

Abstract
Driver somatic mutations are a hallmark of a tumor that can be used for diagnosis and targeted therapy. Mutations are primarily detected from tumor DNA. As dynamic molecules of gene activities, transcriptome profiling by RNA sequence (RNA-seq) is becoming increasingly popular, which not only measures gene expression but also structural variations such as mutations and fusion transcripts. Although single-nucleotide variants (SNVs) can be easily identified from RNA-seq, intermediate long insertions/deletions (indels > 2 bases and less than sequence reads) cause significant challenges and are ignored by most RNA-seq analysis tools. This study evaluates commonly used RNA-seq analysis programs along with variant and somatic mutation callers in a series of data sets with simulated and known indels. The aim is to develop strategies for accurate indel detection. Our results show that the RNA-seq alignment is the most important step for indel identification and the evaluated programs have a wide range of sensitivity to map sequence reads with indels, from not at all to decently sensitive. The sensitivity is impacted by sequence read lengths. Most variant calling programs rely on hard evidence indels marked in the alignment and the programs with realignment may use soft-clipped reads for indel inferencing. Based on the observations, we have provided practical recommendations for indel detection when different RNA-seq aligners are used and demonstrated the best option with highly reliable results. With careful customization of bioinformatics algorithms, RNA-seq can be reliably used for both SNV and indel mutation detection that can be used for clinical decision-making.
Keywords: RNA sequencing, indels, mutation, alignment, variant calling, EGFR
Background
Somatic mutations are a hallmark of a tumor and can be used for diagnosis and targeted therapy selection. These mutations are usually detected from tumor DNA. As dynamic molecules of gene transcription activities, transcriptome profiling by RNA sequence (RNA-seq) is becoming increasingly popular, which not only measures gene expression but also structural variations such as fusion transcripts or mutations. The full utilization of the multilevel genomic information in RNA-seq will facilitate personalized medicine. Although single-nucleotide variants (SNVs) or somatic SNVs when paired tumor/normal RNA samples are available can be easily identified from RNA-seq [1–3], insertions/deletions (indels) longer than two bases cause significant bioinformatics challenges. First, RNA from RNA-seq is not a continuous copy of transcribed genes but of spliced exons, which leads to significant challenges when aligning sequence reads to a reference genome. Several RNA-seq-specific aligners have been developed to deal with the spliced molecules, which take the intron gaps into consideration in the alignment step. This can be achieved by either passing the known exon–intron junctions to the alignment programs or the programs themselves performing junction discovery from read coverage evidence. Some of these short-read alignment programs excel in speed but do not allow gaps in the sequence reads. Second, gapped alignment for indels is a significant bioinformatics challenge that not only slows down alignment speed but also is not easy to find the right positions of the split reads. Lastly, the alignment is the important but not the only step for accurate SNV or indel detection. Variable performances for variant detection in DNA-seq from different variant callers are reported [4–7], and in one particular example of evaluating multiple callers for the same alignment, most programs failed to detect indels longer than two bases [7]. Commonly called indels from different programs were disappointingly low [8, 9], a strong indication that there is much to be improved for accurate indel detection. Data from RNA-seq and combined effect of alignment and variant calling for indels have not been available and rarely investigated.
Despite of the challenges, there is a strong clinical need to detect important and actionable indels. For example, Epidermal Growth Factor Receptor (EGFR) gene mutation is common in multiple cancers, particularly for lung adenocarcinoma in nonsmokers. Over 90% of EGFR mutations in lung adenocarcinoma are in-frame deletions (ranging from 12 to 18 bases) in exon 19 or a point mutation in exon 21 (L858R) [10], and the former alone can account for about 50% of EGFR-mutated lung cancer [11, 12]. Lung cancers with these EGFR mutations are highly sensitive to EGFR tyrosine kinase inhibitors, such as gefitinib and erlotinib [11]. Therefore, it is critical to detect these SNV and indel mutations for clinical decision-making. Furthermore, the less common EGFR mutations such as insertions in exon 20 (about 4%) and point mutations that modify codons G719 (to A, C or S; 3%) and L861 (to Q; 2%) are also useful for guided personal treatment [11, 13].
Motivated by the clinical importance of indels and our experience of underreporting of indels from RNA-seq, we evaluated seven alignment tools for RNA-seq (TopHat, TopHat2, HISAT, HISAT2, STAR, GSNAP and RUM) and six variant/somatic mutation calling programs (GATK HaplotypeCaller, UnifiedGenotyper, FreeBayes, SAMtools/BCFtools and VarScan including VarScan2 for somatic mutations) for indel detection, extensively evaluated for DNA-seq but rarely done for RNA-seq data, in both simulated and real RNA-seq data sets with corresponding exome sequencing data and known indel mutations. We started from the common 15 base deletion in EGFR gene in a representative sample and evaluated the behavior of each aligner and variant caller. We then moved to a simulated RNA-seq data set with many indels with different lengths to obtain the overall picture of the performance of different aligner and variant caller combination. Finally, two lung cancer data sets with validated indels by both exome and other tests were used to demonstrate the reliable results of selected aligner and variant caller combinations.
Materials and methods
Alignment tools
We evaluated seven RNA-seq-specific and one popular generic mapping program BWA [14, 15] for indel alignment of RNA-seq data (Supplementary Table S1). TopHat [16] is the most commonly used alignment program for RNA-seq. The advantages of using TopHat are that it performs splice junction aware alignment; optionally detects fusion transcripts with TopHat-Fusion; and generates alignment that can be streamlined to de novo assembly programs such as Cufflinks [17] or Scripture [18]. The early version of TopHat (TopHat 1) uses Bowtie [19] as the underlying alignment program that performs ungapped global alignment. The upgraded version of TopHat 2 [20] uses Bowtie 2 [21], which can perform gapped extension that uses dynamic programming. HISAT or HISAT2 [22] is the new generation of spliced alignment program for RNA-seq reads based on TopHat 2 with Bowtie 2 with further enhanced performance of faster runtime and low memory usage. STAR is a relatively new ultrafast RNA-seq aligner that performs sequential maximum mappable seed search followed by seed clustering and stitching. It is up to 50× faster than other common aligners like TopHat but still achieves improved alignment sensitivity and precision [23], which makes it an attractive alternative for RNA-seq alignment. GSNAP [24] is another junction aware RNA-seq alignment program that is fast and tolerant for complex variants and splicing. It works with both short (from 14) and long sequence reads. GSNAP uses a successively constrained search process of merging and filtering position lists from a genomic index at the oligomer level and demonstrates better performance for reads with more mismatches or indels up to 30 bases. RUM is an RNA-seq analytical workflow that combines transcriptome and whole genome alignment with Blat [25] to align the unmapped reads to reference genome [26]. As Bowtie 1 is the underlying gapless aligner, incorporation of Blat, which is gapped alignment, presumably gets reads with indels aligned. BWA [14, 15] is the most widely used alignment suite of programs for DNA-seq, which has three algorithms: BWA-backtrack, BWA-SW and BWA-MEM. BWA-backtrack is designed for sequence reads up to 100 bp, whereas the other two are for longer sequences up to 1 Mb. BWA-MEM is the latest addition with the fastest runtime. Although not designed for RNA-seq, we included BWA as a generic alignment tool for its ability to align reads with long gaps so that it could be potentially used for second round of unmapped read alignment after the tools such as TopHat, which is known for ignoring reads with intermediate indels >2 bases.
Variant/mutation calling programs
For variants from single sample, GATK (2.0-35) HaplotypeCaller, UnifiedGenotyper [27], FreeBayes (v0.9.14) [28], SAMtools/BCFtools (v1.2) and VarScan [29] (part of VarScan 2 v2.3.7) were evaluated. Strelka [30] and VarScan 2 [31] (v2.3.7) were the tools for somatic mutation calling from paired tumor/normal samples (Supplementary Table S2). As indels are the primary focus of this article, the commonly used somatic mutation tools such as SomaticSniper [32], JointSNVMix [33] and muTect [34] were not included, as they can only make somatic SNV calls.
Simulated RNA-seq with indels
Two million paired-end reads with incorporated indels were simulated for human chr7 using the BEERS RNA-seq simulation program [26] at 100 bp length. Human RefSeq config files provided by BEERS were used to generate the simulated data. The true indels from the simulated BAM file were evaluated for their depth of coverage and alternative indel allele counts. The positions with simulated reads ≥10× and ≥2 alternative reads were kept as true-positive indels, as many positions can have no reads simulated (more mimic to RNA expression), which gave us 1805 indels and all other positions not overlapping with simulated indels and having ≥10× coverage were treated as true negatives (∼38 million positions). Vast majority of simulated indels from the simulator were between one and two bases and 64 of them were three or more up to nine bases. As the focus of the article was for intermediate indels, these 64 indels were evaluated closely. The generated FASTQ files were processed through different aligner and variant caller combination to make indel discovery. The sensitivity and specificity of the indel calls from each alignment/caller combination were calculated by comparing with the true indels and true-negative positions as defined above.
RNA-seq data with known indel mutations from lung cancer patients
EGFR insertions and deletions in exon 19 and 20 are common mutations in lung adenocarcinoma, particularly in nonsmoker patients. From 27 nonsmoker lung cancer patients with both exome sequence (exome-seq) and RNA-seq [1], we selected five tumors with known typical EGFR indels (3 deletions in 9 and 15 bases and 2 insertions in 6 and 9 bases, also validated by MassARRAY technology, Table 1). The clinical characteristics of these patients were described in our precious work [1]. The RNA-seq data were sequenced at two samples per lane by HiSeq2000 sequencer at 101 cycles with an average of 137–216 million pair-end reads. The data were in high quality as described previously and that study was approved by Mayo Clinic institute review boards [1]. Lu1321A (its paired normal lung is Lu1321C), the tumor with 15 base deletion in exon 19 from a female with stage IB tumor, was examined closely for the performance of aligners and variant callers and underlying causes for missed calls. To evaluate the impact of sequence read length, we also trimmed the reads to 50 bp. The adjacent normal lung tissues of these five tumors were also available and sequenced by both RNA-seq and exome-seq. They were used as reference to detect somatic mutations.
Table 1.
Exome-seq result for known indels in five pairs of samples
| Sample | Known EGFR mutations* | Exome (BWA) | NovoAlign | HaplotypeCaller+ | Strelka |
|---|---|---|---|---|---|
| Lu106 | Chr7_Ex20_9I | ++ (N−) | ++ + (N−) | √ | √ |
| Lu1271 | Chr7_Ex19_9D | ++ + (N−) | ++ + (N−) | √ | √ |
| Lu1321 | Chr7_Ex19_15D | ++ + (N−) | ++ + (N−) | √ | √ |
| Lu1377 | Chr7_Ex20_6I | ++ (N−) | ++ + (N−) | √ | √ |
| Lu1659 | Chr7_Ex19_15D | ++ + (N−) | ++ + (N−) | √ | √ |
Chr7_Ex20_9I: Indel notation by chromosome, exon, indel type, respectively. I/D represents insertion and deletion, and the number before is for the number of bases inserted or deleted. The number of + represents an increasing number of deletion reads aligned. N− represents that there is no deletion reads in the paired normal sample. √ represents that the indel/somatic indel is detected in the respective tumor.
To further test the reliability of the recommended aligner/caller combination for clinical important indels, we also analyzed 77 tumor/normal pairs of lung adenocarcinoma by RNA-seq reported previously [35], among which seven tumors with EGFR deletion of 15 and 18 bases at exon 19 (the similar common deletion as in our samples) were known. The raw data were downloaded from Short Read Archive at the accession# ERP001058. It was sequenced at both ends of 101 cycles. We aligned the data by both STAR and GSNAP and then called indels by GATK HaplotypeCaller, UnifiedGenotyper, FreeBayes, SAMtools/BCFtools and VarScan.
Results
Aligner performance for the well-known 15 base deletion in a clinical sample
As EGFR intermediate deletions are so important clinically and they are often underreported from RNA-seq, we first carefully examined the behavior of each aligner for the common 15 base deletion on exon 19 using one typical paired tumor and normal (Lu1321A, C) with both exome and RNA-seq data. TopHat 1, 2 (with Bowtie 1, 2), Bowtie 2, HISAT, HISAT2, STAR, GSNAP, RUM and BWA were included. We first performed the alignment for the exome-seq data of this sample by both NovoAlign (http://www.novocraft.com/products/novoalign/) and BWA-MEM, and in both the 15 base deletion was clearly seen with approximately 25–30% of support reads (out of 120–150 total reads, Figure 1A for NovoAlign, 1B for BWA-MEM). The EGFR is highly expressed in lung adenocarcinoma, and the RNA of the tumor Lu1321A was sequenced at very high depth over 144 million pair-end reads. At exon 19 where the deletion is, the average coverage is about 100–150×. By examining the aligned output (BAM file), TopHat 1 and 2 did not have any reads with the deletion aligned, which is not surprising as Bowtie 1 is gapless alignment, and in TopHat 2, the local alignment option was not able to be activated, which is available for Bowtie 2 (Figure 1C). Then, we run Bowtie 2 standalone by turning on local alignment option (but without considering splicing); we could see many soft-clipped reads around the deletion region but there were no hard evidence deletions marked in the Concise Idiosyncratic Gapped Alignment Report (CIGAR) string for 50 base reads (Figure 1D). This became better for the 100 base reads where there were eight reads marked with deletion (Figure 1E). STAR also did not mark any reads as deletion but soft clipped many for the 50 bp reads (Figure 2A). However, it aligned a significant number of reads as deletion for the 100 bp reads (Figure 2B). GSNAP demonstrated itself as the most sensitive aligner in this case, as it was able to align the highest number of deletion reads to the correct location with minimal soft clipping for both 50 and 100 base reads (57 and 91, respectively, Figure 2C and D). RUM aligned the reads with the deletion to the correct genomic location but it marked the deletion as splice junctions instead of deletion within the exon. To test if we could recover the reads with deletion in which TopHat did not align, we realigned the unmapped reads from TopHat 1 or 2 by BWA. BWA-backtrack was sensitive to align the 50 base reads with the deletion with gapped alignment (–e 30 option, Figure 3A); however, BWA-MEM and SW aligned most of deletion reads as soft clipping (Figure 3B). Conversely, for 100 base reads, the short-read BWA-backtrack only aligned one deletion read (Figure 3C), whereas BWA-MEM aligned 30 reads with the deletion (Figure 3D). This is a clear indication that the three BWA algorithms just work as they are supposed to do and users need to select the one that works the best according to the sequence lengths. In comparison of nonjunction aware alignment program Bowtie 2 with BWA-MEM for longer reads (≥100 bp) with the deletion, Bowtie 2 appeared inferior to BWA-MEM, as it had far less reads aligned as deletions than BWA-MEM.
Figure 1.

Lu1321A 15 bp deletion at exon 19 by exome-seq and RNA-seq (both at 100 base reads). (A) Exome-seq by NovoAlign. (B) Exome-seq by BWA-MEM. (C) RNA-seq by TopHat 1 and 2. (D) Bowtie 2 local alignment for 50 base reads. (E) Bowtie 2 alignment for 100 base reads. A colour version of this figure is available at BIB online: https://academic.oup.com/bib.
Figure 2.

RNA-seq alignment by STAR and GSNAP for the EGFR 15 base deletion in exon 19. (A) STAR for 50 base reads, all deletion reads soft clipped but no deletion marked. (B) STAR for 100 base reads, 41 reads aligned with deletion but others soft clipped. (C) GSNAP for 50 bp reads, almost all reads with deletion were correctly aligned. (D) GSNAP for 100 bp reads, even more reads with the deletion were correctly aligned, although with some reads as soft clipping. GSNAP has the better sensitivity to align reads with the deletion. A colour version of this figure is available at BIB online: https://academic.oup.com/bib.
Figure 3.

Unmapped reads from TopHat with BWA realignment. (A) Unmapped reads by BWA –e 30 option for 50 base reads. (B) Unmapped reads by BWA-MEM for 50 base reads. (C) Unmapped reads by BWA –e 30 option for 100 base reads. (D) Unmapped reads by BWA-MEM for 100 base reads. A colour version of this figure is available at BIB online: https://academic.oup.com/bib.
The new generation of TopHat aligner, HISAT and HISAT2, is much faster and expected to be more widely used. However, as their predecessors, they still do not support local alignment option, and therefore we would not expect there is a significant enhancement in terms of alignment for reads with intermediate indels. This was indeed the case for HISAT1, as there was no deletion reads aligned for both 50 and 100 base reads (Figure 4A and B). HISAT2 somehow was able to align some reads with the deletion as soft clipped at the deletion edges but no hard evidence deletion for both 50 and 100 base reads (Figure 4C and D). The soft-clipped reads can be potentially used for some variant callers to call the deletion. It is important to note that soft-clipped reads at the deletion edge by gapped or local alignment algorithm increase alignment sensitivity and can potentially be used for some variant calling programs to find structural variants or indels through realignment; however, they cannot be used for many other variant calling programs that depend on hard evidence indels marked in the CIGAR string of the alignment. These soft-clipped reads are generally ignored by these callers. Therefore, the alignment has a significant impact on downstream indel calling.
Figure 4.

HISAT1 and 2 for 15 base deletion alignment. (A) HISAT1 50 base reads, no reads with the deletion were aligned. (B) HISAT1 100 base reads, no reads with the deletion were aligned. (C) HISAT2 50 base reads, part of deletion reads aligned as soft clipping. (D) HISAT2 100 base reads, part of deletion reads aligned as soft clipping. A colour version of this figure is available at BIB online: https://academic.oup.com/bib.
The 15 base deletion detection sensitivity is variable among variant callers
As aligners that are not able to align reads with deletion to reference genome cannot be used for indel detection, our evaluation on indel callers was focused on the alignment by STAR, GSNAP and HISAT2 for both 50 and 100 base reads. As shown in Table 2, whether the deletion was called is dependent on sequence length, aligner and variant caller. For STAR and HISAT2 alignment of 50 base reads and HISAT2 100 base reads, no variant callers except GATK HaplotypeCaller made the correct deletion call, as all the alignments had no hard evidence deletion. GATK HaplotypeCaller conducts local realignment that allows it to use soft-clipped reads for the successful deletion detection. For STAR alignment of 100 base reads and GSNAP of both 50 and 100 base reads, the deletion was detected by all variant calling tools. Noted is that VarScan (or VarScan 2) relies on SAMtools pileup generation. The later version of SAMtools (v1.2) mpileup sometimes does not output the deletion information to pileup file in the plain output format so no deletion call was made at the position. However, it was able to detect the deletion with older version (v0.1.13) of SAMtools pileup. Combined use of SAMtools with BCFtools can make variant calling efficiently through piping mpileup output into BCFtools. VCF or BCF output format needs to be specified for the correct calling.
Table 2.
RNA-seq aligners and indel detection for five tumors with known indels
| Sample | Lu106 | Lu1271 | Lu1321 | Lu1377 | Lu1659 | ||
|---|---|---|---|---|---|---|---|
| Chr_exon_type | Chr7_Ex20_9I | Chr7_Ex19_9D | Chr7_Ex19_15D | Chr7_Ex20_6I | Chr7_Ex19_15D | ||
| Genomic location | chr7:55248998 | chr7:55242465 | chr7:55242465 | chr7:55249013 | chr7:55242464 | Sensitivity | |
| STAR | IGV* | 0‖1 | 1‖1 | 0‖1 | 1‖1 | 0‖1 | |
| GATK HT | 1‖1 | 1‖1 | 1‖1 | 1‖1 | 1‖1 | 1‖1 | |
| GATK UGT | 0‖1 | 0‖1 | 0‖1 | 0‖1 | 0‖1 | 0‖1 | |
| FreeBayes | 0‖0 | 0‖1 | 0‖1 | 0‖1 | 0‖1 | 0‖0.8 | |
| BCFtools | 0‖1 | 0‖1 | 0‖1 | 1‖1 | 0‖1 | 0.2‖1 | |
| VarScan | 0|0‖0|1 | 0|1‖1|1 | 0|0‖0|1 | 0|1‖0|1 | 0|0‖0|1 | 0|0.4‖0.2|1 | |
| GSNAP | IGV* | 0‖1 | 1‖1 | 1‖1 | 1‖1 | 1‖1 | |
| GATK HT | 1‖1 | 1‖1 | 1‖1 | 1‖1 | 1‖1 | 1‖1 | |
| GATK UGT | 0‖1 | 1‖1 | 1‖1 | 0‖1 | 1‖1 | 0.6‖1 | |
| FreeBayes | 0‖0 | 1‖0 | 1‖1 | 0‖1 | 1‖1 | 0.6‖0.6 | |
| BCFtools | 0‖1 | 1‖0 | 1‖1 | 0‖1 | 1‖1 | 0.6‖0.8 | |
| VarScan | 0|0‖1|1 | 1|1‖1|1 | 0|1‖0|1 | 0|0‖1|1 | 0|1‖0|1 | 0.2|0.6‖0.6|1 | |
| TopHat + BWA | IGV* | 0‖0 | 1‖1 | 1‖1 | 0‖1 | 1‖1 | 0.6‖0.8 |
| GATK HT | 0‖0 | 1‖1 | 1‖1 | 0‖1 | 1‖1 | 0.6‖0.8 | |
| GATK UGT | 0‖0 | 0‖1 | 0‖1 | 0‖0 | 0‖0 | 0‖0.4 | |
| FreeBayes | 0‖0 | 1‖1 | 0‖1 | 0‖1 | 0‖1 | 0.2‖0.8 | |
| BCFTools | 0‖0 | 1‖1 | 1‖1 | 0‖1 | 1‖1 | 0.6‖0.8 | |
| VarScan | 0|0‖0|0 | 1|0‖1|1 | 1|0‖1|1 | 0|0‖1|1 | 0|0‖1|1 | 0.4|0‖0.8|0.8 | |
| HISAT2 | IGV* | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 |
| GATK HT | 1‖1 | 1‖1 | 1‖1 | 1‖1 | 1‖1 | 1‖1 | |
| GATK UGT | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | |
| FreeBayes | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | |
| BCFtools | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | |
| VarScan | 0|0‖0|0 | 0|0‖0|0 | 0|0‖0|0 | 0|0‖0|0 | 0|0‖0|0 | 0|0‖0|0 | |
| RUM | IGV* | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 |
| GATK HT | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | |
| GATK UGT | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | |
| FreeBayes | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | |
| BCFtools | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | |
| VarScan | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 | 0‖0 |
HT = HaplotypeCaller; UGT = UnifiedGenotyper. 1 in the table for the indel reported and 0 for not reported. The number before ‖ is for 50 base read and after for 100 base read. For VarScan, the number before the bar is for using SAMtools version v1.2 and after for the SAMtools version v0.1.13. *Only solid indel is considered, not including soft-clipped reads. Lu1321 is used as an example for detailed description in the main text.
Results for other four tumor samples with different indels were similar to the sample Lu1321A and they are summarized in Table 2.
Performance of simulated RNA-seq with indels
To help to select the optimal aligner and variant callers for intermediate indels, it would be useful to use a simulated RNA-seq data with indels incorporated as the ground truths are known. For the simulated data set with 2 million reads from chr7, we aligned the paired-end FASTQ at 100 bp to hg37 reference genome with GSNAP, STAR, TopHat with subsequent BWA-MEM and HISAT2. The aligned BAM files were used to call variants by HaplotypeCaller, UnifiedGenotyper, FreeBayes, BCFtools and VarScan. Figure 5A shows the sensitivity of detection for all 1805 indels regardless of indel lengths. GSNAP and STAR alignments had higher sensitivities for all variant callers than the alignments from combination of TopHat and BWA-MEM and HISAT2. All callers performed better in GSNAP alignment than STAR alignment except BCFtools. For the indels at ≥3 (Figure 5B), the sensitivities of all callers in GSNAP and STAR remained similar to or better than the ones for all indels but they were dramatically reduced in the alignments from TopHat+ BWA-MEM and HISAT2. All alignment and caller combinations had very high specificity (>99.75%) and in some cases, it reached to 100%, for example, there was no false-positive indel calls by UnifiedGenotyper in any of alignments. FreeBayes had the lowest specificity (still at 99.8%) among the all for the GSNAP alignment (Supplementary Figure S1). Overall, we found GSNAP or STAR along with GATK HaplotypeCaller or BCFtools provided the highest sensitivity.
Figure 5.
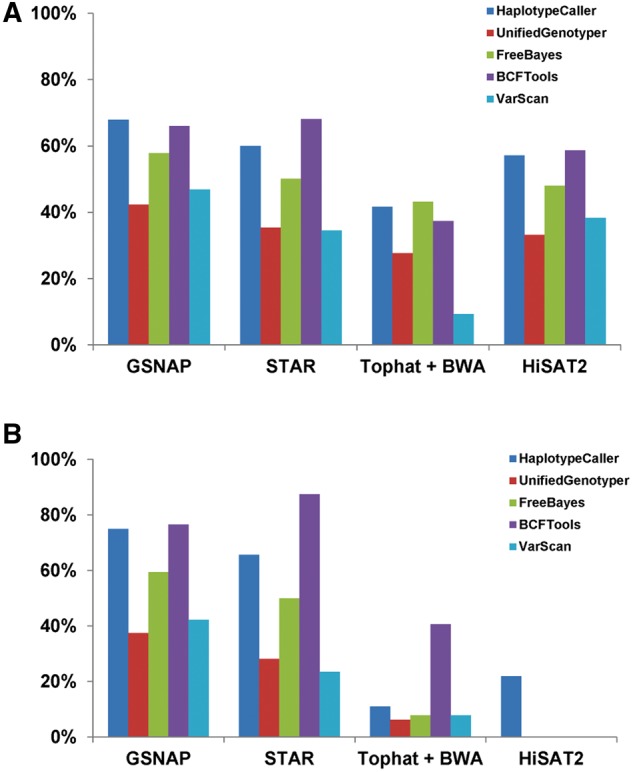
Performance of aligners and variant callers in the simulated RNA-seq data. Panel A is for all simulated 1805 indels with length from 1 to 9. Panel B is for the indels >2 bases (3–9 bases). GSNAP and STAR are the better aligners and HaplotypeCaller and BCFtools are the better choices for indel calling. A colour version of this figure is available at BIB online: https://academic.oup.com/bib.
Strategies for accurate indel detection in RNA-seq using different aligners
Our evaluation demonstrated that intermediate indel detection from RNA-seq highly hinged on aligners and callers. Some aligners do not align reads with longer indels at all and others are more sensitive. Most variant callers need hard evidence deletion clearly marked in the CIGAR string of an alignment file for the deletion calling. The aligners that just soft-clip reads with deletion would miss the deletion entirely for these callers. To recover the deletion, two different strategies can be taken: (1) for the alignment from TopHat families (TopHat 1, 2 and HISAT1 and 2), where only global alignment is conducted, users need to perform the second round of alignment for the unmapped reads by gapped aligners such as BWA-MEM. The newly generated alignment can be merged with the initial alignment for variant calling. As demonstrated, BWA-MEM is sensitive to align longer reads with indels. With the reads with indels correctly mapped, almost all variant calling algorithms could make the correct call. However, this option only serves as a rescue alternative, as it does not work as well as the next option from our simulation. (2) For users who have the flexibility of choosing different aligners for RNA-seq data, STAR or GSNAP would be preferred. Although STAR worked well for sequence reads at 100, it may have a difficulty with shorter reads. In this case, GSNAP is a better choice. The disadvantages of GSNAP are its slower speed compared with STAR and a potential compatibility issue with other downstream analysis programs such as fusion detection and novel transcript assembly.
Optimal combination of aligner and variant/somatic caller for five pairs of tumor/normal samples with known indels
The above evaluation demonstrated that GSNAP and STAR were the choices for indel alignment and GATK HaplotypeCaller (BCFtools is another choice but it cannot use soft-clipped reads) for indel calling in the single-sample mode. To validate this, we used five pairs of samples with more diverse known indels and evaluated the robustness of each aligner and GATK HaplotypeCaller. In addition, we added somatic callers Strelka and VarScan 2 to make somatic indel calls between tumor and its paired normal sample.
The five tumors and normal pairs had both exome-seq and RNA-seq for detailed comparison. All five tumors had either an insertion or deletion ranging from 6 to 15 bases (Table 3). The tumor and normal pair from the same individual allowed us to evaluate somatic indel calling algorithms as well. Exome-seq was aligned with both BWA-MEM and NovoAlign, and in both the expected indels were aligned with strong read support, whereas there were no reads supporting the indels from their paired normal samples. All these indels were correctly called by GATK HaplotypeCaller for the five tumors and by Strelka for somatic indels in the paired tumor and normal comparison.
Table 3.
RNA-seq result for known indels in five pairs of samples
| Sample | Known indels |
GSNAP |
STAR |
||||
|---|---|---|---|---|---|---|---|
| Normal | Tumor | Somatic | Normal | Tumor | Somatic | ||
| Lu106 | Chr7_Ex20_9I | X (−) | √ | √ | X | √ | √ |
| Lu1271 | Chr7_Ex19_9D | X (+) | √ | X | X | √ | √* |
| Lu1321 | Chr7_Ex19_15D | √ ( +++) | √ | X | X | √ | √ |
| Lu1377 | Chr7_Ex20_6I | X (−) | √ | √ | X | √ | √ |
| Lu1659 | Chr7_Ex19_15D | X (+) | √ | √ | X | √ | √ |
√ represents that the deletion is seen/detected; X: the deletion is not detected. −/+ sign in parenthesis for number of deletion reads seen in the BAM files from IGV; − for none; and the number of + represents an increasing number of deletion reads aligned. *The somatic nine base deletion was called in the unfiltered result but filtered out because of low-quality score.
Similarly for RNA-seq, all expected indels in the tumors were called from both GSNAP and STAR alignment (Table 3, results from other callers were provided in Supplementary Table S3); however, for the normal samples, something interesting was observed. Although no single read with indels in the STAR alignment was seen in all but one normal sample (Lu1271A), we observed that at least three normal samples in the GSNAP alignment had indel reads aligned in the same position as its paired tumor or even in some occasions from another tumor sample. Further careful investigation revealed that these normal samples were likely contaminated with tumor samples, as no evidence was seen in any of the respective exome-seq samples and an unexpected indel from a different tumor other than the patient was seen (i.e. >1 indels present in a tumor). The reason GSNAP found more deletions than STAR is that these indel reads sometimes have adapter sequences. GSNAP was able to handle such complex cases with both indels and adapter sequences better than STAR. The contamination could be the result of multiple sources ranging from impure normal sample to library preparation contamination and has been previously reported to be not an uncommon issue [36]. The immediate implication from the contamination is that in paired tumor and normal calling, a true somatic indel may not be called, as the normal sample also contains the indel reads. A wrong mutation may be called in a tumor if tumors are cross-contaminated. As shown in Table 3, sample Lu1271A and Lu1321A had expected mutations detected in the single-sample calling mode but the somatic mutations were missed in paired tumor/normal calling because of the contamination of their matched normal samples in the GSNAP alignment. STAR was less sensitive to align reads with indels; therefore, almost all but one normal sample did not have the contaminated indels, resulting in the correct somatic calls in almost all the cases.
RNA variant concordance with exome-seq data in single-sample calling mode
For the five tumors/normal pairs (10 individual samples), we also compared all SNVs and indel genotype concordances between exome-seq and RNA-seq from either STAR or GSNAP along the GATK HaplotypeCaller. For both exome-seq and RNA-seq, we used the same version of GATK HaplotypeCaller with equivalent parameters (except recommended ones for RNA-seq). As variant calling varies with depth of coverage at genomic locations, we conducted the concordance comparisons from 5× to 80×. As shown in Figure 6 for the 100 base reads, the SNV genotype concordances were generally at 99% (increasing from 98 at 5× to 99.8% at 80×). The indel concordances had a larger range of 95.7–100%. Noted is that exome-seq and RNA-seq may target different regions of the genome, resulting in variability of coverage to make reliable variant calls. The commonly called SNVs were generally less than half of total calls made by either exome-seq (46.2–54% of 27 000–48 888) or RNA-seq (33.4–53.4% of 41,740–46,786) at 5×; the similar observation was reported previously using a different aligner and a variant caller [1]. The common indels were even lower at about one-third of total indels at 5× coverage from either sequencing (30.4–39.7% of 1048–3656 for exome-seq and 11.0–33.5% of 3679–4040 for RNA-seq, Figure 6, right axis). Interestingly, all known indels that were called in exome-seq were reliably detected in RNA-seq.
Figure 6.
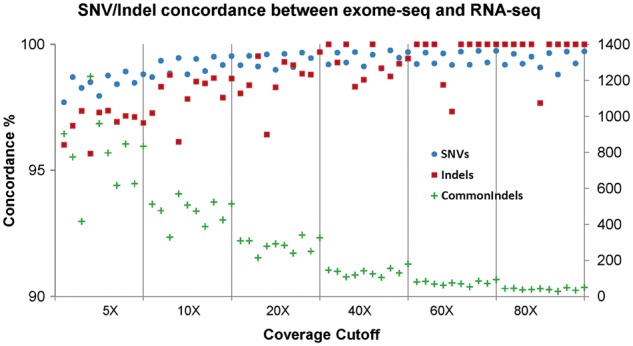
SNV and Indel concordance between exome-seq and RNA-seq at different depths of coverage. X-axis: depth coverage cutoff; Y-axis: the genotyping concordance between exome-seq and RNA-seq by combination of STAR alignment and GATK HaplotypeCaller. There is an overall increase of the concordance with depths of coverage. The green cross (+) indicates the number of common indels made by exome-seq and RNA-seq, which matches the numbers on the right Z-axis (ranging from 416 to 1221 indels at 5X coverage). A colour version of this figure is available at BIB online: https://academic.oup.com/bib.
Somatic mutation comparison of five tumor normal pairs
Using STAR alignment for 100 base reads, we also compared the somatic mutations (both SNVs and indels) between five pairs of tumor and normal between exome-seq and RNA-seq using Strelka as the somatic mutation caller with similar parameter settings. More somatic SNVs were called in RNA-seq than in exome-seq (Table 4). Although the commonly called somatic SNVs accounted for only a small fraction of the total from either, the genotype concordance was 100% for all exome-seq and RNA-seq pairs. Somatic indels (Table 5) were generally less than a couple of dozens from either exome-seq or RNA-seq. Despite the low overlap between the two, the commonly detected indels were all the known and expected. As the normal sample of Lu1271 was contaminated with its tumor deletion reads in the RNA-seq, the somatic deletion was not detected in RNA-seq but the missed call was not because of alignment and calling algorithm but the sample contamination.
Table 4.
Somatic SNVs from exome-seq and RNA-seq
| Sample pair | # SNVs (exome) | # SNVs (RNA) | # Common | % Genotype concordance |
|---|---|---|---|---|
| Lu106 | 445 | 1983 | 12 | 12/12 (100%) |
| Lu1271 | 971 | 2120 | 21 | 21/21 (100%) |
| Lu1321 | 600 | 1522 | 29 | 29/29 (100%) |
| Lu1377 | 210 | 1650 | 11 | 11/11 (100%) |
| Lu1659 | 174 | 1913 | 16 | 16/16 (100%) |
# SNVs: number of somatic SNVs called in exome-seq or RNA-seq; #Common: number of commonly called somatic mutation positions.
Table 5.
Somatic indels from exome-seq and RNA-seq
| Sample pair | # Indels (exome) | # Indels (RNA) | # Common | % Genotype concordance |
|---|---|---|---|---|
| Lu106 | 11 | 16 | 1 | 1/1 (100%) |
| Lu1271 | 25 | 1 | 0 | NA* |
| Lu1321 | 10 | 12 | 1 | 1/1 (100%) |
| Lu1377 | 10 | 11 | 1 | 1/1 (100%) |
| Lu1659 | 6 | 8 | 1 | 1/1 (100%) |
The somatic nine base deletion was detected in both exome-seq and RNA-seq, but it had a low-quality score in RNA-seq and was then filtered out as its paired normal sample contained the same deletion reads, which was likely contained from the tumor or filed effect in the normal sample.
Known indel detection in a public data set
For the seven tumors with known deletions in exon 19, we conducted alignment by both STAR and GSNAP and called variants by five callers. As shown in Table 6, GATK HaplotypeCaller is the only one who consistently performed well for both STAR and GSNAP alignment with perfect sensitivity. However, other callers were dependent on the alignment. In general, UnifiedGenotyper, FreeBayes and VarScan performed better for the GSNAP alignment but BCFtools was more variable. This observation is consistent with the data from the simulated and our internal lung tumor samples.
Table 6.
Known indel detection by different combinations of aligner and caller in a public data set
| Sample ID |
STAR |
GSNAP |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| GATK HT | GATK UGT | FreeBayes | BCFtools | VarScan | GATK HT | GATK UGT | FreeBayes | BCFtools | VarScan | |
| ERR164550 | 1 | 1 | 1 | 0 | 0|1 | 1 | 1 | 1 | 0 | 1|1 |
| ERR164559 | 1 | 1 | 1 | 1 | 0|1 | 1 | 1 | 1 | 1 | 1|1 |
| ERR164560 | 1 | 0 | 0 | 1 | 0|0 | 1 | 1 | 1 | 1 | 1|1 |
| ERR164563 | 1 | 0 | 0 | 0 | 0|0 | 1 | 1 | 1 | 0 | 1|1 |
| ERR164569 | 1 | 0 | 0 | 1 | 0|0 | 1 | 1 | 1 | 0 | 1|1 |
| ERR164585 | 1 | 1 | 1 | 1 | 0|1 | 1 | 1 | 1 | 0 | 1|1 |
| ERR164613 | 1 | 1 | 0 | 1 | 0|1 | 1 | 1 | 1 | 0 | 1|1 |
| Sensitivity | 1 | 0.57 | 0.43 | 0.72 | 0.00|0.57 | 1 | 1 | 1 | 0.29 | 1.00|1.00 |
HT = HaplotypeCaller; UGT = UnifiedGenotyper. 1 in the table for the indel reported and 0 for not reported. For VarScan, the number before the bar is for using SAMtools version v1.2 and after for the SAMtools version v0.1.13.
Discussion
RNA-seq is the most common modality of gene expression profiling and it is becoming increasingly popular in clinical settings for precision medicine, as it measures the dynamic gene activity of the genome for a specific tissue type. Application of RNA-seq to a tumor allows subclassification from expression for treatment selection and outcome prediction, fusion transcript or mutation detection for targeted therapies [37]. Although SNVs are readily detected [1–3], indel detection is much more challenging and is an unexplored territory, mostly because RNA is complicated by alternative splicing and most commonly used RNA alignment programs do not conduct local alignment or perform poorly. In this study, we evaluated both alignment and variant caller impacts on indel detection using both real and simulated samples with known indels. We compared both single-sample variant and paired somatic mutation calling. We found that alignment is a critical step for intermediate indel detection. TopHat family RNA-seq mapping programs do not align reads with intermediate indels or align minimally when HISAT2 is used. Most variant calling programs would miss intermediate indels from these aligners, except the case that there are sufficient soft-clipped reads triggering realignment by HaplotypeCaller (Figure 4C and D, Table 2). To reliably detect intermediate indels, additional alignment by indel-sensitive aligners such as BWA-MEM is needed for unmapped reads from the initial alignment. The combined alignment then can be used for most variant callers. For longer reads (≥100), STAR works almost equally well as GSNAP, but for shorter RNA-seq reads (50 or shorter), GSNAP is much more sensitive and recommended. For indel calling of a single sample, GATK HaplotypeCaller is the most robust one, as it can use soft-clipped reads to infer indels, unlike others that need indels marked and correctly positioned by aligners. Strelka works well in somatic indel detection. Comparisons with exome-seq data show a high concordance and all expected important indels are detectable in RNA-seq, which strongly supports the feasibility of detecting both SNVs and indels from RNA-seq when appropriate alignment and variant calling programs are selected.
In this study, we opted primarily to use the real sequencing data of both exome-seq and RNA-seq with known important indels for the evaluation of various tools and algorithms. This not only allows us to assess the real-life challenges of the data and analysis but also the performance of the tools in detecting the critical and actionable mutations for clinic. Although the simulated data are commonly used, they are generally generated from reference genome and do not reflect the complexity of real data, for example, sample across contamination or adapter sequences. However, simulated data can provide an overall picture of aligner and caller performance as shown in this work.
The RNA-seq sequence reads with indels not mapped to the reference genome not only affect the indel detection but also the downstream gene expression quantification or alternative splicing detection. As shown in our evaluation, when an aligner does not align any reads with intermediate indel, there is a significant coverage drop around the indel region. The deletion reads can account a third to a half of total reads. As they are not counted in gene expression, it may affect downstream differential expression analysis between samples with and without the indel. The significant drop may also potentially lead to a false alternative splicing event at this exon.
The incomplete alignment may have some implications to other sequencing applications. For example, DNA methylation sequencing such as whole genome methylation sequencing, reduced representation bisulfite sequencing (RRBS) or targeted bisulfte sequencing can be used for variant detection or copy number estimation and many data processing pipelines use Bowtie or Bowtie 2 for alignment [38–40]. Similar to what is observed for RNA-seq here, users would not expect to detect any indels from such data sets if Bowtie 1 is used. If most of reads around indel positions are not aligned, it may affect methylation estimate. Combined with the HaplotypeCaller with realignment on the top of the alignment from Bowtie 2, most indels can be detected for sequencing with longer reads, although it may be less sensitive for shorter reads such as RRBS often containing shorter reads.
The study also confirms the observation reported previously that although there is high concordance for SNVs or somatic SNVs between RNA-seq and exome-seq, a significant proportion of private variants is present in either platform [1, 41]. The commonly called variants generally account for one-third of total variants or even less from each for SNVs. This number is even lower for indels. These can be explained largely by different genomic regions that get sequenced, uneven coverage of targeted region or differential expression among different genes in RNA-seq [41]. It is also possible that RNA-seq is noisier than DNA-seq. Despite small fraction, RNA editing may also contribute to the difference.
This study did not evaluate all available RNA-seq alignment and variant calling programs but most common ones with known better performances [23, 24, 42]. As the goal of the study was to detect indel, the common tools that can only detect somatic SNVs at the time of this work were not included such as MuTect, JointSNVMix and SomaticSniper. MutTect and VarScan 2 were reported as more sensitive tools for somatic SNVs [43]. The caveat from these data suggests that VarScan 2 (or VarScan for SNVs/Indels in nonpaired samples) is highly dependent on alignment tools, and intermediate pileup generation by SAMtools and intermediate indels most likely are missed.
The framework of RNA-seq variant detection with sensitive indel detection after STAR or GSNAP alignment has several advantages and can be easily expanded for other genomic features such as fusion transcript detection by STAR-Fusion (https://github.com/STAR-Fusion/STAR-Fusion) and gene expression quantification. Comprehensive and accurate characterization of a tumor would provide critical information for precision medicine. RNA-seq is an excellent tool to achieve that promise with accurate characterization of targetable genomic abnormalities.
Key Points
RNA-seq is the most commonly used sequencing application to monitor gene regulation activity but can be used for mutation, fusion transcript and alternative splicing detection.
Full utilization of these genomic information can maximize the potential of personalized medicine.
Commonly used RNA-seq alignment and variant calling programs perform poorly in detecting intermediate long indels (>2 bases) that are clinically actionable.
Strategies are laid out for indel detection in RNA-seq.
High sensitivity and specificity of these strategies are demonstrated in real RNA-seq samples with known indel mutations.
Funding
This work was supported by Mayo Clinic Center for Individualized Medicine.
Supplementary Material
Zhifu Sun is a Senior Associate Consultant and Associate Professor in Division of Biomedical Statistics and Informatics, Department of Health Sciences Research at Mayo Clinic Rochester, Minnesota. His research focus is genomics and epigenomics.
Aditya Bhagwate is an Informatics Specialist with Master Degree in Bioinformatics in Division of Biomedical Statistics and Informatics, Department of Health Sciences Research at Mayo Clinic Rochester, Minnesota.
Naresh Prodduturi is an Informatics Specialist with Master Degree in Bioinformatics in Division of Biomedical Statistics and Informatics, Department of Health Sciences Research at Mayo Clinic Rochester, Minnesota.
Ping Yang is a Consultant and Professor in Division of Epidemiology, Department of Health Sciences Research at Mayo Clinic Rochester, Minnesota. Her research is lung cancer genetics and epidemiology.
Jean-Pierre A. Kocher is a Consultant and Professor in Division of Biomedical Statistics and Informatics, Department of Health Sciences Research at Mayo Clinic Rochester, Minnesota. His research focus is bioinformatics and system biology.
References
- 1. Sun Z, Wang L, Eckloff BW, et al. Conserved recurrent gene mutations correlate with pathway deregulation and clinical outcomes of lung adenocarcinoma in never-smokers. BMC Med Genomics 2014;7:32.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang C, Davila JI, Baheti S, et al. RVboost: RNA-seq variants prioritization using a boosting method. Bioinformatics 2014;30:3414–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Piskol R, Ramaswami G, Li JB.. Reliable identification of genomic variants from RNA-seq data. Am J Hum Genet 2013;93:641–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. O'Rawe J, Jiang T, Sun G, et al. Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome Med 2013;5:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cornish A, Guda C.. A comparison of variant calling pipelines using genome in a bottle as a reference. Biomed Res Int 2015;2015:456479.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hwang S, Kim E, Lee I, et al. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci Rep 2015;5:17875.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Neuman JA, Isakov O, Shomron N.. Analysis of insertion-deletion from deep-sequencing data: software evaluation for optimal detection. Brief Bioinform 2013;14:46–55. [DOI] [PubMed] [Google Scholar]
- 8. Ghoneim DH, Myers JR, Tuttle E, et al. Comparison of insertion/deletion calling algorithms on human next-generation sequencing data. BMC Res Notes 2014;7:864.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hasan MS, Wu X, Zhang L.. Performance evaluation of indel calling tools using real short-read data. Hum Genomics 2015;9:20.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Politi K, Lynch TJ.. Two sides of the same coin: EGFR exon 19 deletions and insertions in lung cancer. Clin Cancer Res 2012;18:1490–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mitsudomi T, Yatabe Y.. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J 2010;277:301–8. [DOI] [PubMed] [Google Scholar]
- 12. Jang JS, Lee A, Li J, et al. Common oncogene mutations and novel SND1-BRAF transcript fusion in lung adenocarcinoma from never smokers. Sci Rep 2015;5:9755.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yasuda H, Kobayashi S, Costa DB.. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol 2012;13:e23–31. [DOI] [PubMed] [Google Scholar]
- 14. Li H, Durbin R.. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li H, Durbin R.. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 2010;26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Trapnell C, Pachter L, Salzberg SL.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25:1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28:511–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guttman M, Garber M, Levin JZ, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 2010;28:503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009;10:R25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim D, Pertea G, Trapnell C, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013;14:R36.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Langmead B, Salzberg SL.. Fast gapped-read alignment with bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim D, Langmead B, Salzberg SL.. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 2015;12:357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu TD, Nacu S.. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010;26:873–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res 2002;12:656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grant GR, Farkas MH, Pizarro AD, et al. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq unified mapper (RUM). Bioinformatics 2011;27:2518–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garrison E, Marth G.. Haplotype-based variant detection from short-read sequencing. arXiv 2012; preprint arXiv:1207.3907. https://github.com/ekg/freebayes#citing-freebayes. [Google Scholar]
- 29. Koboldt DC, Chen K, Wylie T, et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009;25:2283–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saunders CT, Wong WS, Swamy S, et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28:1811–7. [DOI] [PubMed] [Google Scholar]
- 31. Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22:568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Larson DE, Harris CC, Chen K, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics 2012;28:311–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roth A, Ding J, Morin R, et al. JointSNVMix: a probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics 2012;28:907–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seo JS, Ju YS, Lee WC, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res 2012;22:2109–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Flickinger M, Jun G, Abecasis GR, et al. Correcting for sample contamination in genotype calling of DNA sequence data. Am J Hum Genet 2015;97:284–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mody RJ, Wu YM, Lonigro RJ, et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA 2015;314:913–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bock C. Analysing and interpreting DNA methylation data. Nat Rev Genet 2012;13:705–19. [DOI] [PubMed] [Google Scholar]
- 39. Liu Y, Siegmund KD, Laird PW, et al. Bis-SNP: combined DNA methylation and SNP calling for bisulfite-seq data. Genome Biol 2012;13:R61.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sun Z, Cunningham J, Slager S, et al. Base resolution methylome profiling: considerations in platform selection, data preprocessing and analysis. Epigenomics 2015;7:813–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. O'Brien TD, Jia P, Xia J, et al. Inconsistency and features of single nucleotide variants detected in whole exome sequencing versus transcriptome sequencing: a case study in lung cancer. Methods 2015;83:118–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Engstrom PG, Steijger T, Sipos B, et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods 2013;10:1185–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Q, Jia P, Li F, et al. Detecting somatic point mutations in cancer genome sequencing data: a comparison of mutation callers. Genome Med 2013;5:91.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.