

Circulating methylated TBX2 DNA in plasma is a potential biomarker for hepatocellular carcinoma.
Abstract
Metastases in the later stages of hepatocellular carcinoma (HCC) cause the majority of deaths associated with the disease, making early detection crucial to patient survival. Risk models assessing HCC risk in the general population can be used for risk stratification for further HCC surveillance, however, none have been validated externally. Methylation of circulating DNA shows potential for non-invasive diagnosis of HCC. We conducted a prospective case–control study nested within a community-based cohort. We measured methylation levels in six genes (CDKN2A, RASSF1A, STEAP4, TBX2, VIM and ZNF154) which were identified in our previous work, using pre-diagnostic plasma DNA from 237 HCC cases and 257 matched controls. We found TBX2 hypermethylation was associated with increased HCC risk, with ORs (95% CI) of 3.2 (1.8–6.0). The associations were mainly among high-risk subjects; among subjects infected with HBV/HCV, the OR (95% CI) of TBX2 methylation was 5.3 (2.2–12.7). Among subjects with high risk scores, the ORs (95% CIs) were 7.8 (1.5–38.6) for Wen-HCC model ≥16, 5.8 (2.2–15.5) for Hung-HCC ≥15 and 7.5 (2.2–26.0) for Michikawa-HCC ≥8. Adding TBX2 methylation improved the accuracy of risk models for a high-risk population, with the area under the curve (AUC) of 76% for Wen-HCC score with TBX2 methylation compared with 69% with Wen-HCC alone. The AUCs were 63% for Hung-HCC score plus TBX2 methylation, and 53% for Hung-HCC alone, 65% for Michikawa-HCC score plus TBX2 methylation and 58% for Michikawa-HCC alone. Our findings suggest the potential increase in risk assessment discrimination and accuracy from incorporation of DNA methylation.
Introduction
Worldwide, hepatocellular carcinoma (HCC) is the fifth most common cancer in men, the seventh in women and the third most common cause of death from cancer (1). Although HCC occurs more frequently in less developed regions of the world (1), the incidence of HCC in the USA is increasing at an alarming rate (2,3). Despite great progress in surgical and medical management of the disease, the overall prognosis of HCC patients is still unsatisfactory; the 5-year survival rate is ~17% (2,3). It is estimated that HCC is projected to surpass breast and colorectal cancers to become the third leading cause of cancer-related death in the USA by 2030 (3). Currently only 46% of HCC cases are diagnosed at an early stage and most do not receive curative therapy (3). Surveillance for HCC is recommended in clinical practice in patients who are at increased risk of developing HCC, including those with chronic HBV infection (4,5). HCC surveillance is associated with a 38% reduction in overall mortality partly contributed by early treatment (6).
Many risk models have been developed to assess HCC risk in high-risk individuals, in particular, people with chronic hepatitis B or C virus (HBV/HCV) infection (7–14). Currently, there are only three HCC risk models assessing HCC risk in the general population; Michikawa-HCC score derived from a middle-aged Japanese population incorporates age, sex, alcohol and coffee consumption, body mass index (BMI), diabetes and HBV/HCV infection (15). The Wen-HCC score derived from a private health screening firm in Taiwan includes information such as age, sex, alcohol and smoking habit, hepatitis virus infection status and serum markers [e.g. alanine transaminase (ALT), aspartate transaminase (AST) and α-fetoprotein (AFP)] (16). Hung-HCC score used data (e.g. age, sex ALT, history of chronic liver disease and family history of HCC) from three cohorts involving 12377 Taiwanese adults 20–80 years of age (17). None have been validated in other studies. Risk prediction models for HCC using information regarding HBV/HCV infection status and other risk factors of HCC allow selection of asymptomatic individuals for priority HCC screening (18).
Blood biomarkers can be used to identify at-risk individuals that can be targeted for prevention and early detection of HCC. AFP is the most widely used marker in clinics (19), however, its sensitivity is low (20,21). Therefore, the development of new biomarkers for early HCC detection is important to improve the overall-survival rate.
We previously compared DNA methylation profiles in HCC tumor and adjacent non-tumor tissues and found alterations in DNA methylation frequently occur in HCC (22,23). Analysis of plasma DNA from cases demonstrated that up to 63% of cases had detectable hypermethylated DNA suggesting that measurement of DNA methylation in plasma samples is feasible (22). Emerging studies have examined the role of DNA methylation in selected candidate genes such as CDKN2A and rad association domain family 1A (RASSF1A) as potential blood biomarkers for HCC [reviewed (24)]. Most evidence, however, comes from small clinical studies that are cross-sectional or retrospective, raising concerns about temporality. In our pilot study, we measured DNA methylation in CDKN2A (p16), CDKN2B (p15) and RASSF1A using serum from 50 HCC patients, mainly HBV-related, who provided blood samples before cancer diagnosis and 50 controls without any evidence of HCC (25). We found that compared with controls, cases had a higher prevalence of hypermethylation in these genes. A receiver operating characteristic (ROC) curve that included clinical risk factors such as age, hepatitis B virus surface antigen (HBsAg) status, antibody against hepatitis C virus (anti-HCV) status and hypermethylation biomarkers gave an overall predictive accuracy of 89% with sensitivity and specificity 84 and 94%, respectively (25).
In recent years, an increasing number of HCC-related methylation markers are being discovered through high throughput genome wide data (22,23,26,27). In this study, we prospectively compared methylation of six genes (CDKN2A, RASSF1A, VIM, ZNF154, TBX2 and STEAP4) that we previous found hypermethylated in HCC tumor compared to adjacent non-tumor tissues (22,23) and that were reported in other studies showing hypermethylated in HCC tumors tissues and blood (27–34) in pre-diagnostic plasma collected at baseline (n = 237 HCC cases and 257 age, and sex matched controls). In addition, we assessed the impact of combining data such as DNA methylation in HCC risk models to illustrate the value of using biomarker information in risk assessment.
Materials and methods
Study cohort
Subjects are from the community-based Cancer Screening Program cohort recruited in Taiwan. The cohort characteristics and methods of screening and follow-up have been described in detail previously (35–39). Briefly, individuals who were between 30 and 65 years old and lived in seven townships in Taiwan were recruited between July 1990 and June 1992 with a total of 12,020 males and 11,923 females. Participants were personally interviewed based on a structured questionnaire regarding epidemiological information and donated a 20-ml fasting blood sample at recruitment. Biospecimens were transported on dry ice to a central laboratory at the National Taiwan University and stored at −70°C until transport to Columbia University for analysis. Epidemiological information included socio-demographic characteristics, habits of alcohol intake and cigarette smoking, health history including liver cirrhosis and family history of cancers, including HCC. Habitual cigarette smoking was defined as having smoked >4 days/week for at least 6 months. Information about duration and intensity was also obtained. Habitual alcohol intake was defined as drinking alcohol containing products >4 days/week for at least 6 months.
At enrollment, blood samples were tested in Taiwan for serological markers, including ALT, AST and AFP. HBsAg and AFP were tested by radioimmunoassay (Abbott Laboratories, North Chicago, IL); anti-HCV was tested by enzyme immunoassay using commercial kits (Abbott Laboratories). Both ALT and AST levels were determined with a serum chemistry autoanalyzer (Hitachi Model 736; Hitachi Co., Tokyo, Japan) using commercial reagents (Biomerieus, Mercy I’Etoile, France). Any participants who had an elevated level of ALT (≥45 IU/ml), AST (≥40 IU/ml) or AFP (≥20 ng/ml), was positive for HBsAg or anti-HCV, or had a family history of HCC or liver cirrhosis among first-degree relatives was referred for upper abdominal ultrasonography examination. Suspected HCC cases were referred to teaching medical centers for confirmatory diagnosis by computerized tomography, digital subtracted angiogram, aspiration cytology and pathologic examination. The criteria for HCC diagnosis included a histopathological examination, a positive lesion detected by at least two different imaging techniques (abdominal ultrasonography, angiogram or computed tomography) or by one imaging technique and a serum AFP level >400 ng/ml.
This study was approved by Columbia University’s Institutional Review Board as well as the Research Ethics Committee of the College of Public Health, National Taiwan University. Written informed consent was obtained from all subjects and strict quality controls and safeguards were used to protect confidentiality.
Study subjects
Between February 1991 and June 2008, a total of 237 cases were newly diagnosed with HCC with pre-diagnostic plasma available. Cases were primarily identified through linkage to the national cancer registry and death certification systems. We randomly selected 257 controls from cohort subjects who were not affected with HCC through the follow-up period by matching to each case by age (±5 years), gender, residential township and date of recruitment (±3 months).
DNA extraction and bisulfite treatment
DNAs were extracted from 250 ul plasma using DNeasy Blood & Tissue Kits (Qiagen, Valencia, CA). Aliquots of DNA (500 ng) were bisulfite-treated with the EZ DNA methylation-Gold kit (Zymo Research, Orange, CA) to convert unmethylated cytosines to uracils while leaving methylated cytosines unmodified. The DNA was resuspended in 20 µl of distilled water and stored at −20°C until use. The laboratory investigator who performed the assays was blinded to epidemiologic data.
Pyrosequencing assay
We measured methylation of CDKN2A, VIM, STEAP4 and ZNF154 by pyrosequencing. The sequences of primers for CDKN2A, STEAP4 and ZNF154 have been described in detail previously (22). The primers were GAT GGT TTA GTT GTA AGT TGG TAG TA (FWD), ACC AAA TTA ATT CAA ATC TCA AC—biotin (REV) and AGT TGG TAG TAT TGA GAA (sequence) for VIM. PCR was carried out in a 25-μl reaction mix containing 50 ng bisulfite-converted DNA, 1× PyroMark PCR Master Mix (Qiagen), 1× Coral Load Concentrate (Qiagen) and 0.2 μM forward and reverse primers, using the following PCR program: 95°C for 15 min, then 45 cycles of 94°C for 30 s followed by 56°C for 30 s and 72°C for 30 s, with a final extension at 72°C for 10 min. Each set of amplifications included bisulfite-converted CpGenome universal methylated (EMD Millipore, MA), unmethylated (whole-genome amplified DNA) and non-template controls. Following amplification, the biotinylated PCR products were purified and made single-stranded to act as a template in the pyrosequencing reaction as recommended by the manufacture using the PyroMark Q96 Workstation (Qiagen). Then, 0.3 pmol/μl of pyrosequencing primer was annealed to the purified single-stranded PCR product and pyrosequencing was run on a PyroMark Q96 MD instrument (Qiagen), with subsequent quantitation of methylation levels determined with the PyroMark CpG 1.0.11 software. Non-CpG cytosine residues were used as internal controls to verify efficient sodium bisulfite DNA conversion. Percent methylation was calculated by averaging across all CpG sites interrogated. A plasma DNA sample was considered positive if % methylation was ≥5% since lower values are not reliable, except STEAP4 as most of samples had methylation ≥ 5%.
Real-time PCR
We measured promoter methylation of RASSF1A and TBX2 by PCR. The sequences of primers are GGA GAG GGT TTG ATA GGT AGA AAT (FWD), AAA ACC CCA CTC CTC CTT TAT T (REV) for TBX2 and GTG TTA ACG CGT TGC GTA TC (FWD), AAC CCC GCG AAC TAA AAA CGA (REV) for RASSF1A. The primers only amplify if the target regions are fully methylated. The primers for RASSF1A were used for methylation specific PCR assay in our previous study (25). The primers for TBX2 overlap probe cg21389753 of the Illumina HumanMethylation 450K where we observed hypermethylated in HCC tumors in our previous study (23). PCR was carried out in a 25 μl reaction mix containing 50 ng bisulfite-converted DNA, 1× PyroMark PCR Master Mix (Qiagen), 1× Coral Load Concentrate (Qiagen) and 0.2 μM forward and reverse primers, using the following PCR program: 95°C for 15 min, then 45 cycles of 94°C for 30 s followed by 56°C for 30 s and 72°C for 30 s, with a final extension at 72°C for 10 min. Assays were run on an ABI Prism 7900 Sequence Detection System (Perkin-Elmer, Foster City, CA). A plasma DNA sample was considered positive if the Ct value ≤30.
Statistical methods
We used the χ2 test for categorical variables and Student’s t-test for continuous variables to assess the difference in selected characteristics between cases and controls. To estimate associations with HCC risk, we used conditional logistic regression models stratified on the matching factors to calculate odds ratios (ORs) and 95% confidence intervals (Cls). We modeled the associations adjusting for age (years, continuous), and HBsAg (Yes versus No) in all final models. We calculated Wen-HCC score which ranges from 0 to 39 for each subject based on model 5 (16). Briefly, model 5 assigns score for each variables; sex (1 for male 1, and 0 for female), age (0 for ages 20–39, 2 for ages 40–59 and 6 for ages ≥60), smoking (0 for pack-years ≤9.9 and 1 for pack-year ≥10), alcohol drinking (1 for regular drinker and 0 for none or occasional drinker), diabetes (1 for yes, and 0 for no), AST (0 for <25 IU/l, 4 for 25–39 IU/l, 6 for 40–59 IU/l and 7 for ≥60 IU/l), ALT (1 for ≥25 IU/L, and 0 for <25 IU/l), AFP (0 for <2.5 ng/ml, 2 for 2.5–4.9 ng/ml, 5 for 5.0–9.9 ng/ml and 9 for ≥ 10), HBV (0 for negative and 6 for positive) and HCV (0 for negative and 5 for positive). We calculated Hung-HCC score which ranges from 0 to 28 for each subject based on model 4 (17). The model 4 assigns score for each variables; sex (3 for male 1 and 0 for female), age (0 for ages 20–39, 3 for ages 40–49, 5 for age 50–59 and 7 for ages ≥60), ALT (3 for ≥25 IU/L and 0 for <25 IU/L), smoking (0 for pack-years ≤18 and 1 for pack-year ≥18), prior chronic liver disease (3 for yes and 0 for no), family history of liver cancer (2 for yes, 0 for no) and HBV or HCV (0 for negative, and 9 for positive). The variables in the Michikawa-HCC score are (15): age (40–49 years: 0, 50–59 years: 2 and ≥60 years 3), sex (male: 2 and 0: female), alcohol consumption (never:1, past:0, current <450 g/week of ethanol:0, and current ≥450 g/week of ethanol: 2), BMI (≥25 kg/m2: 1, and <25 kg/m2: 0), diabetes (yes: 1, and no: 0), HBsAg (yes: 4, and no:0) and anti-HCV (yes: 6, and no: 0). We did not collect information on coffee consumption, so could not include score for coffee consumption. We conducted logistic regression modeling using PROC LOGISTIC for ROC analysis. All analyses were performed with SAS software 9.2 (SAS Institute, Cary, NC). All statistical tests were based on two-tailed probability.
Results
Table 1 shows the distributions of subjects’ characteristics at baseline between cases and controls. There are no differences in the distributions of age, BMI and diabetes status nor in habitual smoking and alcohol consumption. As expected, the percents positive for HBsAg and/or anti-HCV were higher in cases than in controls (57.4 versus 43.6% for HBsAg and 29.5 versus 15.2% for anti-HCV). AFP was higher in cases than controls (6.57 ± 8.89 ng/ml for cases and 3.56 ± 5.16 ng/ml for controls). Mean risk scores were statistically significantly higher in cases than controls (15.6 versus 11.1 for Wen-HCC risk score, 15.3 versus 12.9 for Hung-HCC risk score and 8.6 versus 7.1 for Michikawa-HCC risk).
Table 1.
Sociodemographic characteristics of HCC cases and controls
| Cases | Controls | P | |
|---|---|---|---|
| N = 237 | N = 257 | ||
| Age (mean, SD) years | 58.9 (9.2) | 57.6 (8.8) | 0.11 |
| Sex | |||
| Female | 77 (32.5) | 78 (30.4) | 0.61 |
| Male | 160 (67.5) | 179 (69.7) | |
| HBsAg | |||
| Negative | 101 (42.6) | 145 (56.4) | 0.0022 |
| Positive | 136 (57.4) | 112 (43.6) | |
| Anti-HCV | |||
| Negative | 167 (70.5) | 218 (84.8) | 0.0001 |
| Positive | 70 (29.5) | 39 (15.2) | |
| Smoking status | |||
| Never | 146 (61.6) | 166 (64.6) | 0.5 |
| Ever | 91 (38.4) | 91 (35.4) | |
| Alcohol consumption | |||
| Never | 193 (81.4) | 225 (87.6) | 0.17 |
| Ever | 43 (18.1) | 31 (12.1) | |
| Missing | 1 (0.4) | 1 (0.4) | |
| BMI (kg/m2) | |||
| <25 | 128 (54.0) | 157 (61.1) | 0.11 |
| ≥25 | 109 (46.0) | 100 (38.9) | |
| Diabetes | |||
| No | 224 (94.5) | 251 (97.7) | 0.07 |
| Yes | 13 (5.5) | 6 (2.3) | |
| AST (mean, SD), IU/l | 36.1 (34.7) | 28.8 (73.4) | 0.16 |
| ALT (mean, SD), IU/l | 33.3 (42.4) | 27.4 (72.5) | 0.28 |
| AFP (mean, SD), ng/ml | 6.57 (8.89) | 3.56 (5.16) | <0.0001 |
| Wen-HCC risk score (mean, SD) | 15.6 (7.2) | 11.1 (5.9) | <0.0001 |
| Hung-HCC risk score (mean, SD) |
15.3 (5.0) | 12.9 (5.6) | <0.0001 |
| Michikawa-HCC risk score (mean, SD) | 8.6 (3.1) | 7.1 (3.1) | <0.0001 |
The association of DNA methylation markers and HCC risk are presented in Table 2. In a multivariable logistic model adjusting for age at blood draw, HBsAg and anti-HCV status, the OR for those positive for TBX2 methylation compared to those with no methylation was 3.29 (95% CI = 1.80–6.04). In stratified data analysis, we found the association was mainly among subjects with either HBV or HCV infection; the OR was 5.35 (95% CI = 2.23–12.79) for positive for TBX2 methylation.
Table 2.
DNA methylation markers and HCC risk overall and by hepatitis virus status
| Methylation marker status | Cases | Controls | HBV/HCV infected | No HBV/HCV | |
|---|---|---|---|---|---|
| N = 237 | N = 257 | OR (95% CI)a | OR (95% CI)b | OR (95% CI)b | |
| CDKN2A | |||||
| Negative | 185 (78.7) | 203 (79.3) | 1.0 | 1.0 | 1.0 |
| Positive | 50 (21.3) | 53 (20.7) | 1.12 (0.61–2.07) | 0.73 (0.32–1.64) | 1.13 (0.34–3.83) |
| VIM | |||||
| Negative | 205 (86.9) | 230 (90.2) | 1.0 | 1.0 | 1.0 |
| Positive | 31 (13.1) | 25 (9.8) | 1.29 (0.64–2.60) | 1.01 (0.42–2.42) | 1.81 (0.42–7.80) |
| ZNF154 | |||||
| Negative | 89 (39.7) | 111 (43.2) | 1.0 | 1.0 | 1.0 |
| Positive | 135 (60.3) | 146 (56.8) | 1.14 (0.69–1.88) | 1.04 (0.56–1.93) | 1.32 (0.39–4.51) |
| RASSF1A | |||||
| Negative | 216 (91.1) | 240 (93.8) | 1.0 | 1.0 | 1.0 |
| Positive | 21 (8.9) | 16 (6.3) | 1.10 (0.45–2.65) | 1.81 (0.61–5.42) | 0.72 (0.11–4.69) |
| TBX2 | |||||
| Negative | 58 (24.5) | 106 (41.3) | 1.0 | 1.0 | 1.0 |
| Positive | 179 (75.5) | 151 (58.8) | 3.29 (1.80–6.04) | 5.35 (2.23–12.79) | 2.89 (0.76–11.03) |
| STEAP4,% | 19.9 (13.0) | 19.8 (13.1) | 1.00 (0.98–1.02) | 1.00 (0.98–1.02) | 1.00 (0.96–1.05) |
aAdjust for age, HBsAg and anti-HCV.
bAdjust for age.
When we divided subjects based on risk scores, we observed that the effect of TBX2 methylation was mainly among subjects with high risk: the ORs (95% CIs) were 7.85 (1.59–38.69) for Wen-HCC score ≥15, 5.89 (2.23–15.51) for Hung-HCC score ≥15 and 7.59 (2.21–26.07) for Michikawa-HCC ≥8 (Table 3). We then examined the associations of DNA methylation with HCC risk stratified by the difference in time between blood collection and HCC diagnosis (Supplementary Table 1, available at Carcinogenesis Online). HCC risk decreased with increasing time between sample collection and diagnosis with adjusted ORs (95% CI) of 4.97 (2.03–12.18), 3.91 (1.11–13.81) and 0.65 (0.15–2.74) for cases diagnosed with HCC within first year, between 2 and 5 years and more than 6 years after blood collection.
Table 3.
TBX2 methylation markers and HCC risk by risk scores
| Risk score | OR (95% CI) | |
|---|---|---|
| Wen-HCC score | ≥15 | 7.85 (1.59–38.69) |
| <15 | 1.39 (0.69–3.07) | |
| Hung-HCC score | ≥15 | 5.89 (2.23–15.51) |
| <15 | 1.91 (0.66–5.46) | |
| Michikawa-HCC score | ≥8 | 7.59 (2.21–26.07) |
| <8 | 2.24 (0.77–6.54) |
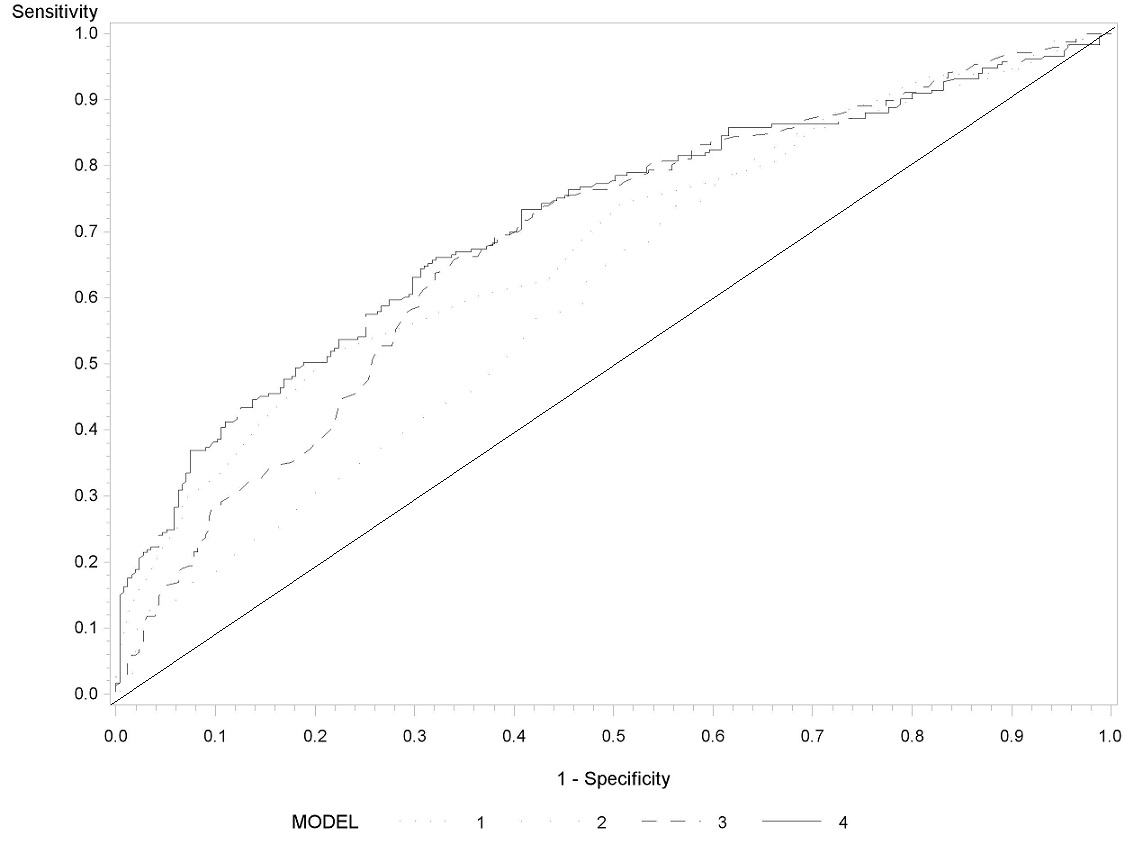
Figure 1 and Supplementary Figure 1, available at Carcinogenesis Online, show the ROC analysis for Wen-HCC risk score and TBX2 methylation. Including TBX2 methylation improved the precision of the risk model, with an area under the curve (AUC) of 68% for Wen-HCC risk score only improving to 71% for both Wen-HCC risk score and TBX2 methylation (Supplementary Figure 1, available at Carcinogenesis Online). We then focused on individuals with Wen-HCC risk score ≥15. The AUC was 76% (95% CI = 70–83%) for Wen-HCC risk score and TBX2 methylation, compared with 69% for Wen-HCC risk score alone (Figure 1).
Figure 1.

Receiver operating characteristics of Wen-HCC risk score and TBX2 methylation marker among subjects with Wen-HCC risk score ≥15. Wen-HCC risk score (model 1: AUC = 69%, 95% CI = 62–76%), TBX2 methylation (model 2: AUC = 66%, 95% CI = 58–74%), TBX2 methylation and HBV/HCV (Model 3: AUC = 68%, 95% CI = 60–76%), and Wen-HCC risk score, and TBX2 methylation (model 4: AUC = 76%, 95% CI = 70–83%).
Figure 2 presents the ROC for Hung-HCC risk score and DNA methylation marker. The AUCs were 66% (95% CI = 61–71%) for Hung-HCC risk score and TBX2 methylation, compared with 62% for Hung-HCC risk score (Supplementary Figure 2, available at Carcinogenesis Online). Among individuals with Hung-HCC risk score ≥16, the AUC with both Hung-HCC risk score and TBX2 methylation increased by 10% compared with AUC with Hung-HCC risk score alone (Figure 2). The AUCs were 67% (95% CI = 63–72%) for Michikawa-HCC risk score and TBX2 methylation, compared with 61% for Michikawa-HCC risk score (Supplementary Figure 3, available at Carcinogenesis Online). The corresponding AUC among individuals with Michikawa-HCC risk score ≥8 were 65 and 58%, respectively (Figure 3).
Figure 2.

Receiver operating characteristics of Hung-HCC risk score and TBX2 methylation marker among subjects with Hung-HCC risk score ≥15. Hung-HCC risk score (AUC = 53%, 95% CI = 47–60%), TBX2 methylation (AUC = 63%, 95% CI = 56–69%), TBX2 methylation and HBV/HCV (AUC = 64%, 95% CI = 57–71%) and Hung-HCC risk score and TBX2 methylation (AUC = 63%, 95% CI = 56–70%).
Figure 3.

Receiver operating characteristics of Michikawa-HCC risk score and DNA methylation marker among subjects with Michikawa-HCC risk score ≥8. Michikawa-HCC risk score (AUC = 58%, 95% CI = 52–65%), DNA methylation (AUC = 61%, 95% CI = 55–68%), TBX2 methylation and HBV/HCV (AUC = 64%, 95% CI = 58–71%) and Michikawa-HCC risk score, and DNA methylation (AUC = 65%, 95% CI = 58–71%).
Discussion
Hepatocellular carcinoma remains one of the leading causes of cancer mortality in the world (1). The early detection of cancer can significantly improve survival (40). However, the early detection of HCC presents a challenge because of the lack of specific biomarkers. AFP, the most commonly used biomarker for patients at risk for HCC, is weakly effective making the development of biomarkers that can help assess risk essential. Aberrant DNA methylation leads to altered gene expression, resulting in cancerous features (41). Emerging evidence, including ours, suggests that aberrant DNA methylation can begin very early in HCC (22,42) and can be detected in body fluids (22,25). In this study, we found that TBX2, a gene that we previously found hypermethylated in HCC tumor tissues is highly methylated in DNA from plasma collected from HCC patients before cancer diagnosis. Our findings from a prospective study provide additional evidence suggesting that DNA methylation shows strong potential for the noninvasive and early diagnosis of HCC.
Circulating cell-free DNA shed from the primary tumor tissue can be retrieved and tested for genetic and epigenetic alterations (43,44). Analyzing plasma DNA from HCC cases, we previously reported that 37–63% of cases had detectable hypermethylated DNA (≥5% methylation) for selected genes including CDKN2A, STEAP4 and ZNF154 that were hypermethylated in the corresponding HCC tumor samples (22). In our previous pilot study, we measured methylation levels in selected genes (CDKN2A, CDKN2B and RASSF1A) in serum DNA of 50 HCC patients who provided repeated blood samples before diagnosis and 50 controls and found methylation markers measured in blood up to 9 years prior to diagnosis (25). In our stratified analysis, we found the association of TBX2 methylation was mainly among patients diagnosis HCC within 5 years after blood collection. This evidence suggests the potential utility of these alterations as surrogate tumor markers.
Practice guidelines from the American Association of the Study of Liver Diseases and the European Association for the Study of the Liver have recommended HCC surveillance for patients at high risk of developing HCC, mainly patients with HBV/HCV or cirrhosis (45,46). The etiology of HCC is not fully understood, although chronic infection with HBV/HCV, resulting in cirrhosis, is the major risk factor (47). Currently, liver biopsy is considered the gold standard for the diagnosis of cirrhosis, but it has major limitations (48). HCC risk prediction models based on major risk factors of HCC can stratify people into different risk categories that can be used to tailor screening decisions and strategies for clinical management of an individual patient (49). Using information from a prospective case–control study nested within a community-based cohort with 16 years of follow-up, our study observed that these three HCC risk models for the general population only have modest discrimination capacity, with AUCs range from 62 to 68%. Integrating biomarkers such as genetic and epigenetic factors into risk models have improved models’ performance in predicting cancer risk (50,51). Evaluating circulating DNA TBX2 methylation levels, we found HCC risk was threefold higher among individuals positive for TBX2 methylation. Our results demonstrate that adding plasma DNA methylation to risk prediction models improves the discrimination for HCC, in particular among a high risk population, with up to a 10% improvement in the AUC in a model that includes Hung-HCC score and TBX2 methylation and 7% for Wen-HCC or Michikawa-HCC models and TBX2 methylation. Our finding suggests that using epigenetic information can improve risk prediction models, facilitate risk stratification and the identification of high risk individuals for cost-effective screening, surveillance and early detection of HCC.
The selection of the six genes analyzed was based on previous tumor studies that found more than 20% hypermethylation in HCC tumor than adjacent non-tumor tissues (34% for CDKN2A, 36% for RASSF1A, 23% for STEAP4, 36% for TBX2, 25% for VIM and 42% for ZNF154) (22,52). However, we found DNA hypermethylation only for TBX2 in plasma of HCC cases compared to controls. The tumor studies consisted of either HBV/HCV-related-HCC, while in the current study about 20% of cases are non-HBV/HCV. As non-alcoholic fatty liver disease (NAFLD) is a risk factor for HCC (53), it is important to use markers that can help in the early detection of all cases whatever the etiology. We observed that the accuracies of the risk models improved only modestly with incorporation of only TBX2 methylation data. More studies are need to determine if a panel of methylation markers related to all HCC etiologies can achieve a more reasonable sensitivity with high specificity in HCC detection. Unlike our pilot study, we did not observe hypermethylation in RASSF1A, CDKN2A (p16) and CDKN2B (p15). Differences in the distribution of risk factors in the control populations might partly explain the conflicting data. A prior study found higher methylation in RASSF1, among cases using controls with no HBV infection; controls with HBV also had a higher prevalence of RASSF1A (58%) methylation (54). In our current study, about 43% of controls are positive for HBsAg, while 20% of controls were positive for HBsAg in our prior study (25). In addition, different methodologies were used in our two studies. In the earlier study methylation specific PCR with gel analysis for detection of methylation was used while in the current study pyrosequencing was used for analysis of CDKN2A methylation and real time PCR for analysis of RASSF1A methylation. Although we did not include spiked-in control DNA to check for bisulfite conversion efficiency, a comparison of different kits including the EZ DNA methylation-Gold kit found high conversion efficiency in all tested kits; the conversion efficiency is 99.7% for EZ DNA methylation-Golden kit (55). In addition, several genes were analyzed in all DNAs with pyrosequencing which internally controls for C to T conversion by analysis of Cs in nonCpG sites as either C or T. All samples showed complete conversion. It is know that pyrosequencing is not that sensitive method compared to PCR-based methods; the detection levels of PCR-based methods is as low as 0.1%, and the sensitivity limitation for pyrosequencing is about 5% (56–58). As the tumor cell-free DNA (cfDNA) represents only small fraction of the total cfDNA (59,60), more studies of quantitative comparison of DNA methylation assays for circulating methylation markers are needed.
Another limitation in our study is we do not have information on stage at HCC diagnosis and it is unclear if TBX2 methylation marker can detect early stage of HCC. The AUC is one of the most common tools used for evaluating the discrimination ability of a risk prediction model (61). Compared to the increased OR in the high-risk groups, we found the diagnostic improvement of adding TBX2 to the ROC curve is only moderate. There have been concerns about poor sensitivity using the changes in AUC as AUC only slightly increases with the addition of a marker with a large OR (50,62–64). Other statistical measurements such as the integrated discrimination improvement and the continuous net reclassification improvement might better evaluate newly added markers in risk prediction model (65).
In this study, we used a prospective study design providing causal evidence for the application of circulating DNA methylation as a biomarker for early detection of HCC. With the largest sample size to date, we examined the associations of DNA methylation and HCC among high and low risk populations. In addition, using epidemiologic data that we collected at baseline, we were able to estimate HCC risk score based on HCC risk models and validate risk models in our population.
If replicated in larger studies, these results suggest that measuring circulating DNA methylation could have a significant impact on HCC risk assessment. Our finding for the improvement of AUCs in models incorporating circulating DNA biomarker, suggests the benefit of interrogating biomarkers in HCC risk assessment.
Supplementary material
Supplementary material is available at Carcinogenesis online.
Funding
This work was supported by National Institutes of Health grants R01ES005116, P30ES009089, and P30CA013696.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors thank the participants of Cancer Screening Program cohort for their contributions to the study.
Conflict of Interest Statement: None declared.
Abbreviations
- AFP
α-fetoprotein
- ALT
alanine transaminase
- anti-HCV
antibodies against hepatitis C virus
- AST
aspartate transaminase
- CI
confidence interval
- HBsAg
hepatitis B virus surface antigen
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- OR
odds ratio
- ROC
receiver operating characteristic
References
- 1. Ferlay J., et al. (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer, 127, 2893–2917. [DOI] [PubMed] [Google Scholar]
- 2. El-Serag H.B. (2011) Hepatocellular carcinoma. N. Engl. J. Med., 365, 1118–1127. [DOI] [PubMed] [Google Scholar]
- 3. Njei B., et al. (2015) Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology, 61, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fitzmorris P., et al. (2015) Surveillance and Diagnosis of Hepatocellular Carcinoma. Gastroenterol. Hepatol. (N. Y)., 11, 38–46. [PMC free article] [PubMed] [Google Scholar]
- 5. Omata M., et al. (2010) Asian Pacific Association for the Study of the Liver consensus recommendations on hepatocellular carcinoma. Hepatol. Int., 4, 439–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mittal S., et al. (2016) Effectiveness of surveillance for hepatocellular carcinoma in clinical practice: A United States cohort. J. Hepatol., 65, 1148–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee M.H., et al. ; R.E.V.E.A.L.-HBV Study Group. (2013) Prediction models of long-term cirrhosis and hepatocellular carcinoma risk in chronic hepatitis B patients: risk scores integrating host and virus profiles. Hepatology, 58, 546–554. [DOI] [PubMed] [Google Scholar]
- 8. Yuen M.F., et al. (2009) Independent risk factors and predictive score for the development of hepatocellular carcinoma in chronic hepatitis B. J. Hepatol., 50, 80–88. [DOI] [PubMed] [Google Scholar]
- 9. Yang H.I., et al. ; REACH-B Working Group. (2011) Risk estimation for hepatocellular carcinoma in chronic hepatitis B (REACH-B): development and validation of a predictive score. Lancet. Oncol., 12, 568–574. [DOI] [PubMed] [Google Scholar]
- 10. Yang H.I., et al. (2010) Nomograms for risk of hepatocellular carcinoma in patients with chronic hepatitis B virus infection. J. Clin. Oncol., 28, 2437–2444. [DOI] [PubMed] [Google Scholar]
- 11. Chang K.C., et al. (2012) A novel predictive score for hepatocellular carcinoma development in patients with chronic hepatitis C after sustained response to pegylated interferon and ribavirin combination therapy. J. Antimicrob. Chemother., 67, 2766–2772. [DOI] [PubMed] [Google Scholar]
- 12. El-Serag H.B., et al. (2014) A new laboratory-based algorithm to predict development of hepatocellular carcinoma in patients with hepatitis C and cirrhosis. Gastroenterology, 146, 1249–55.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang K.C., et al. (2013) Clinical-guide risk prediction of hepatocellular carcinoma development in chronic hepatitis C patients after interferon-based therapy. Br. J. Cancer, 109, 2481–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wong G.L., et al. (2013) Accuracy of risk scores for patients with chronic hepatitis B receiving entecavir treatment. Gastroenterology, 144, 933–944. [DOI] [PubMed] [Google Scholar]
- 15. Michikawa T., et al. ; Japan Public Health Center-based Prospective Study Group. (2012) Development of a prediction model for 10-year risk of hepatocellular carcinoma in middle-aged Japanese: the Japan Public Health Center-based Prospective Study Cohort II. Prev. Med., 55, 137–143. [DOI] [PubMed] [Google Scholar]
- 16. Wen C.P., et al. (2012) Hepatocellular carcinoma risk prediction model for the general population: the predictive power of transaminases. J. Natl. Cancer Inst., 104, 1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hung Y.C., et al. (2015) Development of risk scoring system for stratifying population for hepatocellular carcinoma screening. Hepatology, 61, 1934–1944. [DOI] [PubMed] [Google Scholar]
- 18. Chen C-J, et al. (2015) Hepatocellular carcinoma risk scores: ready to use in 2015? Hepatic Oncol., 2, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sherman M. (2005) Diagnosis of small hepatocellular carcinoma. Hepatology, 42, 14–16. [DOI] [PubMed] [Google Scholar]
- 20. Farinati F., et al. (2006) Diagnostic and prognostic role of alpha-fetoprotein in hepatocellular carcinoma: both or neither? Am. J. Gastroenterol., 101, 524–532. [DOI] [PubMed] [Google Scholar]
- 21. Marrero J.A., et al. (2009) Alpha-fetoprotein, des-gamma carboxyprothrombin, and lectin-bound alpha-fetoprotein in early hepatocellular carcinoma. Gastroenterology, 137, 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shen J., et al. (2012) Genome-wide DNA methylation profiles in hepatocellular carcinoma. Hepatology, 55, 1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shen J., et al. (2013) Exploring genome-wide DNA methylation profiles altered in hepatocellular carcinoma using Infinium HumanMethylation 450 BeadChips. Epigenetics, 8, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mah W.C., et al. (2014) DNA methylation: potential biomarker in hepatocellular carcinoma. Biomark. Res., 2, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y.J., et al. (2007) Predicting hepatocellular carcinoma by detection of aberrant promoter methylation in serum DNA. Clin. Cancer Res., 13, 2378–2384. [DOI] [PubMed] [Google Scholar]
- 26. Song M.A., et al. (2013) Elucidating the landscape of aberrant DNA methylation in hepatocellular carcinoma. PLoS One, 8, e55761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wen L., et al. (2015) Genome-scale detection of hypermethylated CpG islands in circulating cell-free DNA of hepatocellular carcinoma patients. Cell Res., 25, 1250–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lin Q., et al. (2005) Promoter hypermethylation of p16 gene and DAPK gene in sera from hepatocellular carcinoma (HCC) patients. Chin. J. Cancer Res., 17, 250–254. [Google Scholar]
- 29. Yeo W., et al. (2005) High frequency of promoter hypermethylation of RASSF1A in tumor and plasma of patients with hepatocellular carcinoma. Liver Int., 25, 266–272. [DOI] [PubMed] [Google Scholar]
- 30. Wong I.H., et al. (2003) Quantitative analysis of tumor-derived methylated p16INK4a sequences in plasma, serum, and blood cells of hepatocellular carcinoma patients. Clin. Cancer Res., 9, 1047–1052. [PubMed] [Google Scholar]
- 31. Chu H.J., et al. (2004) Detection of aberrant p16INK4A methylation in sera of patients with liver cirrhosis and hepatocellular carcinoma. J. Korean Med. Sci., 19, 83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chang H., et al. (2008) Methylation of tumor associated genes in tissue and plasma samples from liver disease patients. Exp. Mol. Pathol., 85, 96–100. [DOI] [PubMed] [Google Scholar]
- 33. Yamada N., et al. (2016) Genome-wide DNA methylation analysis in hepatocellular carcinoma. Oncol. Rep., 35, 2228–2236. [DOI] [PubMed] [Google Scholar]
- 34. Holmila R., et al. (2017) Targeted deep sequencing of plasma circulating cell-free DNA reveals vimentin and fibulin 1 as potential epigenetic biomarkers for hepatocellular carcinoma. PLoS One, 12, e0174265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee M.H., et al. ; R.E.V.E.A.L.-HCV Study Group. (2011) Community and personal risk factors for hepatitis C virus infection: a survey of 23,820 residents in Taiwan in 1991-2. Gut, 60, 688–694. [DOI] [PubMed] [Google Scholar]
- 36. Pan W-C., et al. (2015) Fine particle pollution, alanine transaminase, and liver cancer: a Taiwanese prospective cohort study (REVEAL-HBV). J. Natl. Cancer Inst., 108. doi:10.1093/jnci/djv341. [DOI] [PubMed] [Google Scholar]
- 37. Wu H.C., et al. (2007) Polycyclic aromatic hydrocarbon- and aflatoxin-albumin adducts, hepatitis B virus infection and hepatocellular carcinoma in Taiwan. Cancer Lett., 252, 104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu H.C., et al. (2007) Urinary 8-oxodeoxyguanosine, aflatoxin B1 exposure and hepatitis B virus infection and hepatocellular carcinoma in Taiwan. Carcinogenesis, 28, 995–999. [DOI] [PubMed] [Google Scholar]
- 39. Wu H.C., et al. (2008) Urinary 15-F2t-isoprostane, aflatoxin B1 exposure and hepatitis B virus infection and hepatocellular carcinoma in Taiwan. Carcinogenesis, 29, 971–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Singal A.G., et al. (2014) Early detection, curative treatment, and survival rates for hepatocellular carcinoma surveillance in patients with cirrhosis: a meta-analysis. PLoS Med., 11, e1001624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kulis M., et al. (2010) 2 - DNA methylation and cancer. In Zdenko H. and Toshikazu U (eds) Advances in Genetics. Academic Press, Atlanta, GA, pp. 27–56. [DOI] [PubMed] [Google Scholar]
- 42. Dong Y., et al. (2014) Aberrant DNA methylation in hepatocellular carcinoma tumor suppression (Review). Oncol. Lett., 8, 963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Elshimali Y.I., et al. (2013) The clinical utilization of circulating cell free DNA (CCFDNA) in blood of cancer patients. Int. J. Mol. Sci., 14, 18925–18958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aarthy R., et al. (2015) Role of circulating cell-free DNA in cancers. Mol. Diagn. Ther., 19, 339–350. [DOI] [PubMed] [Google Scholar]
- 45. European Association for the Study of the L et al. (2012) EASL–EORTC clinical practice guidelines: management of hepatocellular carcinoma. J. Hepatol., 56, 908–943. [DOI] [PubMed] [Google Scholar]
- 46. Bruix J., et al. (2001) Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona-2000 EASL eConference. J. Hepatol., 35, 421–430. [DOI] [PubMed] [Google Scholar]
- 47. Venook A.P., et al. (2010) The incidence and epidemiology of hepatocellular carcinoma: a global and regional perspective. Oncologist, 15 (Suppl. 4), 5–13. [DOI] [PubMed] [Google Scholar]
- 48. Afdhal N.H. (2004) Biopsy or biomarkers: is there a gold standard for diagnosis of liver fibrosis? Clin. Chem., 50, 1299–1300. [DOI] [PubMed] [Google Scholar]
- 49. Della Corte C., et al. (2016) Early diagnosis of liver cancer: an appraisal of international recommendations and future perspectives. Liver Int., 36, 166–176. [DOI] [PubMed] [Google Scholar]
- 50. Gail M.H. (2008) Discriminatory accuracy from single-nucleotide polymorphisms in models to predict breast cancer risk. J. Natl. Cancer Inst., 100, 1037–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu Z., et al. (2013) Epigenome-wide association study of breast cancer using prospectively collected sister study samples. J. Natl. Cancer Inst., 105, 694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shen J., et al. (2013) Exploration of genome-wide circulating microRNA in hepatocellular carcinoma: MiR-483-5p as a potential biomarker. Cancer Epidemiol. Biomarkers Prev., 22, 2364–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kolly P., et al. (2016) Surveillance for hepatocellular carcinoma in patients with NASH. Diagnostics, 6, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chan K.C., et al. (2008) Quantitative analysis of circulating methylated DNA as a biomarker for hepatocellular carcinoma. Clin. Chem., 54, 1528–1536. [DOI] [PubMed] [Google Scholar]
- 55. Holmes E.E., et al. (2014) Performance evaluation of kits for bisulfite-conversion of DNA from tissues, cell lines, FFPE tissues, aspirates, lavages, effusions, plasma, serum, and urine. PLoS One, 9, e93933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lamy P.J., et al. (2015) Next-generation genotyping by digital PCR to detect and quantify the BRAF V600E mutation in melanoma biopsies. J. Mol. Diagn., 17, 366–373. [DOI] [PubMed] [Google Scholar]
- 57. Kristensen L.S., et al. (2009) PCR-based methods for detecting single-locus DNA methylation biomarkers in cancer diagnostics, prognostics, and response to treatment. Clin. Chem., 55, 1471–1483. [DOI] [PubMed] [Google Scholar]
- 58. Wojdacz T.K., et al. (2007) Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res., 35, e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Diehl F., et al. (2008) Circulating mutant DNA to assess tumor dynamics. Nat. Med., 14, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fleischhacker M., et al. (2007) Circulating nucleic acids (CNAs) and cancer–a survey. Biochim. Biophys. Acta, 1775, 181–232. [DOI] [PubMed] [Google Scholar]
- 61. Pencina M.J., et al. (2008) Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat. Med., 27, 157–72; discussion 207. [DOI] [PubMed] [Google Scholar]
- 62. Grund B., et al. (2010) Analysis of biomarker data: logs, odds ratios, and receiver operating characteristic curves. Curr. Opin. HIV AIDS, 5, 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Garcia-Closas M., et al. (2014) Combined associations of genetic and environmental risk factors: implications for prevention of breast cancer. J. Natl. Cancer Inst., 106, dju305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pepe M.S., et al. (2004) Limitations of the odds ratio in gauging the performance of a diagnostic, prognostic, or screening marker. Am. J. Epidemiol., 159, 882–890. [DOI] [PubMed] [Google Scholar]
- 65. Pencina M.J., et al. (2012) Interpreting incremental value of markers added to risk prediction models. Am. J. Epidemiol., 176, 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.