

Abstract
Repositioning of previously approved drugs is a promising methodology because it reduces the cost and duration of the drug development pipeline and reduces the likelihood of unforeseen adverse events. Computational repositioning is especially appealing because of the ability to rapidly screen candidates in silico and to reduce the number of possible repositioning candidates. What is unclear, however, is how useful such methods are in producing clinically efficacious repositioning hypotheses. Furthermore, there is no agreement in the field over the proper way to perform validation of in silico predictions, and in fact no systematic review of repositioning validation methodologies. To address this unmet need, we review the computational repositioning literature and capture studies in which authors claimed to have validated their work. Our analysis reveals widespread variation in the types of strategies, predictions made and databases used as ‘gold standards’. We highlight a key weakness of the most commonly used strategy and propose a path forward for the consistent analytic validation of repositioning techniques.
Keywords: drug repositioning, analytic validation, research reproducibility
Introduction
In recent years, the drug-repositioning field has gained substantial traction with both academics and pharmaceutical companies, both because of the lack of preclinical development and optimization, as well as the substantially reduced risk of unforeseen adverse events [1]. A search for ‘drug repositioning’ in PubMed reveals that the number of publications has grown rapidly from only 11 articles in 2007 to 274 in 2015. Many of the publications in the drug repositioning space have been computational methods; despite advances in high-content screening and robotics, high-throughput in vitro screens are still costly, leading many groups to turn to computational repositioning strategies [2]. Computational repositioning methods have proliferated substantially, using a variety of molecular [3–6], literature-derived [7–10] and clinical [11, 12] data as their core drivers of repositioning hypothesis generation.
All computational repositioning methods promise to prioritize repositioning candidates, and studies describing these methods typically claim superiority over competing methodologies. To do so, such studies perform analytic validation, whereby they compare the computational results of their methods (and competing methods) to existing biomedical knowledge. A successful method is one that consistently identifies known associations between drugs and diseases (and for some, fails to identify ‘wrong’ associations). When examining the repositioning literature, however, it is apparent that there are no consistent best practices for comparing studies and for validation of methods.
In this article, we examine the current trends in validation among studies in the computational repositioning field. We identify three major types of validation, involving the use of case studies, overlap of predictions with known drug indications and sensitivity- or specificity-based methods. All of these methods have drawbacks, trading off between a lack of analytic rigor and the unsatisfying assumption that all repositioning candidates are, a priori, false positives. We propose that the best-case scenario with currently available data is to use ‘overlap-type’ validation and describe a promising next step for the field.
Analytic validation in the computational repositioning literature
To gain a better understanding of validation in the computational repositioning field, we searched PubMed for articles in the computational drug repositioning space that claimed to have performed validation of their methodology or pipeline using a Boolean search [‘(drug repositioning OR drug repurposing) AND (gold standard OR AUC OR receiver operating characteristic OR validation OR validated OR validate)’, performed on 14 June 2016, Figure 1A].
Figure 1.
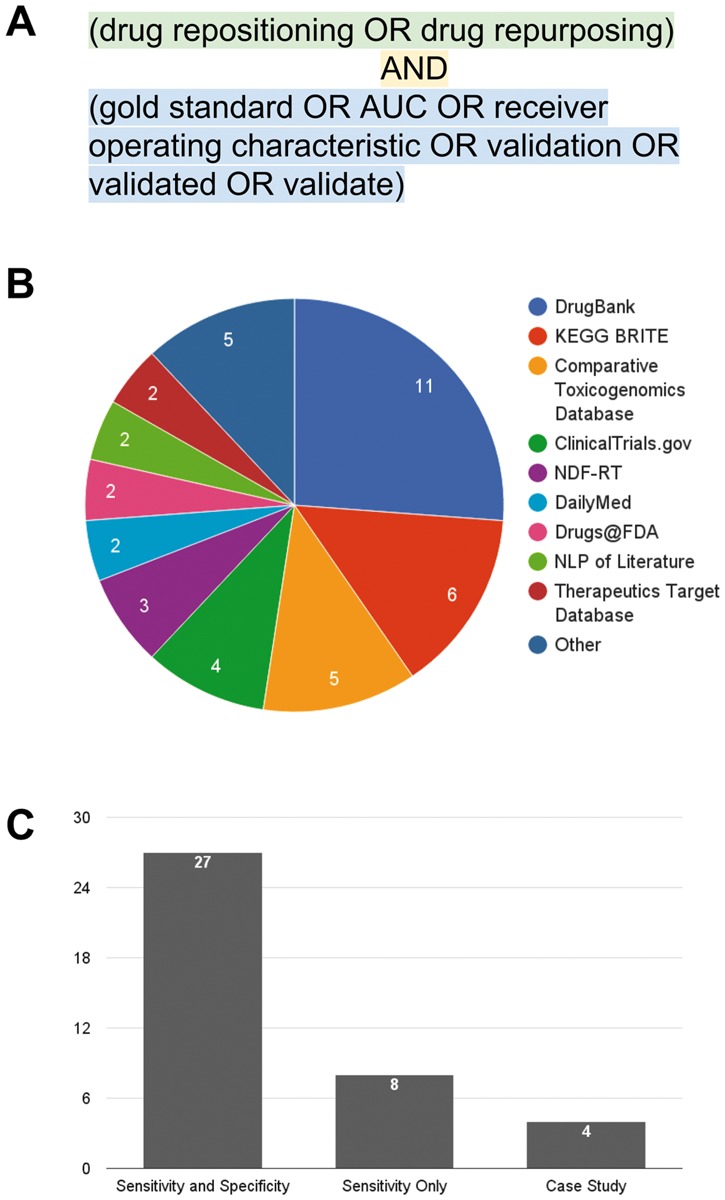
Computational repositioning validation studies. (A) The search term used with PubMed to retrieve articles. (B) Sources of drug-indication annotation data used in studies retrieved in the literature search. (C) Types of validation in studies retrieved in the literature search. See main text for details.
Using this search, we began our analysis with a pool of 213 articles. To further refine our search, we manually reviewed each of the articles, and excluded non-computational papers (e.g. high-throughput drug screens in cell lines or clinical trials), those not in the small molecule/drug field (e.g. articles referring to surgical or dental repositioning), and non-research articles (reviews and book chapters). From the remaining 141 articles, we focused on those that predicted novel indications for drugs. At this point, we excluded 35 articles that focused on target prediction only; in silico target prediction studies aim to predict novel, molecular targets for existing and novel drug candidates. We argue that target prediction is still one step removed from drug repositioning; true or predicted molecular targets can be used as part of repositioning methodologies, but do not themselves provide the full repositioning hypothesis from drug to indication. Furthermore, we note that benchmarking such studies is already possible with the wealth of high-throughput drug–target binding screens [13].
From the studies that predicted new indications for existing drugs, we excluded 67 articles that predicted indications for a single drug or disease, and kept those that made predictions for more than one drug and disease. We excluded these single-drug and disease studies because they were not designed to be applied broadly, and often contained domain-specific knowledge about a particular drug or disease (e.g. Genome Wide Association Studies results for a single disease or structure–association relationship studies for a single drug). This resulted in 39 computational repositioning methods articles with predictions spanning multiple drugs and indications and a clear claim of analytic validation (the full list of captured articles is provided in Supplementary Table S1).
We began our analysis by first examining the types of databases used for analytic validation: we discovered that, although many of the investigators in the 39 studies we examined claim to use a ‘gold standard’, there is substantial heterogeneity in the source of these standards as well as the types of data they contain (Figure 1B). For example, DrugBank [14] contains information about only the FDA-approved indications for drugs, while the Comparative Toxicogenomics Database (CTD) [15] contains literature-annotated links between drugs and both approved and investigational indications. While DrugBank contains a set of true drug-indication annotations, it misses off-label uses and late-stage clinical trials; on the other hand, the CTD relies on literature annotations and contains drug–indication pairs that have subsequently failed to receive FDA approval. This inconsistency in specificity among databases used for validation is detrimental to reproducibility and may lead to claims on extremely high accuracy.
We next examined the types of analytic validation methodologies used by investigators in computational repositioning. We grouped the 39 captured studies into three classes: (1) validation with a single example or case study of a single disease area (CSV), (2) sensitivity-based validation only (SV) and (3) both sensitivity- and specificity-based validation (SSV) (Figure 1C). First, of the three validation types, CSV is the least rigorous; each of the four CSV studies reported one to three clinically justifiable predictions (Supplementary Table S1). For example, Sirota and colleagues [16] identified cimetidine as a potential therapy for lung adenocarcinoma, which was picked from 2664 significant predictions (of >16 000 drug–indication pairs tested) on the basis of tolerability. The investigators provided evidence of its success using both the literature and an in vitro study. The inclusion of in vitro evidence lends additional biological credence to their single case study, but analytic evidence of their method’s overall success is lacking. We note here that we are not arguing that in vitro evidence is inferior to analytic validation; biological validation is a requirement for any individual candidate to be advanced in a drug development pipeline. However, successful biological validation of a single repositioning candidate cannot be extrapolated to all predictions made by a method.
Following CSV, SV provides more analytic rigor by measuring the overlap between currently approved or investigational indications for drugs and the indications predicted by a given repositioning method. In contrast to CSV, SV validation methodologies assess the general ability of repositioning methods to make reasonable claims, rather than selecting a single or several high-ranking predictions to test in depth. For example, Jung and Lee [17] examined the overlap between predictions made by their method and both approved drug indications (from a combination of DrugBank [14], PharmGKB [18] and TTD [19]) and investigational indications from ongoing clinical trials (from ClinicalTrials.gov). SV is appealing because investigators only need to have a set of true positives to which to compare their predictions (e.g. all approved or investigational drug indications). A key drawback of SV is the inability to use traditional two-class machine learning (ML) approaches. An alternative is to train one-class classification algorithms on positive examples only; however, to our knowledge, no methods in the drug repositioning space have used one-class ML approaches. We emphasize, as in any ML exercise, that investigators should perform cross-validation in which algorithms are fine-tuned on a portion of the data and tested on another; testing using an as yet unseen portion of the data is more representative of future performance than training and testing on the full data set [20].
Both CSV and SV validation methods are less popular than SSV. SSV is, in theory, the most rigorous type of validation. For our purposes, SSV-based methods include those that directly report sensitivity and specificity (or reported values for positive or negative predictive value), as well as area under the receiver operating characteristic (AUROC, a commonly used method for determining the predictive value of a method reviewed in [21]). For example, Gottlieb and colleagues [4] used a list of approved drug indications (from DailyMed), and determined how many of their predicted drug indications overlapped with that set (true positives) or did not overlap (false positives); their results are summarized by calculating the AUROC of their predictions. In contrast to sensitivity-only validation methods, methods that rely on both sensitivity and specificity require information about which predicted drug indications are false (false positives). In all of the SSV studies we reviewed, the investigators chose to mark all unannotated drug–indication pairs as false positives. This is troubling for two major reasons. First, the choice of annotation database can substantially impact the sensitivity and specificity estimates. If investigators consistently used a single database of standardized indication information, this issue could be avoided; however, in practice, annotation is derived from a variety of drug information databases and annotation types, from FDA approval, to ongoing clinical trials (Figure 1C). Second, marking unannotated pairs as false suggests that all novel repositioning hypotheses are false positives. This is obviously counterintuitive, as computational repositioning methods should predict novel indications, for which there is no currently annotated association. In addition, this strategy creates a substantial imbalance in the number of true and false positives; such an imbalance has been shown to reduce the accuracy of AUROC and other SSV estimates [22].
It is our opinion that, with currently available data, the best strategy for analytic validation in repositioning studies is SV. Under ideal conditions, in which a database of true positives and true negatives exists, SSV is the optimal choice; currently, however, the field lacks such a database. In contrast, SV does not require true negatives and therefore may be the most practical solution until such a database emerges. We note that there are still two central caveats with using SV for analytic validation: (1) investigators should choose the database to which they should compare their results carefully, potentially corroborating drug–indication pairs between multiple sources and (2) investigators using ML-based methods should test the performance of their methods with cross-validation to prevent over-fitting and limit the reporting of unrealistic predictive power.
The question then becomes the following: where can we go from here for analytic validation? Is there an internally consistent database that could be used with SSV? The way forward for SSV is to develop a set of true negatives; such a set would include drug–indication pairs that were tried in a clinical setting and were proved not to be efficacious or safe. An easily accessible database of this information does not, to our knowledge, currently exist, and creating one would require substantial biomedical, regulatory and legal understanding and resources. Despite these challenges, creating a true ‘gold standard’ that contains both repositioning successes and failures is one way to improve consistency in the field, and allows for equitable comparisons between methods. We believe that such a ‘gold standard’ database can improve the accuracy of drug repositioning methods and increase the probability of success in clinical trials.
Conclusions
We present here a brief review of the computational drug repositioning field, with a focus on strategies for analytically validating such methods. We describe the three types of validation currently in use, and highlight the issues with both consistency and key assumptions made by each. In closing, we propose a strategy for improving the quality of validation in computational repositioning.
Key Points
There are currently three predominate validation methods used for computational repositioning studies: (1) case studies, (2) overlap of predictions with known drug indications and (3) sensitivity- or specificity-based methods.
There is wide variation in the types and sources of annotation data used for performing validation, leading to a lack of consistency in the field.
Despite being rigorous, sensitivity- or specificity-based methods require the use of true negatives, and current studies assume that all unannotated drug–indication pairs are false positives.
While a sensitivity and specificity based method is optimal, we posit that the current best strategy is overlap (sensitivity only) because, despite a lower level of rigor, it does not require contradictory assumptions.
We propose a new direction in repositioning validation through the creation of a repositioning database to promote reproducible calculations of sensitivity and specificity.
Supplementary data
Supplementary data are available online at http://bib.oxfordjournals.org/.
Funding
The National Institutes of Health (NIH) Training grant from the National Human Genome Research Institute (NHGRI; grant number T32HG002295-12 to A.S.B.); the National Institute of Environmental Health Sciences (NIEHS; grant numbers R00 ES023504 and R21 ES025052 to C.J.P.); a gift from Agilent Technologies and a PhRMA fellowship (to C.J.P.).
Supplementary Material
Adam Brown is a PhD candidate in the Biological and Biomedical Sciences program at Harvard Medical School. His research centers around drug repositioning, the process of finding new indications for approved drugs.
Chirag Patel is an assistant professor in the Department of Biomedical Informatics at Harvard Medical School. His research is in translational bioinformatics and development of big data informatics techniques for discovery in biomedicine.
References
- 1. Rodriguez-Esteban R. A drug-centric view of drug development: how drugs spread from disease to disease. PLoS Comput Biol 2016;12:e1004852.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li J, Zheng S, Chen B, et al. A survey of current trends in computational drug repositioning. Brief Bioinform 2016;17:2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lamb J, Crawford ED, Peck D, et al. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006;313:1929–35. [DOI] [PubMed] [Google Scholar]
- 4. Gottlieb A, Stein GY, Ruppin E, et al. PREDICT: a method for inferring novel drug indications with application to personalized medicine. Mol Syst Biol 2011;7:496.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang H, Nguyen T, Ibrahim S, et al. DMAP: a connectivity map database to enable identification of novel drug repositioning candidates. BMC Bioinformatics 2015;16(Suppl 13):S4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brown AS, Kong SW, Kohane IS, et al. ksRepo: a generalized platform for computational drug repositioning. BMC Bioinformatics 2016;17:78.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qu XA, Gudivada RC, Jegga AG, et al. Inferring novel disease indications for known drugs by semantically linking drug action and disease mechanism relationships. BMC Bioinformatics 2009;10(Suppl 5):S4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheung WA, Ouellette BFF, Wasserman WW.. Quantitative biomedical annotation using medical subject heading over-representation profiles (MeSHOPs). BMC Bioinformatics 2012;13:249.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kissa M, Tsatsaronis G, Schroeder M.. Prediction of drug gene associations via ontological profile similarity with application to drug repositioning. Methods 2015;74:71–82. [DOI] [PubMed] [Google Scholar]
- 10. Patchala J, Jegga AG.. Concept modeling-based drug repositioning. AMIA Jt Summits Transl Sci Proc 2015;2015:222–6. [PMC free article] [PubMed] [Google Scholar]
- 11. Ryan PB, Madigan D, Stang PE, et al. Medication-wide association studies. CPT Pharmacometrics Syst Pharmacol 2013;2:e76.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu H, Aldrich MC, Chen Q, et al. Validating drug repurposing signals using electronic health records: a case study of metformin associated with reduced cancer mortality. J Am Med Inform Assoc 2015;22:179–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pahikkala T, Airola A, Pietilä S, et al. Toward more realistic drug–target interaction predictions. Brief Bioinform 2015;16:325–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 2006;34:D668–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Davis AP, Grondin CJ, Lennon-Hopkins K, et al. The comparative toxicogenomics database’s 10th year anniversary: update 2015. Nucleic Acids Res 2015;43:D914–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sirota M, Dudley JT, Kim J, et al. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med 2011;3:96ra77.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jung J, Lee D.. Inferring disease association using clinical factors in a combinatorial manner and their use in drug repositioning. Bioinformatics 2013;29:2017–23. [DOI] [PubMed] [Google Scholar]
- 18. Thorn CF, Klein TE, Altman RB.. PharmGKB: the pharmacogenetics and pharmacogenomics knowledge base. Methods Mol Biol 2005;311:179–91. [DOI] [PubMed] [Google Scholar]
- 19. Zhu F, Shi Z, Qin C, et al. Therapeutic target database update 2012: a resource for facilitating target-oriented drug discovery. Nucleic Acids Res 2012;40:D1128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lever J, Krzywinski M, Altman N.. Points of significance: model selection and overfitting. Nat Methods 2016;13:703–4. [Google Scholar]
- 21. Fawcett T. An introduction to ROC analysis. Pattern Recognit Lett 2006;27:861–74. [Google Scholar]
- 22. Hanczar B, Hua J, Sima C, et al. Small-sample precision of ROC-related estimates. Bioinformatics 2010;26:822–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.