

Both telomere length (TL) and ATM mutations have been associated with cancer susceptibility and ATM participates in the signaling of telomere erosion. We wondered whether carriage of an ATM mutation influences age-related TL shortening and cancer risk in ataxia-telangiectasia families.
Abstract
Recent studies have linked constitutive telomere length (TL) to aging-related diseases including cancer at different sites. ATM participates in the signaling of telomere erosion, and inherited mutations in ATM have been associated with increased risk of cancer, particularly breast cancer. The goal of this study was to investigate whether carriage of an ATM mutation and TL interplay to modify cancer risk in ataxia-telangiectasia (A-T) families.
The study population consisted of 284 heterozygous ATM mutation carriers (HetAT) and 174 non-carriers (non-HetAT) from 103 A-T families. Forty-eight HetAT and 14 non-HetAT individuals had cancer, among them 25 HetAT and 6 non-HetAT were diagnosed after blood sample collection. We measured mean TL using a quantitative PCR assay and genotyped seven single-nucleotide polymorphisms (SNPs) recurrently associated with TL in large population-based studies.
HetAT individuals were at increased risk of cancer (OR = 2.3, 95%CI = 1.2–4.4, P = 0.01), and particularly of breast cancer for women (OR = 2.9, 95%CI = 1.2–7.1, P = 0.02), in comparison to their non-HetAT relatives. HetAT individuals had longer telomeres than non-HetAT individuals (P = 0.0008) but TL was not associated with cancer risk, and no significant interaction was observed between ATM mutation status and TL. Furthermore, rs9257445 (ZNF311) was associated with TL in HetAT subjects and rs6060627 (BCL2L1) modified cancer risk in HetAT and non-HetAT women.
Our findings suggest that carriage of an ATM mutation impacts on the age-related TL shortening and that TL per se is not related to cancer risk in ATM carriers. TL measurement alone is not a good marker for predicting cancer risk in A-T families.
Introduction
Biallelic mutations in the Ataxia-Telangiectasia Mutated (ATM) gene are responsible for Ataxia-Telangiectasia (A-T), a rare autosomal recessive disorder characterized by progressive neuronal degeneration, immunological deficiency, genetic instability, radiosensitivity and an increased risk of cancer. Epidemiological studies on A-T families showed that heterozygous mutation carriers (hereafter called ‘HetAT’ subjects) are at increased risk of cancer (1), particularly of breast cancer (BC) in female relatives (2–4). Moreover, recent studies conducted in familial or early onset BC cases and ethnically similar unrelated controls have confirmed ATM as a BC susceptibility gene in a context independent of a genetic predisposition to A-T (5,6).
The ATM gene encodes a protein kinase, which is a central regulator of the cellular response to DNA damage and also the response to telomere dysfunction. Telomeres are repeats units of TTAGGG nucleotides coated by a six-subunit protein complex named shelterin that comprises TRF1, TRF2, POT1, TPP1, TIN2 and RAP1. The telomeric complex caps the ends of chromosomes to maintain genome integrity by protecting chromosomes from end-to-end fusion and exonuclease degradation (7,8). In normal cells, telomere elongation is telomerase-dependent and is highly regulated by various proteins including ATM which is a positive regulator of telomere lengthening (9,10). The phosphorylation of the shelterin protein TRF1 by ATM diminishes the interaction between TRF1 and telomeric DNA (11) leading to the release of TRF1 and ensuring access for the telomerase enzyme to lengthen telomeres (12). More recently, it was shown that the shelterin complex provides protection of chromosome ends against classical nonhomologous end-joining (NHEJ) and homologous-direct repair (HDR) by repressing the ATM and ATR pathway signaling (13).
Telomeres have long been recognized as a biomarker of cell aging due to their progressive shortening with each cell division (14,15), and a number of studies have demonstrated clear involvement of telomere shortening in aging-related diseases, in particular in the carcinogenesis of several malignancies (16). However, the direction of association between telomere length (TL) and cancer risk appears to be cancer-site dependent. For instance, longer telomeres have been associated with increased risk of gastric (17), lung (18), bladder (19) and esophageal cancers (20), while shorter telomeres have been associated with increased risk of non-Hodgkin lymphoma (21) and melanoma (22). The association between constitutive TL and BC risk remains controversial: some studies have associated shorter telomeres with an increased risk of BC (23,24), other studies have associated this risk with longer telomeres (25,26), while the prospective Sister Study did not support TL as a biomarker for BC risk (27). Results from the EMBRACE study confirmed the null association between TL and breast or ovarian cancer risk in BRCA1 and BRCA2 mutation carriers although longer lymphocyte TL was observed in this high-risk population than in the non-carriers population (28). Furthermore, several studies conducted in the general population have shown a significant association between TL and SNPs located at different loci (29,30), including rs2736100 in TERT (29), rs1317082 in TERC (30), rs2738783 in RTEL1 (30), rs10165485 in ACYP2 (30), rs2320615 in NAF1 (30), rs9257445 in ZNF311 (30) and rs6060727 in BCL2L1 (30). Some of the TL-related SNPs have also been associated with an increased risk of cancer (26,30,31).
Association between the ATM function and TL has been suggested by cytogenetic studies where lymphocytes with biallelic inactivation of ATM were characterized by an abnormal rate of chromosome end-to-end fusions (32,33) suggesting that the disruption of the telomere lengthening might be the cause of genome instability and disorder progression observed in lymphocytes from A-T patients (12). However to our knowledge, no study has examined lymphocyte TL in HetAT subjects. The goal of the present study was to assess whether being a heterozygous carrier of an ATM mutation influences TL, and to examine how ATM mutation status and TL interplay to modify cancer risk in HetAT subjects.
Material and methods
Study participants
Study participants were all related to a child affected with ataxia-telangiectasia (A-T) and were enrolled through one of the three following French studies: the Prospective Cohort of Women Related to an A-T child (CoF-AT) (Lecarpentier J and Andrieu N, personal communication), the Retrospective Study on A-T families (Retro-AT) (34), or the A-T families collection assembled by the Genetic Department of Institut Curie, which is the national reference center for genetic diagnosis of A-T.
Retro-AT was carried out between 1994 and 1997 to assess cancer risk in A-T families living in France (3,34,35). Thirty-four A-T families were identified during this period and 27 of them were subsequently included in the CoF-AT prospective study. CoF-AT is an ongoing national prospective cohort, which was initiated in 2003 to investigate environmental and genetic risk factors for breast cancer (BC) in HetAT and non-HetAT women. All women aged 18 and over are eligible to participate in the study. At inclusion, participants provide a blood sample to determine whether they carry the ATM mutations identified in the affected child in that family. Women are followed up for 10 years after inclusion, and re-contacted every 2 years to provide information on medical data, environmental and lifestyle factors through a detailed epidemiological questionnaire. Three hundred and forty six women (177 HetAT and 169 non-HetAT) belonging to 96 A-T families from CoF-AT were included in the present study. Among these, three CoF-AT families were known to segregate a mutation in the BRCA1 or BRCA2 gene in addition to the familial ATM mutation. Carriers of a mutation in one of these two major BC predisposing genes were excluded from the subsequent analyses (three HetAT and one non-HetAT women). Sixty-seven additional subjects related to an A-T child and who attended the family genetics clinics for molecular diagnosis were also included. Written informed consent was obtained from all participants. The Retro-AT and CoF-AT study protocols were approved by the appropriate ethics committee (CCP Ile-de-France III) and by the French data protection authority (CNIL).
In total, the study was conducted on 458 individuals (284 HetAT and 174 non-HetAT) with available blood DNA sample. Among those, 62 subjects (48 HetAT and 14 non-HetAT) had cancer, and 31 of them were diagnosed after blood sample was collected (incident cases: 25 HetAT and 6 non-HetAT) (Table 1).
Table 1.
Distribution of gender, age, cancer cases and cancer type in the 103 A-T families, by ATM mutation status and sample series
| CoF-AT (N = 346)* | Retro-AT (N = 45) | IC Collection (N = 67) | ||||
|---|---|---|---|---|---|---|
| Non-HetAT (N = 169) | HetAT (N = 177) | Non-HetAT (N = 1) | HetAT (N = 44) | Non-HetAT (N = 4) | HetAT (N = 63) | |
| Gender | ||||||
| Female | 169a | 177a,b | 1 | 15 | 1 | 11 |
| Male | 0 | 0 | 0 | 29 | 3 | 52 |
| Mean age at blood draw (se) | 42.3 (1.2) | 42.7 (1.0) | 35.3 (N/A) | 42.5 (1.5) | 46.1 (8.6) | 43.7 (1.6) |
| Age at blood draw (years) | ||||||
| ≤50 | 120 | 126 | 1 | 37 | 2 | 48 |
| >50 | 49 | 51 | 0 | 7 | 2 | 15 |
| Cancer status | ||||||
| Unaffected females | 155 | 149 b | 1 | 12 | 1 | 7 |
| Affected females | 14 (6) a | 28 (10) a, b | 0 | 3 (2) | 0 | 4 (3) |
| Unaffected males | 0 | 0 | 0 | 22 | 3 | 46 |
| Affected males | 0 | 0 | 0 | 7 (5) | 0 | 6 (5) |
| Cancer Type | ||||||
| Breast | 8 (3)a | 18 (7)a,b | — | 2 (1) | — | 4 (3) |
| Cervix | — | 2 (1) | — | — | — | — |
| Colon-rectum | — | 1 (0) | — | — | — | — |
| Genital organs | — | 1 (0) | — | — | — | — |
| Kidney | 2 (2) | 1 (0) | — | — | — | — |
| Leukemia | — | — | — | — | — | 1 (1) |
| Lung | 1 (0) | — | — | 1 (0) | — | 1 (0) |
| Melanoma | — | 1 (0) | — | — | — | 1 (1) |
| Non-Hodgkin lymphoma | 1 (0) | 1 (0) | — | 1 (1) | — | — |
| Ovarian | 1 (0) | — | — | — | — | — |
| Pancreas | — | 1 (1) | — | — | — | 1 (1) |
| Prostate | — | — | — | 5 (4) | — | 2 (2) |
| Stomach | — | — | — | 1 (1) | — | — |
| Thyroid | — | 2 (1) | — | — | — | — |
| Uterus | 1 (1) | — | — | — | — | — |
The number of cancer cases eligible for the telomere length analysis is indicated in brackets and in italics.
se: Standard error.
aOne woman from this group carried a BRCA1 mutation and was excluded in the subsequent analyses.
bOne woman from this group carried a BRCA2 mutation and was excluded in the subsequent analyses.
DNA preparation and characterization of ATM mutation status
Genomic DNA was extracted from whole blood samples collected from EDTA with the Quickgene 610-L automated system (Fujifilm) according to the manufacturer’s instructions or, for less than 10% of the study participants, using other silica-based DNA purification kits (Macherey Nagel or QIAGEN kits) or standard phenol-chloroform procedure. For all participants the presence of the familial ATM mutation identified in the A-T child was determined according to their parental branch by targeted sequencing on an ABI3500xL DNA analyzer (Applied Biosystems).
Selection of subjects for SNP genotyping and telomere length measurement
While all 458 participants were included in the SNP analysis, TL analysis was restricted to the 427 participants (261 HetAT and 166 non-HetAT) who were unaffected with cancer when biological material was collected (Table 1).
Telomere length measurement
The relative telomere length (RTL) was ascertained by quantitative PCR on the LightCycler480 (Roche) using the protocol developed by Cawthon et al. (36). The RTL was determined as the ratio of detected fluorescence from the amplification of telomere repeat units (TEL) relative to that of a single-copy reference sequence from the housekeeping gene ALB encoding Albumin (REF).
Telomere repeat units and the ALB amplicon were amplified in separate wells on the same 384-well plate to avoid the inter-plate variability. A five-point standard curve was established for each plate with one reference genomic DNA sample diluted in 3-fold series (150 ng, 50 ng, 16.7 ng, 5.55 ng and 1.85 ng per well). The standard curves were then used to determine the TEL and REF amplified DNA amount and corresponding dilution factors, which were used to calculate the TEL/REF ratio representing the RTL. Telomeres and ALB amplifications were performed in duplicate for each individual’s DNA sample. Two blind replicate samples were interspersed with the samples on each plate to assess the inter-plate variability. Samples with failed amplification of telomere repeat units and/or ALB were not repeated. The PCR efficiency for both telomeres and ALB amplifications was greater than 97%.
SNP genotyping
The SNPs rs2736100 (TERT) (29), rs1317082 (TERC) (30), rs2738783 (RTEL1) (30), rs10165485 (ACYP2) (30), rs2320615 (NAF1) (30), rs9257445 (ZNF311) (30) and rs6060727 (BCL2L1) (30) which have been recurrently associated with TL in the literature were selected. These seven SNPs were genotyped for all participants using a standard Taqman protocol and the fluorescence was read on an ABI PRISM7900HT instrument (Applied Biosystems). Taqman probes targeting alleles of the seven SNPs are available from the ThermoFisher Scientific website (http://www.thermofisher.com/order/genome-database/). Ten percent of the samples were randomly duplicated between 384-well plates to validate the results reproducibility. The proportion of successfully genotyped DNA samples was above 95% for all SNPs except for rs2736100 for which the call rate was 90%.
Statistical analysis
Variance analyses (ANOVA) were performed to estimate the intra- and inter-plates assay variability. Linear regressions were used to evaluate the correlation between RTL and age at blood sampling for HetAT and non-HetAT subjects, and a likelihood ratio test was performed to determine if the regression coefficients were significantly different between the two groups.
Logistic regressions were used to obtain the odds ratios (OR) and 95% confidence intervals (CIs) for the association between cancer risk and RTL, as well as for the association between cancer risk and ATM mutation status.
In the SNP analysis, the deviation of the genotypes proportion from Hardy–Weinberg Equilibrium (HWE) was assessed in the unaffected subjects using chi-square test with one degree of freedom and no tested SNPs showed significant HWE deviation with P < 0.05. We used unconditional logistic regression models to estimate ORs and the trend p-value for association between cancer risk and each TL-related SNP, using codominant coding for genotypes (0,1,2) with the homozygote of the common allele as the reference group. The associations between TL and SNPs were assessed using a linear regression with dominant coding for genotypes (no effect allele = 0, at least one effect allele = 1) and grouping subjects with medium and short telomeres together.
All analyses were adjusted on gender (when relevant), age at last interview or age at diagnosis, whichever came first. All statistical tests were two-sided and data was analyzed using STATA, version 14.1 (StataCorp, College Station, TX).
Results
ATM mutation status and cancer risk
Three CoF-AT families were known to segregate a BRCA1 or BRCA2 mutation in addition to the familial ATM mutation. After excluding BRCA1 or BRCA2 mutation carriers, 281 HetAT and 173 non-HetAT participants were included in the genetic study. We confirmed in this study set that carriers of a mutated copy of ATM had a higher overall cancer risk (OR = 2.3, 95% CI = 1.2–4.4, P = 0.01). In particular, women had a 3-fold increase in risk of BC, the most frequent cancer in our study sample (OR = 2.9, 95% CI = 1.2–7.1, P = 0.02) (Table 2). Even though most of the subjects were enrolled prospectively in the study, 22 of the 59 cancer cases were prevalent cases. As a result, the observed association could reflect a measure of susceptibility to both cancer onset and survival. We therefore also performed the analysis on incident cases only, and found similar results (OR = 3.3, 95% CI = 1.3–8.1, P = 0.01 and OR = 3.7, 95% CI = 1.0–13.5, P = 0.04, respectively) (Table 2).
Table 2.
Association between ATM mutation status and cancer risk, for all cancer sites combined and for breast cancer (BC) in females
| All cancer cases | Incident cancer cases | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Outcome | Subjects | Unaffected (N) |
Affected (N) |
ORa | (95% CI) | P | Affected (N) |
ORa | (95% CI) | P |
| Overall cancer risk (all participants) | Non-HetAT | 160 | 13 | 1 | 6 | 1 | ||||
| HetAT | 235 | 46 | 2.3 | (1.2, 4.4) | 0.01 | 31 | 3.3 | (1.3, 8.1) | 0.01 | |
| Overall cancer risk (females only) | Non-HetAT | 157 | 13 | 1 | 6 | 1 | ||||
| HetAT | 167 | 33 | 2.4 | (1.2, 4.7) | 0.01 | 18 | 2.8 | (1.1, 7.4) | 0.03 | |
| BC risk (females only) | Non-HetAT | 157 | 7 | 1 | 3 | 1 | ||||
| HetAT | 167 | 22 | 2.9 | (1.2, 7.1) | 0.02 | 12 | 3.7 | (1.0, 13.5) | 0.04 | |
aOR adjusted for age at last interview/diagnosis, and for gender when relevant.
TL and ATM mutation status
No intra-plate variability was observed within telomere assay duplicates and within ALB assay duplicates (P = 0.07 and P = 0.05, respectively), nor was inter-plate variability observed for the telomere assays or the ALB assays (P = 0.24 and P = 0.20, respectively) using an ANOVA test. Thus, subsequent analyses were performed without adjustment for plate location.
RTL was measured on all unaffected participants and on cases for whom lymphocyte DNA was obtained prior to the diagnosis of cancer (incident cases), as both cancer and treatment may affect telomere elongation. As expected, TL was negatively correlated with age at sampling in the control group of unaffected non-HetAT subjects (R2 = −0.0048, P = 0.003). This tendency was also observed in unaffected HetAT subjects, although the rate of decline was not as steep as for their non-HetAT counterparts (R2 = −0.0015, P = 0.31) (Figure 1). The difference between the slopes for TL with respect to age for HetAT and non-HetAT participants was significant when females and males were included in the analysis (P = 0.0008). Because it has been shown that females have longer telomeres than males (37,38), we next restricted the analysis to females. In this group TL showed a shortening with age but with a less-steep decline in HetAT than in non-HetAT females. However, no statistically significant difference was seen between the two groups (P = 0.24), which could be due to a power lowered by a smaller number of subjects (Figure 1). We could not conduct a similar comparison between unaffected HetAT and non-HetAT males because data for non-HetAT were too scarce. However, as observed in HetAT females, the shortening of the TL with age was not significant in unaffected HetAT males (data not shown). There were too few observations on HetAT and non-HetAT subjects affected with cancer for meaningful analyses to be carried out.
Figure 1.

Correlations between relative telomere length and age at blood draw in unaffected HetAT and non-HetAT subjects. N, number of analyzed subjects. R2 and P values (P) are for the correlation from the linear regression.
We also measured the RTL in a group of 73 A-T children belonging to the CoF-AT and Retro-AT families. As expected we found that they had longer TL than their HetAT and non-HetAT relatives due to their younger age (mean age = 10.6, standard error = 0.92) (data not shown). Therefore making further TL comparison between A-T children and the HetAT studied group was inadequate.
TL status and cancer risk
We first defined tertiles of RTL distribution among all unaffected individuals (Long > 1.2886; Medium: 1.1009–1.2886; Short < 1.1009) and investigated the association between RTL and cancer occurrence using the longest telomere tertile as reference group. No significant association was found with overall cancer risk or with BC risk in women (Table 3). No interaction was evidenced between RTL and ATM mutation status for either overall cancer risk or BC risk (Table 3).
Table 3.
Association between relative telomere length (RTL) and cancer risk, for all cancer sites, and for breast cancer in females
| RTL tertile | Unaffected | Affected | OR | (95% CI) | P trend | ORc | (95% CI) | P trend c | P interaction | |
|---|---|---|---|---|---|---|---|---|---|---|
| (all cancers) | ||||||||||
| All participants | N = 381 |
N = 31 |
||||||||
| 1st (long) | 127 | 7 | 1a | 1d | ||||||
| 2nd (medium) | 127 | 13 | 1.8 | (0.7, 4.7) | 1.9 | (0.7, 4.9) | 0.68 | |||
| 3rd (short) | 127 | 11 | 1.5 | (0.5, 3.9) | 0.48 | 1.5 | (0.5, 3.9) | 0.49 | 0.72 | |
| (all cancers) | ||||||||||
| Females only | N = 312 |
N = 21 |
||||||||
| 1st (long) | 104 | 6 | 1b | 1e | ||||||
| 2nd (medium) | 104 | 9 | 1.5 | (0.5, 4.4) | 1.68 | (0.6, 5.0) | 0.75 | |||
| 3rd (short) | 104 | 6 | 0.9 | (0.3, 3.0) | 0.91 | 0.96 | (0.3, 3.1) | 0.94 | 0.57 | |
| (breast cancer) | ||||||||||
| Females only | N = 312 |
N = 14 |
||||||||
| 1st (long) | 104 | 4 | 1b | 1e | ||||||
| 2nd (medium) | 104 | 6 | 1.5 | (0.4, 5.5) | 1.6 | (0.4, 6.0) | 0.93 | |||
| 3rd (short) | 104 | 4 | 1.0 | (0.2, 4.1) | 0.99 | 1.0 | (0.2, 4.0) | 0.97 | 0.99 |
aAdjusted on gender and age at blood draw.
bAdjusted on age at blood draw.
c ATM status was included in the model.
dAdjusted on gender, age at blood draw and ATM status.
eAdjusted on age at blood draw and ATM status.
We next classified the subjects into four groups according to their ATM mutation status and RTL. Due to the limited number of observations in some groups, subjects with medium or short telomeres were pooled together; non-carriers with long telomeres were used as the reference group. Although results did not reach statistical significance, we found that HetAT subjects with shorter telomeres were more prone to develop cancer at any site than the other groups (Ptrend = 0.03). The same trend was seen when restricting the analysis to BC risk (Ptrend = 0.05) (Table 4).
Table 4.
Cancer risk according to ATM mutation status and RTL
| Group | Unaffected | Affected | OR | (95% CI) | P | P trend | |
|---|---|---|---|---|---|---|---|
| (all cancers) | |||||||
| All participants | N = 381 |
N = 31 |
|||||
| Non-HetAT + Long RTL | 53 (13.9%) | 1 (3.2%) | 1a | ||||
| Non-HetAT + Medium/Short RTL | 103 (27.0%) | 5 (16.1%) | 2.5 | (0.3, 22.3) | 0.40 | ||
| HetAT + Long RTL | 74 (19.4%) | 6 (19.4%) | 3.8 | (0.4, 33.3) | 0.22 | ||
| HetAT + Medium/Short RTL | 151 (39.7%) | 19 (61.3%) | 5.8 | (0.7, 45.5) | 0.10 | 0.03a | |
| (all cancers) | |||||||
| Females only | N = 312 |
N = 21 |
|||||
| Non-HetAT +Long RTL | 53 (17.0%) | 1 (4.8%) | 1b | ||||
| Non-HetAT + Medium/Short RTL | 100 (32.0%) | 5 (23.8%) | 2.3 | (0.3, 20.8) | 0.44 | ||
| HetAT + Long RTL | 56 (18.0%) | 5 (23.8%) | 4.2 | (0.5, 37.7) | 0.20 | ||
| HetAT + Medium/Short RTL | 103 (33.0%) | 10 (47.6%) | 5.0 | (0.6, 39.9) | 0.13 | 0.06b | |
| (breast cancer) | |||||||
| Females only | N = 312 |
N = 14 |
|||||
| Non-HetAT + Long RTL | 53 (17.0%) | 1 (7.2%) | 1b | ||||
| Non-HetAT + Medium/Short RTL | 100 (32.0%) | 2 (14.3%) | 1.0 | (0.1, 11.8) | 0.97 | ||
| HetAT + Long RTL | 56 (18.0%) | 3 (21.4%) | 2.8 | (0.3, 27.7) | 0.39 | ||
| HetAT + Medium/Short RTL | 103 (33.0%) | 8 (57.1%) | 4.1 | (0.5, 33.5) | 0.19 | 0.05b |
aAdjusted on age at last interview/diagnosis and gender adjustment.
bAdjusted on age at last interview/diagnosis.
Finally, we examined whether telomere-related SNPs were associated (i) with RTL in order to evaluate how those SNPs could serve as surrogate of RTL measurement in our population sample, and (ii) with cancer risk.
Since age-related shortening of TL was different between HetAT and non-HetAT subjects, we tested the association between SNPs and RTL in the two groups separately. Surprisingly, none of the tested SNPs showed association with RTL in the non-HetAT group (all the 162 non-HetAT subjects are females but three), while suggestive association between rs9257445 and RTL was observed in the HetAT group where both 162 females and 76 males were included in the analysis (P = 0.03) (Supplementary Table 1). In addition to rs9257445, two other SNPs, rs2320615 and rs10165485 showed significant association with RTL when analyses were conducted on the HetAT male subgroup (P = 0.02, P = 0.04 and P = 0.02, respectively).
Regarding the contribution of the seven selected SNPs and cancer risk, only rs6060627 in BCL2L1 showed suggestive association with overall cancer risk, with a significant trend when the analysis was focused on females (Ptrend = 0.03) (Table 5). No association between the seven SNPs and cancer risk was found when HetAT and non-HetAT subjects were analyzed separately (data not shown).
Table 5.
Associations between telomere-related SNPs and cancer risk
| SNP | Genotype | Unaffected | Affected |
OR | (95%CI) | P | P trend c | |
|---|---|---|---|---|---|---|---|---|
| (all cancers) | ||||||||
| All participants | rs2736100 (TERT) | CC | 99 | 13 | 1a | |||
| CA | 155 | 24 | 1.18 | (0.57, 2.46) | 0.66 | |||
| AA | 91 | 10 | 0.9 | (0.37, 2.17) | 0.81 | 0.84 | ||
| rs1317082 (TERC) | AA | 214 | 30 | 1a | ||||
| AG | 130 | 18 | 0.87 | (0.45, 1.66) | 0.67 | |||
| GG | 18 | 6 | 2.39 | (0.88, 6.53) | 0.09 | 0.38 | ||
| rs2738783 (RTEL1) | TT | 255 | 43 | 1a | ||||
| TG | 102 | 11 | 0.67 | (0.33, 1.35) | 0.26 | |||
| GG | 12 | 2 | 1.03 | (0.22, 4.79) | 0.97 | 0.41 | ||
| rs10165485 (ACYP2) | TT | 308 | 42 | 1a | ||||
| TC | 52 | 12 | 1.78 | (0.87, 3.62) | 0.11 | |||
| CC | 7 | 1 | 1.09 | (0.13, 9.13) | 0.93 | 0.20 | ||
| rs2320615 (NAF1) | GG | 203 | 33 | 1a | ||||
| GA | 144 | 20 | 0.91 | (0.50, 1.66) | 0.75 | |||
| AA | 24 | 3 | 0.82 | (0.23, 2.89) | 0.76 | 0.68 | ||
| rs9257445 (ZNF311) | GG | 206 | 36 | 1a | ||||
| GC | 141 | 13 | 0.48 | (0.24, 0.95) | 0.035 | |||
| CC | 22 | 7 | 1.58 | (0.61, 4.13) | 0.35 | 0.54 | ||
| rs6060627 (BCL2L1) | CC | 146 | 29 | 1a | ||||
| CT | 173 | 22 | 0.61 | (0.33, 1.12) | 0.11 | |||
| TT | 52 | 5 | 0.49 | (0.18, 1.35) | 0.17 | 0.07 | ||
| Females only | (all cancers) | |||||||
| rs2736100 (TERT) | CC | 82 | 7 | 1b | ||||
| CA | 127 | 21 | 1.93 | (0.79, 4.76) | 0.15 | |||
| AA | 72 | 9 | 1.47 | (0.52, 4.16) | 0.47 | 0.48 | ||
| rs1317082 (TERC) | AA | 175 | 24 | 1b | ||||
| AG | 104 | 13 | 0.91 | (0.44, 1.87) | 0.80 | |||
| GG | 17 | 5 | 2.12 | (0.71, 6.27) | 0.18 | 0.43 | ||
| rs2738783 (RTEL1) | TT | 210 | 33 | 1b | ||||
| TG | 85 | 9 | 0.67 | (0.30, 1.45) | 0.31 | |||
| GG | 7 | 2 | 1.68 | (0.33, 8.59) | 0.54 | 0.65 | ||
| rs10165485 (ACYP2) | TT | 250 | 32 | 1b | ||||
| TC | 44 | 11 | 1.93 | (0.91, 4.12) | 0.09 | |||
| CC | 6 | 0 | N/A | N/A | N/A | N/A | ||
| rs2320615 (NAF1) | GG | 171 | 24 | 1b | ||||
| GA | 110 | 17 | 1.10 | (0.57, 2.15) | 0.77 | |||
| AA | 22 | 3 | 1.0 | (0.28, 3.61) | 1.00 | 0.86 | ||
| rs9257445 (ZNF311) | GG | 170 | 28 | 1b | ||||
| GC | 115 | 10 | 0.53 | (0.25, 1.13) | 0.10 | |||
| CC | 16 | 6 | 2.27 | (0.82, 6.29) | 0.12 | 0.91 | ||
| rs6060627 (BCL2L1) | CC | 115 | 24 | 1b | ||||
| CT | 144 | 17 | 0.56 | (0.29, 1.10) | 0.09 | |||
| TT | 44 | 3 | 0.33 | (0.09, 1.14) | 0.08 | 0.03 | ||
| Female only | (breast cancer) | |||||||
| rs2736100 (TERT) | CC | 82 | 4 | 1b | ||||
| CA | 127 | 11 | 1.77 | (0.55, 5.76) | 0.34 | |||
| AA | 72 | 6 | 1.71 | (0.46, 6.32) | 0.42 | 0.43 | ||
| rs1317082 (TERC) | AA | 175 | 11 | 1b | ||||
| AG | 104 | 11 | 1.68 | (0.70, 4.01) | 0.24 | |||
| GG | 17 | 4 | 3.74 | (1.07, 13.05) | 0.039 | 0.10 | ||
| rs2738783 (RTEL1) | TT | 210 | 19 | 1b | ||||
| TG | 85 | 8 | 1.04 | (0.44, 2.48) | 0.93 | |||
| GG | 7 | 0 | N/A | N/A | N/A | N/A | ||
| rs10165485 (ACYP2) | TT | 250 | 22 | 1b | ||||
| TC | 44 | 4 | 1.03 | (0.34, 3.18) | 0.96 | |||
| CC | 6 | 0 | N/A | N/A | N/A | N/A | ||
| rs2320615 (NAF1) | GG | 171 | 15 | 1b | ||||
| GA | 110 | 11 | 1.14 | (0.51, 2.57) | 0.75 | |||
| AA | 22 | 1 | 0.52 | (0.07, 4.13) | 0.54 | 0.83 | ||
| rs9257445 (ZNF311) | GG | 170 | 18 | 1b | ||||
| GC | 115 | 6 | 0.49 | (0.19, 1.28) | 0.15 | |||
| CC | 16 | 3 | 1.77 | (0.47, 6.66) | 0.40 | 0.72 | ||
| rs6060627 (BCL2L1) | CC | 115 | 15 | 1b | ||||
| CT | 144 | 10 | 0.53 | (0.23, 1.23) | 0.14 | |||
| TT | 44 | 2 | 0.35 | (0.08, 1.59) | 0.17 | 0.08 |
aAdjusted with age at last interview/diagnosis and gender.
bAdjusted with age at last interview/diagnosis.
c P value of the trend test.
Discussion
Few studies have examined whether TL modified cancer risk in the context of a familial predisposition involving inherited mutations with incomplete penetrance. One study has assessed the association between cutaneous malignant melanoma and TL in melanoma-prone families with and without CDKN2A mutations. Authors found that longer telomeres increased the melanoma risk in non-carrier cases (39). Regarding familial breast cancer, Pooley et al. showed that carriers of a mutation in BRCA1 or BRCA2 have longer telomeres than their non-carrier relatives, but BC risk for carriers was not modified by the TL28.
To our knowledge, this is the first exploratory study investigating TL as a modifier of cancer risk for individuals carrying an ATM mutation. One asset of our study design is the homogeneity of the study sample, given that HetAT and non-HetAT participants were all related to an A-T child. Consequently only ‘true’ pathogenic ATM mutations, i.e. base substitutions, insertions or deletions that generate premature termination codons or splicing abnormalities were considered, thus avoiding variant misclassification. Moreover, most of the study participants were recruited prospectively, with blood samples collected prior to cancer diagnosis for 50% of the cases. Such a design is better able to address the question of whether telomeres become shorter before cancer diagnosis, and are therefore predictive of cancer development. Finally, all DNA samples were processed, stored and analyzed using the same workflow thus eliminating biases when measuring RTL.
Consistent with previous reports, and in particular with the French retrospective study conducted on 34 A-T families (3,34,35), we found in this larger family series that HetAT individuals had a 2- to 3-fold increased overall cancer risk as compared to non-HetAT individuals. Because BC was the most frequent cancer in the study population composed of 81.7% of women, we could only estimate risk for this specific cancer separately. We found that HetAT women had a 3-fold increase risk of developing BC and this was even higher when considering incident cases only, confirming previous findings in A-T families (3,4,35). About 1% of the general population and nearly 3% of hereditary breast and ovarian cancer patients are estimated to carry a deleterious ATM variant (5,6). It is therefore important to better estimate the risk of cancer associated with a mutation in this gene by taking into account other genetic and environmental/lifestyle factors which could modulate this risk, as well as potential genotype-phenotype correlations.
Given the functional link between ATM and telomere maintenance, we hypothesized that TL and telomere-related SNPs could act as modifiers of cancer risk for HetAT subjects. Interestingly, we observed a sex-dependent effect of some SNPs on RTL in HetAT subjects. Indeed, the β cœfficient for rs10165485, rs2320615 and rs9257445 was 2- to 5-fold higher in males that in females. Such a comparison in non-HetAT subjects was not possible due to the limited number of non-HetAT males in the study population (Supplementary Table 1). This difference in gender was not observed in the general population as reported in Codd et al. 2013 (29). We have no obvious explanation for this sex-dependent effect in ATM carriers, and larger studies are warranted to confirm this finding.
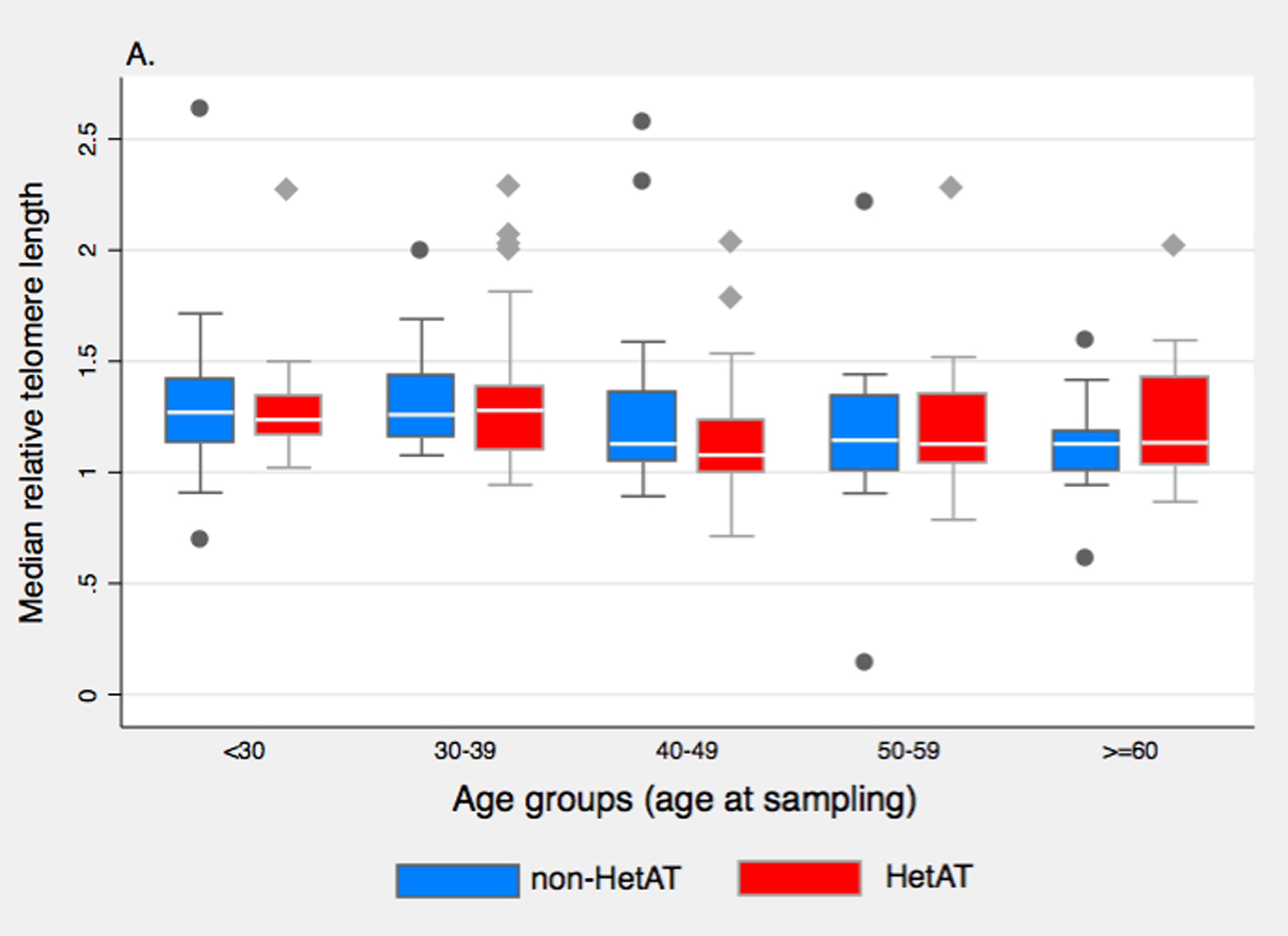
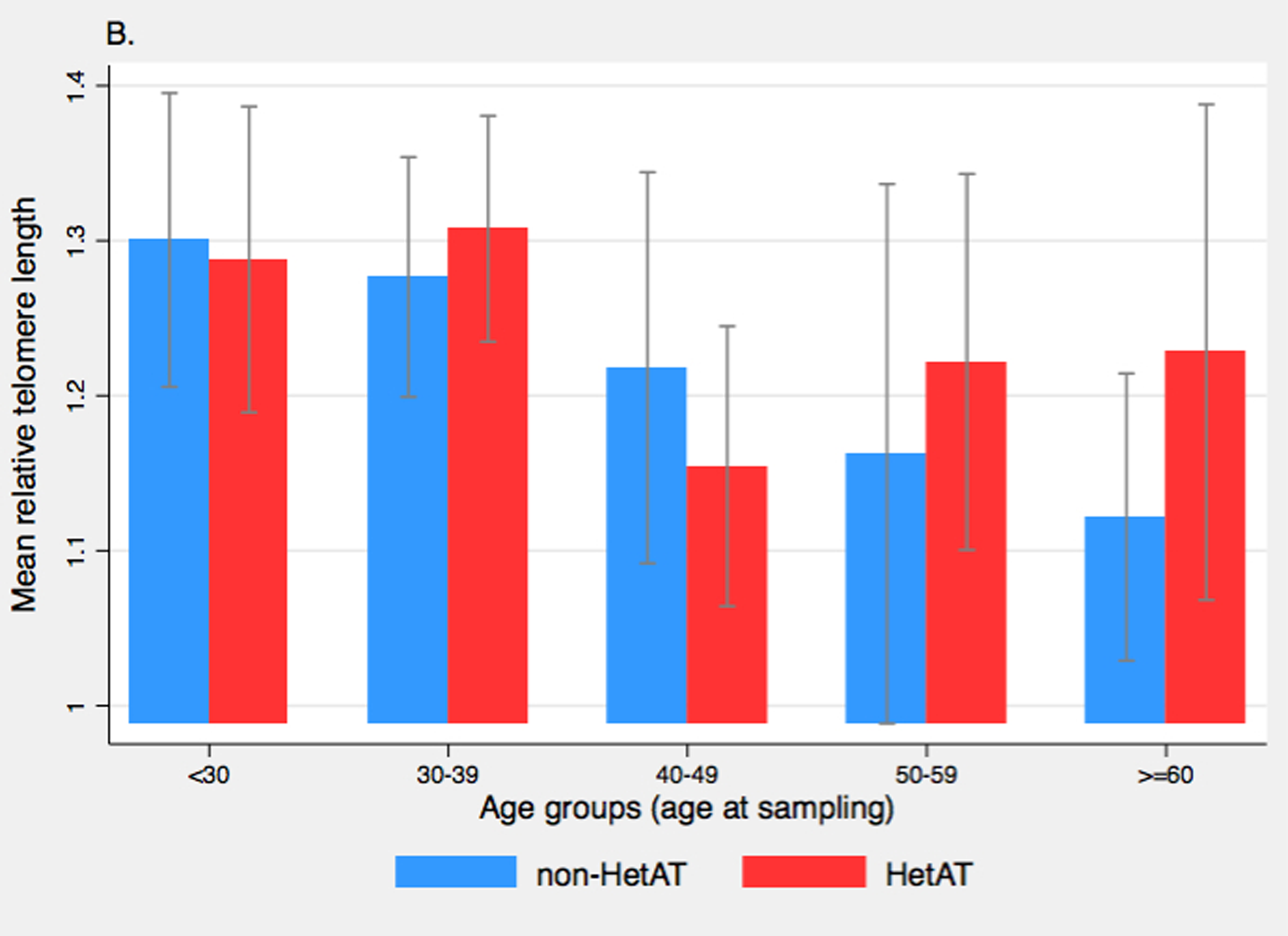
Regarding the possible modifier role of TL on cancer risk, we found that RTL, as measured with the Q-PCR assay, was not a risk factor for cancer susceptibility in our study population. In the SNP analysis, only rs6060627 in BCL2L1 showed association with overall cancer risk although this intronic SNP was not associated with TL. Therefore a mechanism other than telomere maintenance involving BCL2L1 could explain the observed association here. There are several limitations to this study that should be noted. First, the power to detect SNPs with small effect sizes may be low due to the relatively small number of cancer cases included in this study because of the uniqueness of the studied population. Indeed for SNP rs10165485 with a low MAF in controls (10%), the power of our study for detecting an association between this candidate SNP and cancer could reach 80% only for an OR of 2.5 or higher (alpha < 0.05). For SNP rs2536100 with a MAF in controls of 50%, our study had a power of 90% for detecting an association with an OR equals 1.40 (alpha = 0.05). Second, due to the limited number of cases, the main analyses were conducted considering all cancer sites together, which could mask association with some specific cancer sites. Indeed, risk of non-Hodgkin lymphoma (40), lung (18) and prostate (41) cancers have been associated with longer telomeres, whereas melanoma (22), thyroid (42) and ovarian (43) cancers have been associated with shorter telomeres. No significant association between BC risk and RTL was found here when sorting RTL into tertiles; moreover the association between ATM mutation status and cancer was not contingent with RTL (i.e. no statistical interaction was found). However, classifying subjects according to their ATM mutation status and Long vs. Medium/Short telomeres suggested that HetAT individuals with shorter telomeres have an increased overall cancer risk and an increased BC risk as compared to HetAT individuals with Long telomeres. This observation is in agreement with the hypothesis that shorter telomeres increased genomic instability, as observed in A-T children. Indeed, studies on lymphocytes and fibroblasts from A-T children have shown an accelerated shortening of telomeres, a deregulation probably involved of the chromosome end-to-end fusion and genomic instability characteristic of the disorder (44,45). The accelerated telomere shortening ascertained for A-T children and HetAT cancer cases could be explained by the involvement of ATM in two mechanisms of telomere lengthening regulation. The first pathway involves the phosphorylation of TFR1 by ATM which is required to release TRF1 from telomeric DNA, ensuring the formation of replication forks that allow telomerase recruitment (12,46). In the second TRF1-independent pathway, ATM facilitates the telomerase assembly by phosphorylation of currently unknown proteins (9). Thus, in HetAT individuals a reduction in the quantity of functional ATM protein might cause a lower efficiency to induce replication fork formation and telomerase assembly, leading to an accelerated telomere shortening and an increased genomic instability. Despite these observations, no statistically significant interaction between ATM status and telomere length was found in cancer risk but observations are too few to exclude this possibility. The finding that unaffected ATM mutation carriers have longer TL than their non-carrier relatives was unexpected. In order to assess the possibility of a different dispersion of RTL measures in HetAT and non-HetAT subjects, we further explored median and mean RTL in the two groups according to age (Supplementary Figure 1). No difference in median RTL was seen between non-HetAT and HetAT subjects in each age group but the decline in mean RTL according to age was more marked for non-HetAT subjects than for HetAT subjects, which is in accordance with our observation that telomere shortening is slower in HetAT subjects (Figure 1).
One possible explanation for HetAT subjects having longer TL could be that the quantitative PCR assay used also amplifies extra-chromosomal telomeric DNA, thus explaining the surplus of telomere signals. Linear or circular extra-chromosomal telomeric DNA is characteristic of alternative lengthening of telomeres (ALT) a mechanism independent of the telomerase (47). The formation of such elements was observed in fibroblasts from A-T children (48). Hence, the ALT mechanism might participate in telomere lengthening in cells from HetAT subjects, as previously shown in BRCA2−/+ cell lines (49) and could open new lines of research into the function of ATM. This deregulation might be due to some environmental exposures like radiation or to other unidentified genetic factors involved in TL maintenance.
Taken together, our findings suggest that TL may be modulated by the carriage of an ATM mutation but it does not seem itself directly related to increased cancer risk in carriers. TL measurement alone is not a good marker for predicting cancer risk in A-T families or for predicting ATM mutation status of an individual.
Supplementary Material
Supplementary data are available at Carcinogenesis online.
Funding
This work was supported by Inserm and Ministère de la Recherche (01P0751 - 01P0752 - 01P0753 - 01P0754 - 01P0755), Electricité de France (conseil scientifique de Radioprotection d’EDF, grant EP 2002–03, EP 2004-03 RB 2016–22), Fondation de France (grants 2001009761 and 2005011201), La Ligue (grants PRE04/NA, PRE07/NA and PRE2015 LNCC/NA), La Ligue Comité du Maine et Loire, MGEN Union, ITMO Santé Publique d’AVIESAN (grant AAP12-COH-110), Institut Curie (CEST NC2013-015), Institut National du Cancer (grant INCa-9578) and the European Network of Excellence DoReMi (grant 249689). A.L.R. was the recipient of a fellowship from the Fondation ARC pour la recherche sur le cancer.
Authors contribution
Conceived and designed the experiments: A.L.R., J.H., N.A., F.L. Performed the experiments: A.L.R., N.M., C.D.E. Analyzed the data: A.L.R., N.A., F.L. Contributed reagents/materials/analysis tools: J.L., C.D.E., A.L., M.G.D. Coordinated the CoF-AT and Retro-AT studies: D.S.L., N.A., J.H. Included CoF-AT participants, collected data: E.C., D.L.G., M.L. Invited A-T family members: G.L., D.L., L.G., C.A., M.F., L.F., B.G.-D., A.L., J.-P.F., K.D., J.-O.B., M.L., B.B., P.V.†, N.J., H.Z., P.B., A.C., I.C., D.S.L. Wrote the paper: A.L.R., F.L. All authors read and approved the final version of the manuscript.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We are most grateful to the families who so willingly participated into the study. We acknowledge José-Arturo Londono Vallejo for helpful discussions regarding the set-up of experiments for measuring telomere length. We thank all the CoF-AT collaborating cancer clinics: Institut Curie, Paris: Dominique Stoppa-Lyonnet, Bruno Buecher, Antoine De Pauw and Sophie Lejeune-Dumoulin; Hôpital Arnaud de Villeneuve, Montpellier: Isabelle Coupier; ICM Val d’Aurel, Montpellier: Isabelle Coupier; CHU de Nîmes: Audrey Combès; Centre François Baclesse, Caen: Pascaline Berthet; Hôpital Saint-Joseph, Marseille: Hélène Zattara; Clinique Universitaire Saint-Luc, Bruxelles: Nicolas Janin and Karin Dahan; Centre Oscar Lambret, Lille: Philippe Vennin and Claude Adenis; Institut Bergonié, Bordeaux: Michel Longy; Centre Jean Perrin, Clermont-Ferrand: Jacques-Olivier Bay; Centre Jean-Paul Strauss, Strasbourg: Jean-Pierre Fricker; Centre Catherine de Sienne, Nantes: Alain Lortholary; CHU de Poitiers, Poitiers: Brigitte Gilbert-Dussardier; Hôpital d’Enfants, Dijon: Laurence Faivre and Caroline Jacquot; Centre Antoine Lacassagne, Nice: Marc Frenay; Institut Claudius Regaud, Toulouse: Laurence Gladieff; CHU de Grenoble, Grenoble: Dominique Leroux; CHU de Lyon, Lyon: Gaétan Lesca; Gustave Roussy, Villejuif: Agnès Chompret†; Centre Léon Bérard, Lyon: Christine Lasset; Hotel-Dieu, Chambéry: James Lespinasse; CHI Toulon La Seyne-sur-Mer, Toulon: Xavier Tchiknavorian; Centre Eugène Marquis, Rennes: Catherine Dugast†; CHU du Mans, Le Mans: Dominique Martin-Coignard; CHRU Hôpital Caremeau, Nîmes: Jean Chiesa; Hôpital Universitaire Dupuytren, Limoges: Laurence Venat-Bouvet; Centre René Gauducheau, Nantes: Capucine Delnatte; Hôpital Porte-Madeleine, Orléans: Sonia Nizard; CHU de Nancy, Nancy: Bruno Leheup; Hôpital Sainte-Musse, Toulon: Patrick Collignon; Polyclinique Courlancy, Reims: Liliane Demange†; CHU de Besançon-Hôpital Jean Minjoz, Besançon: Pierre Rohlrich; CHU Vaudois, Lausanne: Florence Fellmann; CHU d’Angers, Angers: Isabelle Pellier; Hôpitaux de Rouen, Rouen: Julie Tinat. We wish to thank more specifically our late colleague Josué Feingold who has been instrumental in the set-up of the CoF-AT study. †Deceased prematurely.
Conflict of Interest Statement: None declared.
Abbreviations
- ALB
Albumin
- ATM
Ataxia-Telangiectasia Mutated
- A-T
Ataxia-Telangiectasia
- BC
breast cancer
- REF
Reference
- RTL
relative telomere length
- TEL
telomere repeat units
- TL
telomere length
References
- 1. Swift M., et al. (1976) Malignant neoplasms in the families of patients with ataxia-telangiectasia. Cancer Res., 36, 209–215. [PubMed] [Google Scholar]
- 2. Børresen A.L., et al. (1990) Breast cancer and other cancers in Norwegian families with ataxia-telangiectasia. Genes. Chromosomes Cancer, 2, 339–340. [DOI] [PubMed] [Google Scholar]
- 3. Andrieu N., et al. (2005) Ataxia-telangiectasia genes and breast cancer risk in a French family study. J. Dairy Res., 72 Spec No, 73–80. [DOI] [PubMed] [Google Scholar]
- 4. Thompson D., et al. (2005) Cancer risks and mortality in heterozygous ATM mutation carriers. J. Natl. Cancer Inst., 97, 813–822. [DOI] [PubMed] [Google Scholar]
- 5. Renwick A., et al. ; Breast Cancer Susceptibility Collaboration (UK). (2006) ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet., 38, 873–875. [DOI] [PubMed] [Google Scholar]
- 6. Tavtigian S.V., et al. ; Australian Cancer Study; Breast Cancer Family Registries (BCFR); Kathleen Cuningham Foundation Consortium for Research into Familial Aspects of Breast Cancer (kConFab). (2009) Rare, evolutionarily unlikely missense substitutions in ATM confer increased risk of breast cancer. Am. J. Hum. Genet., 85, 427–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blackburn E.H. (1991) Structure and function of telomeres. Nature, 350, 569–573. [DOI] [PubMed] [Google Scholar]
- 8. Cervantes R.B., et al. (2002) Mechanisms of chromosome-end protection. Curr. Opin. Cell Biol., 14, 351–356. [DOI] [PubMed] [Google Scholar]
- 9. Tong A.S., et al. (2015) ATM and ATR Signaling Regulate the Recruitment of Human Telomerase to Telomeres. Cell Rep., 13, 1633–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Verdun R.E., et al. (2006) The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell, 127, 709–720. [DOI] [PubMed] [Google Scholar]
- 11. McKerlie M., et al. (2012) ATM regulates proteasome-dependent subnuclear localization of TRF1, which is important for telomere maintenance. Nucleic Acids Res., 40, 3975–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu Y., et al. (2007) MRE11-RAD50-NBS1 and ATM function as co-mediators of TRF1 in telomere length control. Nat. Struct. Mol. Biol., 14, 832–840. [DOI] [PubMed] [Google Scholar]
- 13. Sfeir A., et al. (2012) Removal of shelterin reveals the telomere end-protection problem. Science, 336, 593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harley C.B., et al. (1990) Telomeres shorten during ageing of human fibroblasts. Nature, 345, 458–460. [DOI] [PubMed] [Google Scholar]
- 15. Lindsey J., et al. (1991) In vivo loss of telomeric repeats with age in humans. Mutat. Res., 256, 45–48. [DOI] [PubMed] [Google Scholar]
- 16. DePinho R.A. (2000) The age of cancer. Nature, 408, 248–254. [DOI] [PubMed] [Google Scholar]
- 17. Hou L., et al. (2009) Telomere length in peripheral leukocyte DNA and gastric cancer risk. Cancer Epidemiol. Biomarkers Prev., 18, 3103–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rode L., et al. (2013) Short telomere length, lung function and chronic obstructive pulmonary disease in 46,396 individuals. Thorax, 68, 429–435. [DOI] [PubMed] [Google Scholar]
- 19. Broberg K., et al. (2005) Constitutional short telomeres are strong genetic susceptibility markers for bladder cancer. Carcinogenesis, 26, 1263–1271. [DOI] [PubMed] [Google Scholar]
- 20. Risques R.A., et al. (2007) Leukocyte telomere length predicts cancer risk in Barrett’s esophagus. Cancer Epidemiol. Biomarkers Prev., 16, 2649–2655. [DOI] [PubMed] [Google Scholar]
- 21. Lan Q., et al. (2009) A prospective study of telomere length measured by monochrome multiplex quantitative PCR and risk of non-Hodgkin lymphoma. Clin. Cancer Res., 15, 7429–7433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anic G.M., et al. (2013) Telomere length and risk of melanoma, squamous cell carcinoma, and basal cell carcinoma. Cancer Epidemiol., 37, 434–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shen J., et al. (2009) Telomere length, oxidative damage, antioxidants and breast cancer risk. Int. J. Cancer, 124, 1637–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qu S., et al. (2013) Association of leukocyte telomere length with breast cancer risk: nested case-control findings from the Shanghai Women’s Health Study. Am. J. Epidemiol., 177, 617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gramatges M.M., et al. (2010) Longer relative telomere length in blood from women with sporadic and familial breast cancer compared with healthy controls. Cancer Epidemiol. Biomarkers Prev., 19, 605–613. [DOI] [PubMed] [Google Scholar]
- 26. Pellatt A.J., et al. (2013) Telomere length, telomere-related genes, and breast cancer risk: the breast cancer health disparities study. Genes. Chromosomes Cancer, 52, 595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim S., et al. (2011) Telomere length in peripheral blood and breast cancer risk in a prospective case-cohort analysis: results from the Sister Study. Cancer Causes Control, 22, 1061–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pooley K.A., et al. ; EMBRACE. (2014) Lymphocyte telomere length is long in BRCA1 and BRCA2 mutation carriers regardless of cancer-affected status. Cancer Epidemiol. Biomarkers Prev., 23, 1018–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Codd V., et al. ; CARDIoGRAM consortium. (2013) Identification of seven loci affecting mean telomere length and their association with disease. Nat. Genet., 45, 422–427, 427e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pooley K.A., et al. (2013) A genome-wide association scan (GWAS) for mean telomere length within the COGS project: identified loci show little association with hormone-related cancer risk. Hum. Mol. Genet., 22, 5056–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bojesen S.E., et al. ; Australian Cancer Study; Australian Ovarian Cancer Study; Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab); Gene Environment Interaction and Breast Cancer (GENICA); Swedish Breast Cancer Study (SWE-BRCA); Hereditary Breast and Ovarian Cancer Research Group Netherlands (HEBON); Epidemiological study of BRCA1 & BRCA2 Mutation Carriers (EMBRACE); Genetic Modifiers of Cancer Risk in BRCA1/2 Mutation Carriers (GEMO). (2013) Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat. Genet., 45, 371–384, 384e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kojis T.L., et al. (1991) The cytogenetics of ataxia telangiectasia. Cancer Genet. Cytogenet., 56, 143–156. [DOI] [PubMed] [Google Scholar]
- 33. Pandita T.K., et al. (1995) Chromosome end associations, telomeres and telomerase activity in ataxia telangiectasia cells. Cytogenet. Cell Genet., 71, 86–93. [DOI] [PubMed] [Google Scholar]
- 34. Janin N., et al. (1999) Breast cancer risk in ataxia telangiectasia (AT) heterozygotes: haplotype study in French AT families. Br. J. Cancer, 80, 1042–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cavaciuti E., et al. (2005) Cancer risk according to type and location of ATM mutation in ataxia-telangiectasia families. Genes. Chromosomes Cancer, 42, 1–9. [DOI] [PubMed] [Google Scholar]
- 36. Cawthon R.M. (2002) Telomere measurement by quantitative PCR. Nucleic Acids Res., 30, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gardner M., et al. ; Halcyon study team. (2014) Gender and telomere length: systematic review and meta-analysis. Exp. Gerontol., 51, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lapham K., et al. (2015) Automated Assay of Telomere Length Measurement and Informatics for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics, 200, 1061–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burke L.S., et al. (2013) Telomere length and the risk of cutaneous malignant melanoma in melanoma-prone families with and without CDKN2A mutations. PLoS One, 8, e71121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Machiela M.J., et al. (2016) Genetically predicted longer telomere length is associated with increased risk of B-cell lymphoma subtypes. Hum. Mol. Genet., 25, 1663–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Julin B., et al. (2015) Circulating leukocyte telomere length and risk of overall and aggressive prostate cancer. Br. J. Cancer, 112, 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. He M., et al. (2013) Telomere length is shorter in affected members of families with familial nonmedullary thyroid cancer. Thyroid, 23, 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mirabello L., et al. (2010) Leukocyte telomere length in a population-based case-control study of ovarian cancer: a pilot study. Cancer Causes Control, 21, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Metcalfe J.A., et al. (1996) Accelerated telomere shortening in ataxia telangiectasia. Nat. Genet., 13, 350–353. [DOI] [PubMed] [Google Scholar]
- 45. Xia S.J., et al. (1996) Reduced telomere length in ataxia-telangiectasia fibroblasts. Mutat. Res., 364, 1–11. [DOI] [PubMed] [Google Scholar]
- 46. Sfeir A., et al. (2009) Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell, 138, 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Henson J.D., et al. (2002) Alternative lengthening of telomeres in mammalian cells. Oncogene, 21, 598–610. [DOI] [PubMed] [Google Scholar]
- 48. Hande M.P., et al. (2001) Extra-chromosomal telomeric DNA in cells from Atm(-/-) mice and patients with ataxia-telangiectasia. Hum. Mol. Genet., 10, 519–528. [DOI] [PubMed] [Google Scholar]
- 49. Bodvarsdottir S.K., et al. (2012) Dysfunctional telomeres in human BRCA2 mutated breast tumors and cell lines. Mutat. Res., 729, 90–99. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.