastrol-supplemented diet with a difference of 10% at both 16 weeks (4 months) and 24 weeks (6 months) of age (P = 0.0076 and P = 0.0156, respectively) (Figure 2C). Importantly, these differences in clinical manifestations were also evident at gross necropsy with celastrol suppression of the significant, progressive mucosal thickening found in the large intestines of DKO mice at 4 and 6 months of age (Figure 3A). Gross necropsy of large intestines of DKO mice receiving celastrol-supplemented diet was largely normal in appearance at 4 months of age (Figure 3A), and at 6 months, the average colon weight per unit length (g/cm) as a measure of colon thickness in DKO mice receiving celastrol-supplemented diet was half that of DKO mice receiving control diet (0.055 vs. 0.109 g/cm) (P = 0.0273) (Figure 3B). Furthermore, DKO mice display prominent, progressive enlargement of the proximal duodenum that is also greatly attenuated in DKO mice receiving celastrol-supplemented diet (Figure 3C). Histological cross-sections stained with hematoxylin and eosin (H&E) in DKO colon and duodenum reveal gross enlargement and interruption of tissue architecture in the organs of DKO mice receiving control diet, while tissue architecture is greatly preserved in DKO mice receiving celastrol-supplemented diet (Figure 3D). These results indicate that celastrol administration in the diet of DKO mice at 12.5 ppm beginning at 6 weeks of age profoundly suppresses carcinogenesis and delays presentation of overt CAC.
Figure 2.

Celastrol improves survival and weight maintenance in DKO mouse model for spontaneous CAC. (A) Experimental scheme for treatment of DKO mice. At 6 weeks of age, DKO mice were divided into groups receiving control diet or a diet containing 12.5 ppm celastrol. Cohorts were euthanized and evaluated at 4 and 6 months of age. (B) Mortality, n = 15 for control diet and n = 13 for celastrol diet. (C) Percent weight defined as follows: percentage = (final weight – weight at 6 weeks) / weight at 6 weeks × 100, n = 22 for control diet and n = 24 for celastrol diet. Results represent mean for each treatment. *P < 0.05, **P < 0.01.
Figure 3.

Celastrol suppresses colitis-associated colon cancer in DKO mouse model for spontaneous CAC. (A) Representative photographs of excised colons from DKO mice on control diet or celastrol diet, euthanized at 4 or 6 months of age. (B) Colon weight per length (g/cm) of 4 and 6 month-old DKO mice on control and celastrol diets, n = 5 mice per group at 4 months and n = 8 mice per group at 6 months. *P<0.05. (C) Representative photographs of excised stomach (black arrow) and proximal duodenum (red asterisk) of DKO mice on control diet or celastrol diet, euthanized at 4 or 6 months of age. (D) Hematoxylin and eosin staining for colon and duodenal cross-sections in 4 month-old DKO mice on control or celastrol diet. Scale bar = 500 μm. Magnified inset is ×5 original.
To examine the effect of celastrol on mediators of inflammation associated with CAC in the DKO model, we evaluated epithelial scrapings from the mucosal surface of the colons of DKO mice receiving control or celastrol-supplemented diets. A high level of iNOS expression was detected in the colon of DKO mice on control diet at both 4 and 6 months as seen by Western blot, however chronic dietary administration of celastrol suppressed iNOS expression at both time points (Figure 4A). It is notable that the profound suppression of iNOS by celastrol was detected as early as 4 months, while suppression of COX-2 expression and phosphorylation of IκB were largely unchanged at that time point, but then significantly suppressed after the 6 month time point (Figure 4A). These data are corroborated by an evaluation of mRNA from the colon mucosal surface at both time points, which again shows significant suppression of iNOS transcripts at both the 4 and 6 month time points while suppression of other mediators of inflammation, including COX-2, IL-6 and IL-1β, was significant only after the 6 month time point (Figure 4B). These data indicate an increased sensitivity for iNOS for suppression by celastrol relative to other inflammatory mediators earlier in the carcinogenic process in the DKO model for chronic inflammation and spontaneous CAC. This interpretation is supported by analyses of circulating NO production in sera collected from DKO mice receiving either control or celastrol-supplemented diets which also demonstrated significantly decreased NO production in the serum of mice on celastrol-supplemented diet at 4 and 6 months, indicating that the down-regulation of iNOS by celastrol was systemic (Figure 4C). Thus, celastrol profoundly suppresses both iNOS expression in colon and circulating NO during both early and late-stage carcinogenesis, while its effect on other mediators of inflammation is mainly during late-stage carcinogenesis.
Figure 4.

Celastrol decreases iNOS at 4 and 6 months, and other mediators of inflammation in DKO colon at 6 months. (A) Expression of iNOS, COX-2 and phosphorylated-IκB in lysates from colon epithelia of DKO mice on control diet or celastrol diet at 4 and 6 months of age, as detected by Western blot. (B) Expression of iNOS, COX-2 and cytokines in colon epithelia of DKO mice on control diet or celastrol diet at 4 and 6 months of age, relative to age-match wild type tissue, as measured by RT-PCR, n = 5 mice per group at 4 months and n = 8 mice per group at 6 months. (C) Concentration of nitric oxide in serum of DKO mice on control diet or celastrol diet at 4 and 6 months of age, n = 4 or 5 mice per group at 4 months and n = 7 or 8 mice per group at 6 months. (D) Expression of iNOS in colon sections from 4 month old DKO mice treated with control or celastrol diet. Scale bar = 100 μm and insets are ×4 original magnification. B and C results are mean ± SEM. *P < 0.05.
iNOS has a known role in fighting pathogens by expression in immune cells infiltrating an area where tissue damage is present (18). However, many studies have also evaluated the role of iNOS expression in colonic epithelial cells in the context of IBD (19). To examine whether iNOS expression and its suppression by celastrol was localized to the immune cell or colonic epithelial cell compartments, colon sections from 4 month-old DKO mice were evaluated for iNOS expression in tissue by immunohistochemistry. High iNOS expression was observed primarily in DKO colon epithelial cells, and was detected to a lesser extent in the infiltrating immune cell population. iNOS suppression by celastrol administration in the diet was evident in colon tissue as observed by immunohistochemistry (Figure 4D). The result suggests that attenuation of inflammation and spontaneous CAC in DKO mice by celastrol is mediated by suppression of iNOS in colonic epithelial cells and infiltrating immune cells.
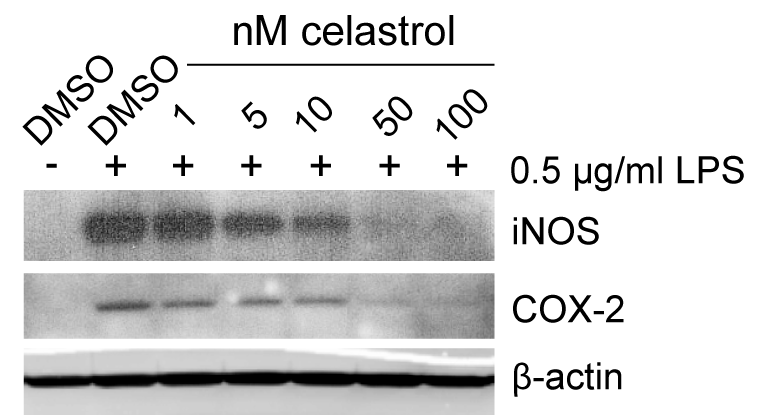
To define the dose-dependent nature of iNOS suppression by celastrol in cell culture, we evaluated the ability of celastrol to inhibit LPS-induced inflammation in peritoneal mouse macrophages. Macrophages were activated with 0.5 μg/ml LPS and co-treated with 1 to 100 nM celastrol for 24 h. As observed by Western blot, iNOS and COX-2 expression were both dose-dependently suppressed by celastrol, showing significant reduction at 100 nM celastrol (Supplementary Figure S2). Interestingly, in peritoneal macrophages, iNOS was also moderately suppressed at low nanomolar concentrations (5 and 10 nM), at which we did not observe suppression of COX-2, indicating that iNOS is more sensitive to suppression by celastrol, as was also observed in vivo in the DKO mouse model.
Dietary supplementation with celastrol suppresses carcinogen-induced colitis-associated colon cancer
To support our findings in the DKO model, we examined the effect of dietary celastrol in the well-established mouse model of azoxymethane/dextran sodium sulfate (AOM/DSS) inflammation-associated CAC. Mice (C57BL/6, 8 weeks of age) received 10 mg/kg AOM by IP injection, followed by exposure to 2% DSS in drinking water for 5 days. A total of 3 cycles of 2% DSS were completed with 9 days normal water provided between cycles. Mice were maintained on either a control diet or a celastrol-supplemented diet containing 12.5 ppm celastrol beginning with the first cycle of DSS and continuing throughout the experiment. Mice were sacrificed 31 days after the final administration of DSS (Figure 5A). Mice receiving celastrol-supplemented diet experienced zero mortality while survival dropped slightly to 84% in mice receiving control diet 47 days after AOM administration (P = 0.2042) (Figure 5B). Weight was measured 3 days per week during DSS cycles and 2 days per week thereafter. Mice receiving celastrol-supplemented diet demonstrated early and significantly enhanced recovery in bodyweight loss in response to DSS treatment when compared to mice receiving control diet. Notably, weight maintenance in mice receiving celastrol-supplemented diet compared to mice receiving control diet was statistically significant during the first DSS recovery phase (Day 11 to Day 15) and during subsequent DSS treatment phases (Day 18 to Day 22 and Day 34, respectively). The difference in weight change between diets was also significant at the time the experiment was terminated at Day 68 (Figure 5C).
To evaluate animal appetites and ingestion of celastrol, diet intake was monitored weekly as weight of diet (g) per mouse per day throughout the experiment and normalized to average body weights for each treatment group. Diet intake per g body weight was similar between groups, except for two occasions (Day 14 and Day 42) when suppressed intake in the control group was presumed to be due to compromised appetite from recent DSS exposure (Supplementary Figure S3). When the average dose of celastrol intake was calculated for the celastrol treated group, it was determined that mice received a range of 1.6 to 2.1 mg/kg celastrol per day (data not shown).
To determine the effect of celastrol on disease presentation in the AOM/DSS model for CAC, we evaluated colon characteristics in mice receiving control versus celastrol diet. In this model, colon length decreases due to stress, inflammation and ulceration. Further, colon weight is increased consequent to inflammation-induced swelling and tumor incidence. We observed that colon length was significantly longer in mice receiving celastrol-supplemented diet (7.5 ± 0.2 cm) compared to mice receiving control diet (6.8 ± 0.2 cm) (P = 0.0250) (Figure 5D). This trend is also apparent in the representative photographs of colons from mice on control and celastrol diets (Figure 5E). Colon weight was also significantly decreased in mice receiving celastrol in the diet (0.24 ± 0.01 g) compared to the colon weight in mice receiving control diet (0.28 ± 0.01 g) (P = 0.0326) (Figure 5D). The ratio of those values, as a reflection of overall colon thickness, shows a highly significant difference in colon weight per unit length (g/cm) between mice receiving celastrol diet (0.032 ± 0.002) and mice receiving control diet (0.040 ± 0.001) (P = 0.0004) (Figure 5D).
It was further determined that celastrol administration in the diet of mice treated with AOM/DSS led to suppression of tumorigenesis. In this model, DSS administration fosters an IBD-like environment and drives the incidence of AOM-induced tumors. Mice receiving celastrol-supplemented diet demonstrated significant protection from AOM/DSS-induced tumor formation, which can be observed in the representative gross necropsy (Figure 5E), and in histological longitudinal colon sections stained with H&E (Figure 5F). While tumor formation in the colon was observed in 100% of the mice on control diet, only 40% of mice (4 out of 10) receiving celastrol-supplemented diet had tumor incidence in the colon (Figure 5G). Colon tumor multiplicity in mice receiving celastrol-supplemented diet was markedly decreased with mice harboring, on average, less than one tumor per mouse (0.6 ± 0.3), compared to an average of 4.6 ± 0.6 tumors per mouse in mice receiving control diet (P < 0.0001) (Figure 5H). Interestingly, 50% of the tumors in mice receiving celastrol diet were smaller than 3 mm in diameter and tumors larger than 5 mm in diameter were only observed in mice receiving control diet (Supplementary Figure S4). These results indicate that administration of celastrol in the diet suppresses carcinogen-induced CAC and attenuates tumorigenesis in the AOM/DSS mouse model.
Celastrol suppresses colon epithelial expression of inflammatory mediators and γH2AX in the AOM/DSS model of CAC
We evaluated the colon epithelial surface for expression of inflammatory mediators in mice on either control or celastrol-supplemented diet. Multiple mediators of inflammation were suppressed by celastrol administration in the diet. Similar to results in the DKO model, celastrol significantly abrogated iNOS production in the colon as observed by both Western blot (Figure 6A) and quantitative RT-PCR (Figure 6B). We also observed moderate suppression of IκB phosphorylation by Western blot, and significant suppression of COX-2 and multiple inflammatory cytokines, including IL-1β, IL-6 and TNFα by RT-PCR in colon mucosa of AOM/DSS mice receiving celastrol-supplemented diet (Figure 6A and B). These results indicate that celastrol attenuates CAC in the AOM/DSS model by suppression of the inflammatory response, with particularly robust suppression of iNOS expression in colon mucosa.
Figure 6.

Celastrol suppresses CAC in AOM/DSS model by inhibition of inflammatory mediators and DNA damage. (A) Expression of iNOS and phosphorylated-IκB lysates from colon epithelia of AOM/DSS-treated mice receiving control diet or celastrol diet, as detected by Western blot. (B) Expression of iNOS, COX-2 and cytokines in colon epithelia of AOM/DSS-treated mice receiving control diet or celastrol diet, relative to untreated age-match wild type tissue, as measured by RT-PCR, n = 11 (control) and n = 10 (celastrol). Results are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. (C) Expression of iNOS and gamma-H2AX in colon sections from AOM/DSS mice on control diet or celastrol diet. Scale bar = 100 μm and insets are ×4 original magnification. (D) Expression of iNOS and gamma-H2AX in lysates from colon epithelia of AOM/DSS-treated mice on control diet or celastrol diet, as detected by Western blot.
Further evaluation of colon tissues for iNOS by immunohistochemistry revealed that robust iNOS expression was localized to colon epithelial cells in AOM/DSS mice on control diet, while abundant iNOS expression in infiltrating immune cells was not observed. In comparison, a significant suppression of iNOS expression was apparent in tissues from AOM/DSS mice receiving celastrol-supplemented diet (Figure 6C). These results demonstrate that in the AOM/DSS model, iNOS expression is elevated in colon epithelial cells and potently suppressed by administration of celastrol in diet. Shaked et al. have previously explored the implications of increased iNOS expression in intestinal epithelial cells, consequent to constitutively active IκB kinase β (IKKβ), in mice prone to intestinal tumorigenesis due to loss of adenomatous polyposis coli (APC) function. It was suggested that enhanced DNA damage response due to nitrosative damage to intestinal epithelial cell DNA resulted in increased loss of heterozygosity and resultant β-catenin+ foci and intestinal tumorigenesis (20). Additionally, DNA damage induced by inflammation has been shown to contribute to carcinogenesis in AOM/DSS-induced CAC (21). To determine whether cancer suppression by celastrol could result from iNOS suppression and consequent inhibited nitrosative damage to DNA in colonic epithelial cells, tissues from AOM/DSS mice receiving either control or celastrol diet were examined for phosphorylated H2AX (γH2AX), a marker for double-stranded DNA damage (22). By immunohistochemistry, robust γH2AX was localized to epithelial cells where iNOS was also highly expressed (Figure 6C). Similar to suppression of iNOS, γH2AX was also significantly decreased in tissues from AOM/DSS-treated mice receiving celastrol-supplemented diet (Figure 6C). In mucosal scrapings from the colon, increased γH2AX was also observed by Western blot, correlating with high expression of iNOS in mice receiving control diet, while γH2AX was undetected in mice receiving celastrol-supplemented diet (Figure 6D). These results indicate that in the AOM/DSS mouse model for CAC, celastrol activity is manifest in the resident epithelial cell population and that by suppressing iNOS, celastrol potentially inhibits downstream nitrosative damage to DNA in intestinal epithelial cells.
Discussion
This work indicates that dietary celastrol is well tolerated on a long-term basis in rodents and acts to suppress the inflammatory response in mouse models for acute LPS infection as well as chronic inflammation-driven colon carcinogenesis. This is the first demonstration of the suppression of spontaneous CRC by administration of celastrol. Importantly, the genetics and development of spontaneous inflammation-driven colon carcinogenesis described in the DKO mouse model are highly relevant to CAC in humans, in whom the reduced expression of p27KIP1 is associated with increased disease recurrence or death (23) and in whom SMAD4 mutations are predisposing to development of gastrointestinal cancer (24,25). Formerly, our lab discovered the necessity of Smad4 signaling in T cells in the suppression of gastrointestinal carcinogenesis in mice (Smad4TKO) (16) and subsequently demonstrated the value of the Smad4TKO mouse as a model for testing potential cancer chemopreventives, unraveling a unique mechanism for CAC suppression by a synthetic triterpenoid CDDO-Me (8). Carcinogenesis and disease progression in the colon of DKO mice takes place on a more rapid time frame relative to Smad4TKO mice (submitted manuscript), allowing for timely assessment of preventive and therapeutic drug effects. In this preclinical model, we demonstrate both the tolerance and safety of long-term daily administration of dietary celastrol and the tremendous capacity for cancer chemoprevention of spontaneous CAC in this genetic model of CAC. To confirm the effects we observed in the DKO model, we utilized the traditional AOM/DSS model for inflammation-driven CRC. We observed a similarly potent suppressive effect on disease progression and outcome, as well as inflammatory mediators in the AOM/DSS model, indicating similar effects and mechanistic targets in multiple models for CAC. Our findings in the AOM/DSS model corroborate recently published data by Lin et al. However, here we provide direct evidence in both spontaneous and carcinogen-induced models of CAC of the potency of celastrol as a cancer chemopreventive acting through direct suppression of inflammation that precedes the onset of overt disease, as opposed to the principal focus of Lin et al. on the putative effects of celastrol on progression and metastatic potential of established primary tumors (26).
Our observations regarding celastrol-mediated repression of the carcinogenic process by inhibition of mediators of inflammation implicate an importance of the particularly robust suppression of iNOS expression and circulating NO. iNOS has been established as a critical mediator in the progression of IBD and colon cancer (19,27,28) and has long been predicted to play a role in the initiation and promotion of carcinogenesis by driving production of reactive oxygen and nitrogen species (RONS) (29,30). Accordingly, we showed in AOM/DSS mice that celastrol suppression of iNOS in the epithelial compartment correlated with suppressed H2AX phosphorylation, a known marker for DNA damage. Expression of iNOS is regulated primarily at the transcription level. As a primary transcription factor for iNOS expression, NFκB is recognized as a central target for both activators and inhibitors of iNOS expression and is inhibited by celastrol via blockage of IκB kinase activity (31,32). Thus, we were intrigued that in the cohort of 4 month-old DKO mice receiving celastrol diet, both colonic iNOS expression and circulating NO were significantly suppressed in the absence of any change in phosphorylation of IκB, compared to mice receiving control diet. Thus we conjecture that iNOS suppression by celastrol in the early stages of carcinogenesis in the DKO mouse model may be independent of an inhibitory effect on NFκB signaling. Alternative mechanisms have been suggested by studies demonstrating a role for the cellular chaperone, heat shock protein 90 (Hsp90) in enhancing iNOS activity and enabling iNOS mRNA synthesis (33,34). Regarding the latter, it was shown that Hsp90 was required for binding of active transcription factors, NFκB and signal transducers and activators of transcription 1 (STAT1), to DNA elements prior to iNOS mRNA transcription (33). As celastrol is a known inhibitor of Hsp90 (35), it is plausible that iNOS suppression by celastrol in the absence of inhibition of IκB phosphorylation could be via Hsp90 inhibition and consequent interruption of transcription complex binding to the iNOS promotor, disabling mRNA synthesis. Moreover, celastrol has also been identified as an inducer of Nuclear factor-erythroid 2 (NF-E2)-related factor 2 (Nrf2) driven antioxidant and cytoprotective signaling. Indeed, the progression of inflammation-induced CRC has been shown to be significantly more severe than in mice deficient in Nrf2 signaling (36–39). Similarly, celastrol induction of antioxidant gene expression has been shown to be protective in a murine model for DSS-induced colitis (40,41). More recently, Nrf2 has been shown to directly suppress transcription of proinflammatory cytokine genes (42), suggesting that multiple mechanisms underlie cancer chemoprevention by celastrol. Further studies will be necessary to more fully elucidate the mechanistic role of celastrol in its robust suppression of inflammation, particularly iNOS suppression, in inflammation-driven colon carcinogenesis.
Beyond the potent effect we have demonstrated for the suppression of inflammation-driven carcinogenesis by celastrol, there is a significant amount of literature detailing anti-cancer activities of celastrol. Such activities include inhibition of proliferation and induction of caspase 3 activity and apoptosis in multiple cancer cell lines, as well as suppression of tumor growth in xenograft models and anti-cancer effects in non-colon cancer models (14,43–45). The effect of celastrol in CRC models without underlying inflammatory bowel disease remains to be evaluated and will ultimately be a valuable contribution to this field. Prior findings in cancer cells and animal studies lend to the prediction that celastrol would exhibit anti-proliferative and anti-metastatic properties in such models. Notwithstanding, the underlying role of inflammation in driving cancer development even in non-inflammation driven CRC (46) would predict inflammatory mediators to also be likely targets of celastrol in the tumor microenvironment in either inflammation-driven or sporadic circumstances. Indeed, our in vitro data in immune cells (Supplementary Figure S2) indicates that celastrol exhibits anti-inflammatory activities at decreased doses from those observed for anti-proliferative activities in cancer cell lines (44). This is not an uncommon phenomenon for other triterpenoids as well (47). As a potent compound with multiple identified cellular targets, celastrol is likely to impact several stages of cancer development, dependent upon dose and tissue exposure. The nature of the triterpenoid class of molecules to influence more than one cellular pathway is an aspect which makes them particularly appealing candidates in both chemoprevention and cancer therapy (47).
In summary, dietary celastrol is protective against LPS-induced inflammation in mice and suppresses inflammatory mediators and disease progression in chronic inflammation-driven colon carcinogenesis in both spontaneous and carcinogen-induced models for CAC. An important implication of the present study is that dietary supplementation of the well tolerated natural triterpenoid, celastrol, may offer potential advantage over available NSAIDs and other anti-inflammatory drugs currently in use as therapeutics in IBD and as pre-emptive agents for CAC in populations at increased risk. Oral bioavailability will be an important consideration for future clinical studies. A few reports have demonstrated systemic absorption of celastrol when administered orally to animals in pure form or in Tripterygium wilfordii extract to animals or humans (48,49). Bioavailability of celastrol was calculated to be 17.06% in rats receiving 1 mg/kg celastrol by oral gavage with increased absorption when administered as Tripterygium wilfordii extract instead of pure form (49). Interestingly, Zhang et al. also observed improved absorption in female versus male rats (49). Further preclinical studies will be necessary to examine tissue exposure, absorption, metabolism, distribution and excretion of celastrol when administered on a continual basis in the diet. Preclinical demonstration of safety and effectiveness of dietary celastrol will pave the way for future studies in clinical settings and in human trials in inflammatory-driven cancers and other inflammatory diseases.
Supplementary material
Supplementary data are available at Carcinogenesis online.
Funding
National Institutes of Health (R01 CA168586 to J.J.L., F31 GM093585 and T32CA059366 to E.C.B and R01 CA157735 to G.P.T.); The Reuter Foundation.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We would like to acknowledge Janet K. Robinson for her work in making available the Smad4co/co;Lck-crep27Kip1-/- (DKO) mice for this study. We also acknowledge Adam Kresak in the Tissue Resources Core Facility in the Comprehensive Cancer Center at Case Western Reserve University for his assistance with immunohistochemistry. We would further like to acknowledge use of the Leica SCN slide scanner in the Light Microscopy Imaging Facility at Case Western Reserve University made available through the Office of Research Infrastructure (NIH-ORIP) Shared Instrument Grant (S10RR031845). Dr. Letterio would like to acknowledge the generous support of the Jane and Lee Seidman Chair in Pediatric Cancer Innovation.
Conflict of Interest Statement: None declared.
Abbreviations
- AOM
azoxymethane
- CAC
colitis-associated CRC
- COX-2
cyclooxygenase-2
- CRC
colorectal cancer
- DKO
double knockout
- DSS
dextran sodium sulfate
- IBD
inflammatory bowel disease
- iNOS
inducible nitric oxide synthase
- LPS
lipopolysaccharide
- NSAIDs
non-steroidal anti-inflammatory drugs
References
- 1. Danese S., et al. (2011)Colitis-associated cancer: the dark side of inflammatory bowel disease. Gut, 60, 1609–1610. [DOI] [PubMed] [Google Scholar]
- 2. Munkholm P. (2003)Review article: the incidence and prevalence of colorectal cancer in inflammatory bowel disease. Aliment Pharmacol Ther., 18 (suppl. 2), 1–5. [DOI] [PubMed] [Google Scholar]
- 3. Bernstein C.N., et al. (2001)Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer, 91, 854–862. [DOI] [PubMed] [Google Scholar]
- 4. Asano T.K., et al. (2004)Non steroidal anti-inflammatory drugs (NSAID) and Aspirin for preventing colorectal adenomas and carcinomas. Cochrane Database Syst Rev., 2, CD004079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fink S.P., et al. (2014)Aspirin and the risk of colorectal cancer in relation to the expression of 15-hydroxyprostaglandin dehydrogenase (HPGD). Sci. Transl. Med., 6, 233re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang D., et al. (2013)The role of anti-inflammatory drugs in colorectal cancer. Annu. Rev. Med., 64, 131–144. [DOI] [PubMed] [Google Scholar]
- 7. Todoric J., et al. (2016)Targeting Inflammation in Cancer Prevention and Therapy. Cancer Prev. Res. (Phila)., 9, 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choi S.H., et al. (2014)Synthetic triterpenoid induces 15-PGDH expression and suppresses inflammation-driven colon carcinogenesis. J. Clin. Invest., 124, 2472–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dzubak P., et al. (2006)Pharmacological activities of natural triterpenoids and their therapeutic implications. Nat. Prod. Rep., 23, 394–411. [DOI] [PubMed] [Google Scholar]
- 10. Kannaiyan R., et al. (2011)Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett., 303, 9–20. [DOI] [PubMed] [Google Scholar]
- 11. Corson T.W., et al. (2007)Molecular understanding and modern application of traditional medicines: triumphs and trials. Cell, 130, 769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chang J. (2000)Medicinal herbs: drugs or dietary supplements?Biochem. Pharmacol., 59, 211–219. [DOI] [PubMed] [Google Scholar]
- 13. Madka V., et al. (2013)Anti-inflammatory phytochemicals for chemoprevention of colon cancer. Curr. Cancer Drug Targets, 13, 542–557. [DOI] [PubMed] [Google Scholar]
- 14. Yang H., et al. (2006)Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res., 66, 4758–4765. [DOI] [PubMed] [Google Scholar]
- 15. Li H., et al. (2005)Beneficial effect of tripterine on systemic lupus erythematosus induced by active chromatin in BALB/c mice. Eur. J. Pharmacol., 512, 231–237. [DOI] [PubMed] [Google Scholar]
- 16. Kim B.G., et al. (2006)Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature, 441, 1015–1019. [DOI] [PubMed] [Google Scholar]
- 17. Kiyokawa H., et al. (1996)Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell, 85, 721–732. [DOI] [PubMed] [Google Scholar]
- 18. MacMicking J., et al. (1997)Nitric oxide and macrophage function. Annu. Rev. Immunol., 15, 323–350. [DOI] [PubMed] [Google Scholar]
- 19. Kolios G., et al. (2004)Nitric oxide in inflammatory bowel disease: a universal messenger in an unsolved puzzle. Immunology, 113, 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shaked H., et al. (2012)Chronic epithelial NF-κB activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation. Proc. Natl. Acad. Sci. U. S. A., 109, 14007–14012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meira L.B., et al. (2008)DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Invest., 118, 2516–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rogakou E.P., et al. (1999)Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol., 146, 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chu I.M., et al. (2008)The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer, 8, 253–267. [DOI] [PubMed] [Google Scholar]
- 24. Howe J.R., et al. (1998)Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science, 280, 1086–1088. [DOI] [PubMed] [Google Scholar]
- 25. Howe J.R., et al. (2002)Common deletion of SMAD4 in juvenile polyposis is a mutational hotspot. Am. J. Hum. Genet., 70, 1357–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin L., et al. (2015)Celastrol Ameliorates Ulcerative Colitis-Related Colorectal Cancer in Mice via Suppressing Inflammatory Responses and Epithelial-Mesenchymal Transition. Front. Pharmacol., 6, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ahn B., et al. (2001)Suppression of intestinal polyposis in Apc(Min/+) mice by inhibiting nitric oxide production. Cancer Res., 61, 8357–8360. [PubMed] [Google Scholar]
- 28. Beck P.L., et al. (2004)Paradoxical roles of different nitric oxide synthase isoforms in colonic injury. Am. J. Physiol. Gastrointest. Liver Physiol., 286, G137–G147. [DOI] [PubMed] [Google Scholar]
- 29. Ohshima H., et al. (1994)Chronic infections and inflammatory processes as cancer risk factors: possible role of nitric oxide in carcinogenesis. Mutat. Res., 305, 253–264. [DOI] [PubMed] [Google Scholar]
- 30. Wiseman H., et al. (1996)Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem. J., 313 (Pt 1), 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee J.H., et al. (2006)Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem. Pharmacol., 72, 1311–1321. [DOI] [PubMed] [Google Scholar]
- 32. Pautz A., et al. (2010)Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide, 23, 75–93. [DOI] [PubMed] [Google Scholar]
- 33. Luo S., et al. (2011)Obligatory role of heat shock protein 90 in iNOS induction. Am. J. Physiol. Cell Physiol., 301, C227–C233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yoshida M., et al. (2003)Heat shock protein 90 as an endogenous protein enhancer of inducible nitric-oxide synthase. J. Biol. Chem., 278, 36953–36958. [DOI] [PubMed] [Google Scholar]
- 35. Hieronymus H., et al. (2006)Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell, 10, 321–330. [DOI] [PubMed] [Google Scholar]
- 36. Khor T.O., et al. (2006)Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res., 66, 11580–11584. [DOI] [PubMed] [Google Scholar]
- 37. Khor T.O., et al. (2008)Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev. Res. (Phila)., 1, 187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Osburn W.O., et al. (2007)Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. Int. J. Cancer, 121, 1883–1891. [DOI] [PubMed] [Google Scholar]
- 39. Seo W.Y., et al. (2011)Celastrol induces expression of heme oxygenase-1 through ROS/Nrf2/ARE signaling in the HaCaT cells. Biochem. Biophys. Res. Commun., 407, 535–540. [DOI] [PubMed] [Google Scholar]
- 40. Shaker M.E., et al. (2014)Celastrol ameliorates murine colitis via modulating oxidative stress, inflammatory cytokines and intestinal homeostasis. Chem. Biol. Interact., 210, 26–33. [DOI] [PubMed] [Google Scholar]
- 41. Itoh K., et al. (2010)Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid. Redox Signal., 13, 1665–1678. [DOI] [PubMed] [Google Scholar]
- 42. Kobayashi E.H., et al. (2016)Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun., 7, 11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chang F.R., et al. (2003)Antitumor agents. 228. five new agarofurans, Reissantins A-E, and cytotoxic principles from Reissantia buchananii. J. Nat. Prod., 66, 1416–1420. [DOI] [PubMed] [Google Scholar]
- 44. Kannaiyan R., et al. (2011)Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis, 16, 1028–1041. [DOI] [PubMed] [Google Scholar]
- 45. Chang W., et al. (2016)Protective effects of Celastrol on diethylnitrosamine-induced hepatocellular carcinoma in rats and its mechanisms. Eur. J. Pharmacol., 784, 173–180. [DOI] [PubMed] [Google Scholar]
- 46. Grivennikov S.I., et al. (2012)Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature, 491, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liby K.T., et al. (2007)Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat. Rev. Cancer, 7, 357–369. [DOI] [PubMed] [Google Scholar]
- 48. Ouyang X.K., et al. (2008)Development and validation of a liquid chromatography coupled with atmospheric-pressure chemical ionization ion trap mass spectrometric method for the simultaneous determination of triptolide, tripdiolide, and tripterine in human serum. J. Anal. Toxicol., 32, 737–743. [DOI] [PubMed] [Google Scholar]
- 49. Zhang J., et al. (2012)Oral bioavailability and gender-related pharmacokinetics of celastrol following administration of pure celastrol and its related tablets in rats. J. Ethnopharmacol., 144, 195–200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.