

CD38 overexpression was found in human lung cancer cell lines and tissues. CD38 deletion inhibited oncogenesis in vitro and in vivo. It is expected that anti-CD38 treatment may have therapeutic potential in lung cancer.
Abstract
The ectodomain of the plasma membrane ectoenzyme CD38 functions as both an NAD glycohydrolase and an ADP-ribosyl cyclase by catalyzing, respectively, the conversion of NAD to nicotinamide and ADP-ribose or cyclic ADP-ribose. CD38 is attracting particular attention in cancer therapy. An anti-CD38 monoclonal antibody (daratumumab) was approved for treatment of patients with multiple myeloma. However, the role of CD38 in non-hematological malignancies has not been explored. Previously, we reported that ADP-ribose-acceptor hydrolase (ARH)-1 deficiency in mice was associated with tumor development. In the present study, we found that in wild-type and ARH1-deficient mice deletion of the CD38 gene reduced tumor formation. Significant reductions in tumor number were observed in lymphomas, adenocarcinomas and hemangio/histolytic sarcomas. Consistent with a role for CD38 in tumorigenesis, CRISPR/Cas9-based knockout of CD38 in A549 human adenocarcinoma cells inhibited anchorage-independent cell growth, cell invasion and xenograft growth in nude mice. CD38 mRNA and protein expression were evaluated in human lung cancer cell lines and in human lung cancer specimens. CD38 overexpression in tumor cells was identified in 11 of 27 patient samples. In addition, some human lung cancer cell lines had dramatically higher CD38 mRNA and protein expression than normal cells. Consistent with these observations, search of the Oncomine database showed that some human lung adenocarcinomas had higher CD38 mRNA levels compared to normal lung tissues. In total, our data are consistent with the conclusion that CD38 plays a role in murine and human lung tumorigenesis and that anti-CD38 treatment may have therapeutic potential in lung cancer.
Introduction
According to World Health Organization (WHO), every year about 1.50 million deaths globally can be attributed to lung cancer (1). In the United States, an estimated 158000 people died of lung cancer in 2015; approximately 80–85% of all cases of lung cancer are non-small-cell lung cancer (NSCLC) (2). When diagnosed at early stages, surgery with or without chemotherapy and radiation may be curative. However, most patients with NSCLC will experience relapse, and the 5-year survival rate has remained poor at 17.4% (2). Better understanding of lung cancer biology has led to the finding of more than 10 druggable targets in NSCLC, e.g. epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), ROS1, vascular endothelial growth factor receptor (VEGFR) (3–5). Targeted agents are becoming standard therapeutics for advanced lung cancers, and may improve progression-free survival rates and quality of life in highly selected patients (6–9). However, molecular drivers in a large proportion of NSCLC still have not been identified.
Mono-ADP-ribosylation is a posttranslational protein modification, which involves the transfer of the ADP-ribose moiety from β-nicotinamide adenine dinucleotide (NAD) to specific acceptors, e.g. arginine, proteins (10). ADP-ribosylation of arginine appears to be a reversible modification of proteins. ADP-ribosyltransferases (ARTs) catalyze the stereo-specific formation of α-anomeric ADP-ribosyl-arginine, while ADP-ribose-aceptor hydrolase 1 (ARH1) cleaves α-ADP-ribose-(arginine) protein, regenerating the unmodified protein (11,12). Thus far, several proteins have been reported to be ADP-ribosylated on arginine, with functional effects (13–15). Human neutrophil peptide 1, isolated from airways of patients with idiopathic pulmonary fibrosis and asthma, exhibits decreased antimicrobial and cytotoxic activities after ADP-ribosylation (14). In vitro ADP-ribosylation of P2X7 by ART2.2 in the presence of NAD leads to rapid apoptotic death of native murine T lymphocytes (15). Modification by cholera toxin of arginine on Gsα, the α subunit of Gs protein, inhibits its GTPase activity, while ARH1 counteracts these effects by de-ADP-ribosylating the protein (13). Modified Gsα is increased in intestinal loops of ARH1-deficient mice injected with cholera toxin, as is fluid accumulation and total ADP-ribosylarginine content (13). Maintenance of an ADP-ribosylation cycle also appears critical for proper regulation of cell proliferation and disruption is associated with tumor formation (16,17). For example, deletion of the ARH1 gene in mice promotes spontaneous tumor development and ARH1-knockout mouse embryonic fibroblasts (MEFs) exhibit malignant potential (17). Compared to their wild-type counterparts, ARH1KO MEFs grow faster, form more colonies and produce larger tumors in nude mice. Transformation of ARH1KO MEFs with wild-type ARH1 gene completely abolishes their neoplastic properties, while the gene encoding an enzymatic inactive mutant did not have these effects (17). ARH1KO MEFs transformed with mutant ARH1 genes, which were identified in tumors from ARH1 heterozygous mice and MEFs, exhibit reduced ARH1 catalytic activity compared to wild-type MEFs, with altered cell proliferation and clonogenic ability (16). Interestingly, ARH1 mutations observed in mouse tumors are similar in location to those found in human cancers according to COSMIC cancer database (16) and tend to be in exons encoding the catalytic site. These data are consistent with the hypothesis that a functional ARH1 gene suppresses tumor development.
CD38 is a multifunctional protein with several enzymatic activities (18). Using NAD as a substrate, CD38 catalyzes the formation of nicotinamide (NAM) and ADP-ribose; NAD can also be converted to cyclic ADP-ribose (cADPR) with release of NAM (19). Cyclic ADP-ribose can also be hydrolyzed to ADP-ribose (20). At acidic pH and in the presence of free nicotinic acid, CD38 catalyzes a base-exchange reaction that replaces the nicotinamide group of NADP with nicotinic acid to generate nicotinic acid adenine dinucleotide phosphate (NAADP) (21). Both cADPR and NAADP regulate calcium mobilization (21). CD38 also binds hyaluronic acid, a component of the extracellular matrix (22,23), and serves as a cell surface receptor for CD31, a member of the immunoglobulin (Ig) superfamily, which regulates leukocyte adhesion and transmigration (24). Signal cascades triggered by CD38 rely on its localization to lipid rafts and association with signaling complexes in immune cells (25). CD38 and the B-cell–receptor complex colocalize within membrane rafts and mediate survival signals in IL-2-treated B-cell chronic lymphocytic leukemia (CLL) cells (26).
CD38 expression has been extensively investigated in hematologic malignancies. Most malignant plasma cells in all stages of multiple myeloma (MM) strongly express CD38 (27), leading to the use of anti-CD38 monoclonal antibodies as a targeted therapy for patients with MM (28). CD38 is aberrantly expressed in retinoic acid-treated acute promyelocytic leukemia (APL) cells. Expression of CD38 enhances inflammatory cytokine production and apoptosis of lung endothelial cells (29,30). Increased expression of CD38 is associated with activation of CLL cells and more aggressive disease, e.g., shorter survival rates, higher incidence of lymphadenopathy, poor response to therapy (31). CD38 enzymatic activities are necessary for growth and trafficking of malignant cells in CLL (32). CD38+ CLL cells contain lower NAD and higher cADPR levels than CD38− cells. CLL cells with forced wild-type CD38 expression exhibit improved chemotactic responses, adhesion and MMP-9 secretion as well as being more aggressive in xenograft models than those with enzymatically inactive mutants (32). In breast cancer, the presence of CD38 in tumor cells appears to induce NAD+ hydrolysis, while NAD/NADH imbalance caused by NAD consumption may promote metastasis and disease progression (33).
The above evidence demonstrates that both the ARH1 and CD38 genes may play pivotal roles in regulating carcinogenesis. In the current report, we generated double-knockout mice to examine potential synergistic effects of CD38 and ARH1 in tumor development.
Material and methods
Generation of ARH1 and CD38 double-knockout mice
Animal protocol (ASP-H0172) was approved by the Animal Care and Use Committee (ACUC) of the National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD. ARH1 mice were backcrossed 10 times using C57BL/6J mice to generate ARH1KO mice (13). CD38KO mice of C57BL/6J background were prepared as described (34). ARH1/CD38 double KO mice were generated by The Jackson Laboratory using rederived C57BL/6J ARH1KO and CD38KO mice. In brief, female/male ARH1 heterozygous mice and male/female CD38 heterozygous mice pairs were used to produce compound ARH1/CD38 heterozygous mice. These F1 heterozygous mice were backcrossed to ARH1 or CD38 single heterozygous mice to create ARH1 homozygous/CD38 heterozygous mice, which were mated with each other to generate ARH1/CD38 double-homozygous mice. Double homozygous mouse matings were then used to expand and maintain this strain. Mouse genotypes were identified using DNA from tail cuts and Western blot analysis of mouse tissues.
Reagents
Rabbit antibodies against full-length recombinant mouse ARH1 were purchased from Cocalico Biological, Inc. (Reamstown, PA); rabbit monoclonal antibody against C-terminal region of human CD38 was from Abcam, Inc. (Cambridge, MA); FITC-conjugated mouse CD38 antibody from BD bioscience (San Jose, CA); and cell culture medium from Invitrogen Life Technologies (Paisley, United Kingdom). All other reagents were from Sigma unless stated otherwise.
Cell culture
Cell lines included in the study were obtained from American Type Culture Collection (ATCC, Manassas, VA). Frozen aliquots were used in experiments within 6 months of culture period, after the first thawing of the cells. All cell lines have been tested and authenticated. Lung cancer cell lines including A549, H2126, H2028, Calu-6, H358, H1299, H1734, H520, H82, SK-LU-1 and H841 were maintained in RPMI 1640 medium supplemented with 10% of fetal bovine serum and 1% of penicillin–streptomycin. Primary Small Airway Epithelial Cells (SAEC) were cultured in serum-free Airway Epithelial Cell Basal Medium. All cell lines were grown in a humidified atmosphere with 5% of CO2 at 37°C.
Clinical specimens
Human lung tumors samples were obtained from Dr. David Schrump of the Thoracic and GI Oncology Branch, National Cancer Institute, NIH. All human samples used in this study were approved for research by The NCI Institutional Review Board (protocol 06-C-0014) and written, informed consent was obtained from patients.
NAD+ measurement
Male C57/BL6J mice, 5–6 month of age, were used for NAD+ measurement. After euthanizing of mice with carbon dioxide, tissues were rapidly excised and immediately frozen in liquid nitrogen. NAD+ levels were measured using NAD/NADH Quantification Colorimetric Kit (BioVison) according to manufacturer’s instructions.
Immunohistochemistry and stained sample evaluation
Tissues were fixed with 4% paraformaldehyde, embedded in paraffin and cut into 5 μm sections. Deparaffinization was routinely performed using a Leica ST5010 Autostainer XL system. Antigen retrieval was performed in citrate buffer (pH 6.0) in a microwave. After blocking with 10% normal serum for 2 h, slides were incubated with primary anti-CD38 antibodies overnight at 4°C, then with 0.3% H2O2 in phosphate-buffered saline for 15 min to block endogenous peroxidase. The signal was detected using VECTASTAIN® ABC Kit and DAB Peroxidase (HRP) Substrate Kit (Vector Laboratories, CA). As negative controls, sections were stained either without primary antibody or with an isotype-matched IgG. CD38 expression was evaluated by senior pathologists who were blinded to the clinical outcomes. The stained lung samples were scored according to cell stained at each intensity. We divided the staining intensity of the cell into four categories, ranging from 0 to 3: No staining, 0; weak staining, 1 (light brown staining); intermediate staining, 2; strong staining, 3 (dark brown staining) (35).
Flow cytometry
Mice were euthanized via carbon dioxide and the spleens were rapidly harvested. Splenocytes were preincubated with anti-mouse CD16/32 (5 μg/ml; BD-Biosciences) and then stained with the following fluorochrome-conjugated antibodies: anti-CD19 (BD Biosciences), anti-CD38 (eBioscience) and Aqua live/dead kit (Life Technologies/Molecular Probes). Cells were analyzed with a FACSCanto II (BD Biosciences) and FlowJo v10.2 (FlowJo LLC).
SDS-PAGE and western blot
Tissues were homogenized in lysis buffer (2% SDS in 20 mM Tris–HCl, pH 7.4) and the lysates were subjected to Bis-Tris SDS-PAGE, followed by transfer to nitrocellulose membranes (Invitrogen). After blocking with 5% non-fat dry milk in TBS for 1 h at RT, the blots were incubated with primary antibody overnight at 4°C and then with secondary antibody. SuperSignal West Pico Chemiluminescent Substrate (PIERCE) was used for detection.
Quantitative real-time PCR (qRT-PCR)
For gene expression profiling, RNA was converted to cDNA using iScript cDNA synthesis kit (Bio-Rad). qRT-PCR was performed according to standard Taqman quantitative real-time RT-PCR protocol with commercial CD38 primers (ABI #Hs01120071_m1) (Thermo Fisher Scientific). cDNA from A549 was used to generate the relative standard curves for CD38 and β-actin. Samples were standardized by dividing the copy number of the target gene by that of the endogenous reference gene, β-actin.
Generation of stable CD38 knockout cell lines
CD38 CRISPR/Cas9 KO plasmid or control CRISPR/Cas9 Plasmid (Santa Cruz, TX) was transfected into A549 cell lines using DNA-InTM transfection reagent (VitaScientific, MD). The puromycin-N-acetyl transferase gene in the same vector provides selection in the presence of puromycin. Cell colonies resistant to puromycin were selected and seeded for expansion. CD38 protein level was determined by Western blot.
Cell proliferation assay
Cells (2 × 103) were seeded on 96-well plates. Cell numbers were determined at indicated times using CCK-8 kit (Dojindo, Japan) according to the manufacturer’s instructions by measuring absorbance at 450 nm (SpectraMax M5 Microplate Reader).
Colony formation in soft agar
Soft agar clonogenic assay was performed to assess anchorage-independent growth of A549 cells. Cells (2 × 103) in 0.5 ml of 0.375% (w/v) agar in 10% fetal bovine serum Dulbecco's modified Eagle's medium were added to a 24-well plate with each well containing 0.5 ml of 0.4% (w/v) agar (Difco) in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. Plates were incubated (37°C, 5% CO2) under standard conditions for 28 days before colony number was quantified microscopically (MZFL III; Leica).
Cell invasion assay
Cell invasion assays were performed in triplicate in 24 trans-well units with 8-µm filters coated with Matrigel (BD Biosciences). Each well was loaded with 1 × 105 cells. After incubation for 24 h, cells passing through the filters into bottom wells were fixed in 100% methanol and stained with crystal violet. Images were taken under microscope (IX51, Olympus) and cell numbers were counted using Image J.
Xenograft tumor growth in nude mice
Lung cancer cells (2 × 106) in 200 μl Dulbecco's modified Eagle's medium were harvested and injected subcutaneously into both female and male athymic mice (two injection sites per mice, five mice per group). Tumor growth was quantified twice a week with a digital caliper and tumor volume was calculated as length × width2 × 0.5. Mice were euthanized when tumor length reached 2 cm and study was stopped when more than half of the mice were sacrificed.
Statistical analysis
All statistical analyses were performed using GraphPad Prism. Significance was determined using Chi-square test and one-way ANOVA with post hoc Bonferroni test. Maximum tumor volume was used to compare tumor growth among cell lines. Data are presented as means ± SEM of values from the indicated number of experiments.
Results
Effects of CD38 on tumor formation in ARH1-deficient mice
As described previously (17), ARH1 deficiency contributes to spontaneous tumor formation in multiple organs. ARH1/CD38 double-knockout mice were generated to evaluate the effect of CD38 on tumorigenesis. In each instance, ARH1 and CD38 genotype and protein expression were confirmed (Figure 1A–C). CD38−/−, ARH1−/− and ARH1−/−/CD38−/− mice were viable and fertile.
Figure 1.

Generation of ARH1 and CD38 double-knockout mice. (A) Genotyping of mice. ARH1 primers amplified a 0.8-kb fragment from the wild-type allele and a 1.0-kb fragment from the knockout allele (solid arrow). CD38 primers amplified a 0.4-kb fragment from the wildtype allele and a 0.5-kb fragment from the knockout allele (dotted arrow). Two mice were used for each genotype. (B) CD38 expression by splenocytes from wild-type, ARH1−/−, CD38−/− and ARH1−/−/CD38−/− mice as assessed by flow cytometry. Data are representative of two independent experiments and are shown as histograms with an overlay of CD38 expression by wild-type splenocytes (gray peak). (C) ARH1 expression in mouse brain lysates detected by Western blot (n = 3).
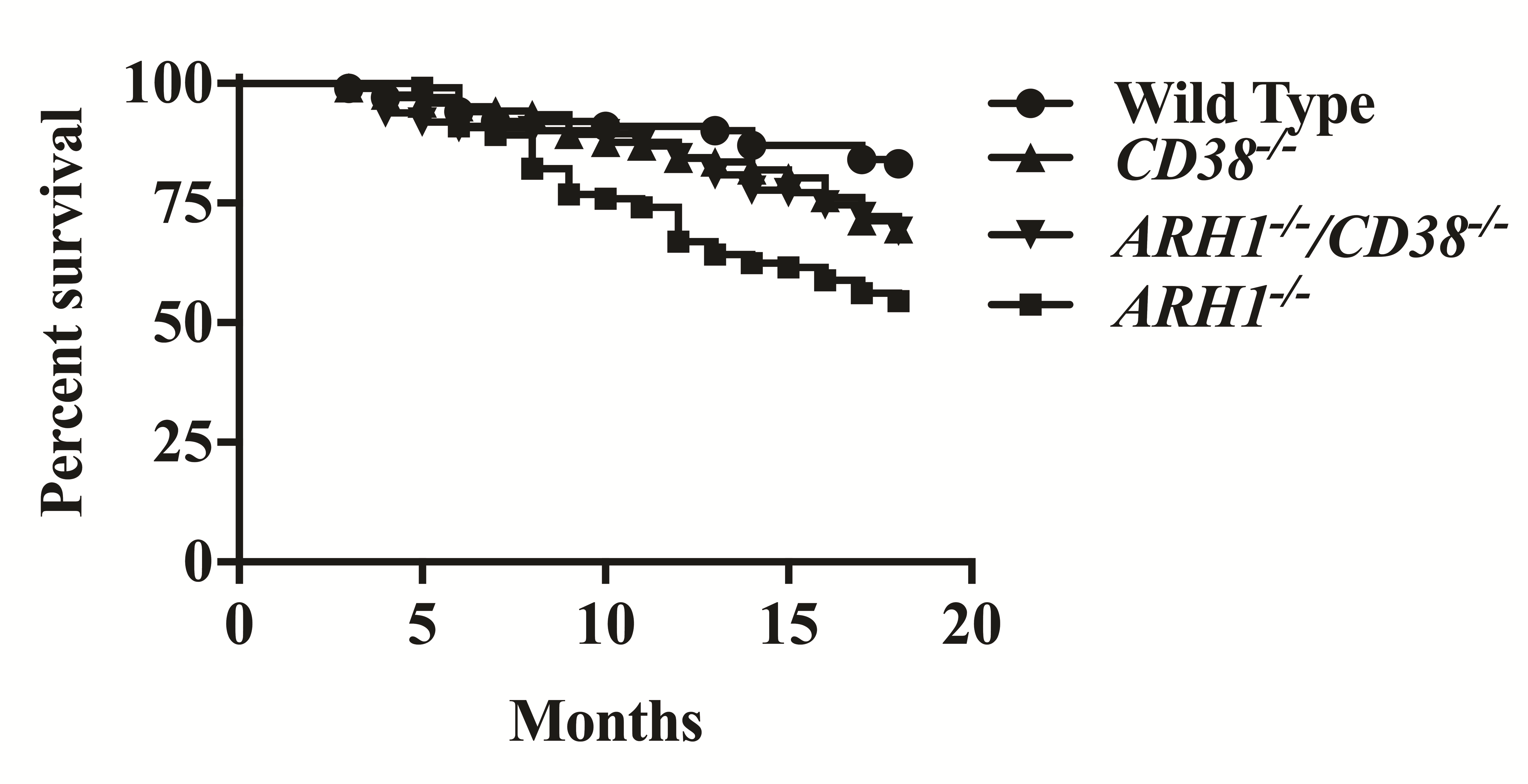
Tumor development in WT, ARH1−/−, CD38−/− and ARH1−/−/CD38−/− mice was monitored for as long as 18 months. As expected, based on prior studies (17), ARH1−/− mice developed a variety of malignancies (lymphoma, adenocarcinoma and hemangio/histolytic sarcoma) (Table 1). Tumors were seen in both male and female WT (0.99 versus 6.93%), ARH1−/− (3.57 versus 18.75%), CD38−/− (0.81% versus 0) and ARH1−/−/CD38−/− (1.23 versus 1.23%) mice. The total tumor occurrence was significantly higher in ARH1−/− mice than in their wild-type (WT) counterparts (22.32 versus 7.92%, P < 0.05). Both CD38−/− (0.81%) and ARH1−/−/CD38−/− (2.46%) showed significant reductions in spontaneous tumor formation compared to ARH1−/− and WT mice. Our results suggested that CD38 deletion suppressed tumor development in WT or ARH1−/− mice. In addition, survival of ARH1−/− mice was significantly shorter than that of wild-type, as well as CD38−/− and ARH1−/−/CD38−/− mice (Supplementary Figure 1, available at Carcinogenesis Online). CD38−/− and ARH1−/−/CD38−/− mice both has a shorter lifespan than wild-type mice, while no significant difference was found in their survival (Supplemental Figure 1, available at Carcinogenesis Online).
Table 1.
Effect of mouse genotype on incidence and type of spontaneous tumors
| Genotype | Number of mice | Incidence, n (%) | ||||
|---|---|---|---|---|---|---|
| Total | Lymphoma | Adenocarcinoma | Hemangiosarcoma | Histolytic sarcoma | ||
| Wild type | 101 | 8 (7.92) | 5 (4.46) | 0 (0.0) | 0 (0.0) | 3 (2.97) |
| ARH1 −/− | 112 | 25 (22.32)# | 13 (11.61) | 5 (4.46) | 2 (1.79) | 5 (4.46) |
| CD38 −/− | 123 | 1 (0.81)* | 1 (0.81) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| ARH1 −/−/CD38−/− | 162 | 4 (2.46)* | 2 (1.23) | 0 (0.0) | 0 (0.0) | 2 (1.23) |
Cumulative number and histopathological distribution of spontaneous tumors by genotype.
Pairwise comparisons of tumor incidence were considered as significant at P < 0.05. #P < 0.05 versus wild type, CD38−/− and double knockout; *P < 0.05 versus wild type.
CD38 deficiency upregulated NAD+ level in mice
CD38 is believed to be the major NAD glycohydrolase (NADase) in mammalian cells, as indicated by the markedly decreased NADase activity (34) and increased β-NAD (36) levels in CD38-deficient mice. We investigated the association between NAD level and CD38 deficiency. Compared to wild-type mice, CD38-/- mice as well as ARH1−/−/CD38−/− mice have, in multiple organs, significantly higher levels of NAD+ (Figure 2, P < 0.05, n = 3–5 for each group) including brain (342.7 ± 53.1%, 410.0 ± 50.49 versus 100 ± 18.1%), heart (232.4 ± 13.6%, 174.5 ± 10.85 versus 100 ± 20.7%) and lung (322.7 ± 89.6%, 292.4 ± 39.1 versus 100 ± 25.6%). No significant differences were found in NAD+ levels between wild-type and ARH1−/− mice (brain 75.2 ± 43.1%, heart 68.7 ± 10.1% and lung 98.9 ± 35.8%).
Figure 2.

Increased NAD+ level in CD38 knockout and ARH1/CD38 double knockout mice NAD+ levels were measured in mouse brain(A), heart (B) and lung (C) of wild-type, ARH1−/−, CD38−/− and ARH1-/-/CD38−/− mice. NAD+ levels are shown as percentage of those in wild-type mice (mean ± SEM, n = 3–5). *P < 0.05 versus WT, #P < 0.05 versus ARH1−/−.
CD38 expression by human lung carcinoma cell lines and specimens
We performed comprehensive examination of CD38 expression from transcript to protein level in both human lung cancer cell lines and paired human lung cancers and normal lung samples. A panel of lung cancer cell lines was examined for CD38 mRNA expression. As shown in Figure 3A, most lung cancer cell lines had higher copy numbers of CD38 mRNA than normal human respiratory epithelial cell lines. We next searched for CD38 and ARH1 gene expression using the Oncomine database (37). Most studies showed that CD38 mRNA levels were significantly upregulated in human lung carcinoma compared to normal lung tissues, while ARH1 mRNA was down-regulated significantly in patients with lung cancer (Supplemental Table 1, available at Carcinogenesis Online). Enhanced expression of CD38 protein in several lung cancer cell lines was confirmed by immunoblotting (Figure 3B). In addition, CD38 protein expression was examined in human lung carcinoma samples. The clinical information of patients used in the study is summarized in Supplemental Table 2, available at Carcinogenesis Online. Samples from 27 lung cancer patients, including 12 males and 15 females, were collected for analysis of CD38 expression. Compared to adjacent normal tissues, all the tumor tissues were infiltrated with strong CD38-positive cells such as macrophages, plasma cells and lymphocytes (Figure 3C). CD38 overexpression in tumors cells were detected in 11 of 27 patients, including adenocarcinoma and squamous cell carcinoma (Figure 3C, Supplemental Table 3, available at Carcinogenesis Online). These data together indicated that in general, CD38 was overexpressed in human lung carcinomas.
Figure 3.

CD38 expression in human lung cancer samples and cell lines. (A) Quantitative real-time RT-PCR analysis of CD38 mRNA expression in multiple non-small and small cell lung cancer cell lines. (B) Immunoblotting of CD38 in lung cancer cell lines. (C) Representative HE staining images show the typical morphology of lung adenocarcinoma and adjacent normal tissue. Serial sections from the same sample were used for immunohistochemically analysis of CD38. CD38-positive lymphocytes/macrophages (dotted arrow) were seen in all sections, with enlarged images in upper-right square. CD38-positive tumor cells (solid arrow) were detected only in some samples, enlarged images in bottom-right square. Scale bar: 50 μm. Experiment was repeated three times with similar results. HBEC, human bronchial epithelial cells; NHBE, normal human bronchial epithelial cells; SEAC, human small airway epithelial cells.
Knockout of CD38 inhibits lung cancer growth in vitro and in vivo
To explore the possible role of CD38 in human tumorigenesis, we generated CD38KO A549 cell lines using CD38 CRISPR/Cas9 KO plasmids. Protein expression was completely ablated in CD38KO A549 cells (Figure 4A). Two control, two knockout and parental cell lines were used for further experiments. Compared to A549 parental cells, cell proliferation did not differ in CD38KO cell lines (Figure 4B), while CD38 deletion led to reduced anchorage-independent colony formation in soft agar (Figure 4C and D) and suppressed serum-induced cell invasion in transwell invasion assays (Figure 4E and F). To further evaluate the role of CD38 in tumor progression, we injected the above cells subcutaneously into nude mice. Tumor xenograft growth of CD38-deficent cells (KO1 and KO2) were remarkably slower than those of control cell lines (Con1 and Con2) in both female (Figure 5A) and male (Figure 5B) nude mice. Based on the above results, we conclude that CD38 knockout may suppress tumor progression.
Figure 4.

Knockout of CD38 in A549 cells inhibited anchorage-independent growth and invasion. (A) CD38 expression was examined by Western blot in A549 cell lines after transfection with CD38 CRISPR/Cas9 KO plasmid or control CRISPR/Cas9 plasmid. (B) Cell proliferation assay; (C) typical images of A549 cells grown in soft agar and observed using an inverted microscope; (D) quantification of total colony area from triplicate wells of 24-well plates for each group. (E) Cell invasion ability in Transwell assay; (F) cell numbers calculated from at least three random fields. All experiments were repeated three times with similar results (mean ± SEM). *P < 0.05 versus Parental, Con1 and Con2, respectively.
Figure 5.

Knockout of CD38 in A549 cells suppressed in vivo tumorigenesis. Two control cell lines (Con1, Con2), 2 knockout cell lines (KO1, KO2) and parental cells, were injected subcutaneously into nude mice. After injection, the tumor masses were measured every two days until they reached 2 cm. Tumor growth curves of A549 cells in both female and male nude mice are presented in panels (A) and (B). (A) Multiple comparisons test showed that xenograft tumors generated by KO1 (◊) and KO2 (×) cells grew significantly more slowly than did parental (●), Con1 (▲) and Con2 (▼) cells in female nude mice (n = 5, P < 0.05). (B) Similarly, in male nude mice, tumor masses in the KO1 (◊) and KO2 (×) groups were also smaller than those of parental (●), Con1 (▲) and Con2 (▼) groups (n = 5, P < 0.05).
Discussion
Previously, it was shown that ARH1-deficient mice developed tumors (e.g. tumors in lung, liver, spleen and lymph nodes) much more frequently than did wild-type mice (17). Metastasis and multi-tumor mice were seen in more than 10% of ARH1−/− mice. Tumors occurred mostly within the age range of 6–16 months. The most frequently seen tumor type was lymphoma, followed by adeno- and hepatocellular- carcinomas. Survival of ARH1−/− mice was significantly shorter than that of ARH+/+ mice. In brief, these malignancies are diverse types, affect multiple organs/tissues and lead to death (17). Consistently, we found frequency of tumor development in ARH1−/− mice was three times higher than in wild-type mice.
Originally documented as a T cell activation/proliferation marker, CD38 at present is a multifunctional protein contributing to cancer progression (18). CD38-positive CLL cells have enhanced propensity to migrate in response to chemokine (C-X-C Motif) Ligand 12 (CXCL12), an essential chemokine in the recirculation of neoplastic cells between blood and lymphoid organs (38,39), which could be inhibited by blocking anti-CD38 monoclonal antibody (SUN-4B7) in vitro (40). With forced expression of wild-type CD38, Mec-1 cells, a CCL-like cell line, showed increased disease aggressiveness in NSG mice. However, this was not seen in cells carrying an enzymatic inactive mutant (31). Expansion of glioma cells was significantly attenuated in CD38-deficient mice compared to wild-type mice, and CD38 deficiency also prolonged the life span of glioma-bearing mice (41). Consistent with these results, we demonstrated here that tumors occurred less frequently in CD38−/− than in wild-type mice, and in CD38−/−/ARH1−/− than in ARH1−/− mice. Our results suggest that CD38 deficiency may directly inhibit tumor development in mice. ARH1 mutation seemed to affect cell cycle progression and led to more rapid proliferation (17). CD38 may induce G1 to S phase transition, as evidenced by induced S phase accumulation in cervical cancer cells with overexpressed CD38, and promote cell proliferation (42). CD38 overexpression also suppressed cell senescence and reduced apoptosis of cervical cancer cells (42). Therefore, tumor suppression in ARH1KO mice via CD38 knockout may result from dysregulation of cell cycle and cell apoptosis.
CD38 generates several compounds that regulate migration, cellular homeostasis and metabolism through consumption of extracellular NAD+ and generation of cyclic ADP-ribose and ADP-ribose (25). For example, CD38, through its production of cADPR and ADPR, regulates calcium signaling through G protein coupled chemokine receptors (43,44) and controls the migration of normal cells (45) as well as tumor cells (39) to both homeostatic and inflammatory chemokines. This may explain, at least in part, why the CD38-deficient tumor cells exhibited decreased migration in the serum invasion assays. Furthermore, we found that NAD+ levels were elevated in multiple tissues of CD38-deficient mice and mice deficient in both CD38 and ARH1 relative to WT controls. NAD+ is an essential compound for many enzymatic processes. Using biosensor, Cambronne et al. (46) could directly measure the NAD pool within subcellular compartments. Their results demonstrated that genetic deletion of single enzyme involved in NAD+ biosynthesis is sufficient to change NAD concentrations in nucleus, cytosol and mitochondria (46). How CD38 regulates the intracellular NAD+ level is unclear. Recently, Liu et al. (47) observed that a new type of CD38, topologically amenable to cytosolic regulation, is functionally active in producing cellular cADPR, and therefore maybe responsible for cytosolic NAD consumption. Elevated intracellular NAD may result in an environment that is unfavorable for tumor formation. Prior studies demonstrated that expression of yeast NADH dehydrogenase Ndi1 in human breast cancer cells increased NAD/NADH ratio and mitochondrial activity, inhibiting tumor growth and metastasis (33). Furthermore, strategies to enhance NAD+ content or NAD+/NADH ratio may inhibit cancer progression as knockdown of nicotinamide phosphoribosyltransferase (Nampt), which regulates NAD synthesis, renders breast cancer cells more aggressive (33). In contrast, in MMTV-PYMT mice, treatment with nicotinamide (NAM), an NAD+ precursor, inhibited tumor metastasis in xenograft models as well as spontaneous breast cancer progression (33). As reported recently, CD38 deficiency also upregulated mitochondrial function and NAD/NADH ratio, as well as enhancement of SIRT3 activity during aging in mice (48). Similar to our findings, CD38−/− and CD38−/−/SIRT3−/− mice have higher mitochondrial NAD+ levels compared to WT counterparts (48). Previous studies recognized SIRT3 as the major mitochondrial deacetylase that functions as a tumor suppressor (49). However, mechanisms underlying this role still need to be elucidated.
CD38 protein is poorly expressed on most mature resting lymphocytes, while increased expression of CD38 is reported in a subset of hematological malignancies (50), particularly on the surface of plasma cells of patients with MM. Majority of MM cells were CD38 positive and this immunophenotype did not differ between previously treated and untreated patients (27). Here, we performed a comprehensive examination of CD38 transcripts and protein expression in human lung cancer. CD38 mRNA and protein levels in certain lung cancer cells was much higher than in normal cells. Immunostaining of human lung cancer tissues showed that CD38 overexpression in tumor cells were detected in 11 of 27 samples. Data from Oncomine database confirmed that the CD38 mRNA level was significantly upregulated in lung cancer patients. In contrast, ARH1 mRNA level was down-regulated, which is consistent with the previous report that ARH1 might function as a tumor suppressor gene.
CD38 has been proposed as a biomarker of poor prognosis in CLL (51). Based on the percentage of CD38+ leukemic cells within a CLL clone, CLL patients were classified into CD38-positive and -negative groups, with the cutoff value between 20% and 30% (31,52). CD38+ CLL patients had shorter event-free survival and overall survival and required continuous chemotherapy (51,53). Strong expression of CD38 is also correlated with poor outcome of patients with natural killer/T cell lymphoma (54). Based on the above, we propose that CD38 overexpression may be a factor in predicting lung cancer prognosis.
Further, inflammatory cells are commonly seen in the vicinity or admixed with human lung cancer. CD38 overexpression is common in several immunosuppressive cell types in human lung cancer (55,56). Consistently, CD38-positive lymphocytes were abundant in all tumor sections as shown in our study. Regulatory T cells (Tregs) are known to have host-immune dampening effects in many tumors and are associated with increased frequency of tumor recurrence (57). CD38-high Tregs possessed greater immunosuppressive activity than CD38-low Tregs (56). CD38 has been identified as a novel marker for myeloid-derived suppressor cells (MDSCs), which possess enhanced capacity to suppress activated T cells as a result of increased inducible nitric oxide synthase (iNOS) production, thereby promoting tumor growth (55). CD38 expression can be activated by several cytokines including IFNγ, TNFα, IL-6, insulin-like growth factor-binding protein 3 (IGFBP3) and Chemokine (C-X-C motif) ligand 16 (CXCL16), which are often produced during chronic inflammation and are essential to establish an immunosuppressive microenvironment within tumors (25,55). CD38-positive MDSCs expanded in the peripheral blood of advanced stage lung cancer (55). Overall, CD38-high cells in lung cancer may provide a microenvironment for tumor cell survival and are suitable for targeted treatment.
Cancer immunotherapy with antibodies has shown increased promise in a number of malignances following advances in technologies for the development of therapeutic antibodies (3,58). Overexpression of CD38 in a majority of malignant plasma cells of MM patients made it a reasonable target for cancer immunotherapy, which resulted in the successful application and approval of the anti-CD38 monoclonal antibody daratumumab (DARA) for treatment of this disease (28). DARA is efficient in killing MM cells by enhancing antibody-dependent, cell-mediated cytotoxicity, complement-dependent cytotoxicity and macrophage-mediated phagocytosis (28). Besides directly annihilating the tumor cells, cross-linking with anti-CD38 antibody impairs the survival of CD38-positive MDSCs that are increased in cancer and thereby suppresses tumor progression (55). Similarly, we propose that CD38 may be involved in regulating growth of human lung cancers. Consistent with this model, CD38 knockout in A549 cells lead to decreased invasiveness, reduction of clonogenicity as well as reduced tumor growth in nude mice.
Considering the expression and role of CD38 in lung cancer, an anti-CD38 monoclonal antibody might hold great promise for treatment of solid malignancies, especially for those selected based on CD38 expression. However, CD38 is ubiquitously expressed and potential adverse effects caused by anti-CD38 therapy should be carefully considered. A phase II trial (NCT01985126) of daratumumab in MM showed that in patients who received a median of five prior lines of therapy the overall response rate to DARA was 29.2% and the median duration of response was 7.4 months (58). The most frequently reported adverse events included fatigue, nausea, back pain, pyrexia, cough and upper respiratory tract infection (58). Serious adverse reactions were reported in 33% of patients, with the most frequent serious adverse reactions being pneumonia (6%), general physical health deterioration (3%) and pyrexia (3%) (58). Therefore, in patients with MM, anti-CD38 therapy (DARA) approached its therapeutic goals with acceptable adverse effects (58).
In summary, we demonstrate that CD38 deletion inhibited oncogenesis in vitro and in vivo. CD38 overexpression was found in human lung cancer cells lines and tissues. Due to the encouraging effects of DARA in MM, it is expected that CD38 monoclonal antibodies might improve the prognosis of lung cancer in patients that overexpress CD38.
Supplementary material
Supplementary data can be found at Carcinogenesis online.
{kind=link}
Funding
This research was supported by the Intramural Research Program of the National Institutes of Health, National Heart, Lung, and Blood Institute and National Cancer Institute.
Acknowledgments
We thank Dr. Zu-Xi Yu, NHLBI Pathology Core, for guidance on the immunohistochemistry and Betty Mousseau and Sara L. Stone for assistance with flow cytometry.
Conflict of Interest Statement: None declared.
Abbreviations
- ARH
ADP-ribose-acceptor hydrolase
- CLL
chronic lymphocytic leukemia
- DARA
daratumumab
- MEF
mouse embryonic fibroblasts
- MM
multiple myeloma
- NSCLC
non-small-cell lung cancer
- NAD
nicotinamide adenine dinucleotide
References
- 1. Torre L.A., et al. (2015)Global cancer statistics, 2012. CA. Cancer J. Clin., 65, 87–108. [DOI] [PubMed] [Google Scholar]
- 2. Howlader N., et al. (2016)SEER Cancer Statistics Review, 1975–2013. Bethesda MD: National Cancer Institute. [Google Scholar]
- 3. Adams G.P., et al. (2005)Monoclonal antibody therapy of cancer. Nat. Biotechnol., 23, 1147–1157. [DOI] [PubMed] [Google Scholar]
- 4. Minguet J., et al. (2016)Targeted therapies for treatment of non-small cell lung cancer–Recent advances and future perspectives. Int. J. Cancer, 138, 2549–2561. [DOI] [PubMed] [Google Scholar]
- 5. Ye M., et al. (2016)ALK and ROS1 as targeted therapy paradigms and clinical implications to overcome crizotinib resistance. Oncotarget, 7, 12289–12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cufer T., et al. (2013)Systemic therapy of advanced non-small cell lung cancer: major-developments of the last 5-years. Eur. J. Cancer, 49, 1216–1225. [DOI] [PubMed] [Google Scholar]
- 7. Polanski J., et al. (2016)Quality of life of patients with lung cancer. Onco. Targets. Ther., 9, 1023–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saito M., et al. (2016)Gene aberrations for precision medicine against lung adenocarcinoma. Cancer Sci., 107, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tsao A.S., et al. (2016)Scientific advances in lung cancer 2015. J. Thorac. Oncol., 11, 613–638. [DOI] [PubMed] [Google Scholar]
- 10. Laing S., et al. (2011)ADP-ribosylation of arginine. Amino Acids, 41, 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moss J., et al. (1985)Reversibility of arginine-specific mono(ADP-ribosyl)ation: identification in erythrocytes of an ADP-ribose-L-arginine cleavage enzyme. Proc. Natl. Acad. Sci. USA, 82, 5603–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Okazaki I.J., et al. (1996)Mono-ADP-ribosylation: a reversible posttranslational modification of proteins. Adv. Pharmacol., 35, 247–280. [DOI] [PubMed] [Google Scholar]
- 13. Kato J., et al. (2007)Enhanced sensitivity to cholera toxin in ADP-ribosylarginine hydrolase-deficient mice. Mol. Cell. Biol., 27, 5534–5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paone G., et al. (2002)ADP ribosylation of human neutrophil peptide-1 regulates its biological properties. Proc. Natl. Acad. Sci. USA, 99, 8231–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seman M., et al. (2003)NAD-induced T cell death: ADP-ribosylation of cell surface proteins by ART2 activates the cytolytic P2X7 purinoceptor. Immunity, 19, 571–582. [DOI] [PubMed] [Google Scholar]
- 16. Kato J., et al. (2015)Mutations of the functional ARH1 allele in tumors from ARH1 heterozygous mice and cells affect ARH1 catalytic activity, cell proliferation and tumorigenesis. Oncogenesis, 4, e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kato J., et al. (2011)ADP-ribosylarginine hydrolase regulates cell proliferation and tumorigenesis. Cancer Res., 71, 5327–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Quarona V., et al. (2013)CD38 and CD157: a long journey from activation markers to multifunctional molecules. Cytometry B. Clin. Cytom., 84, 207–217. [DOI] [PubMed] [Google Scholar]
- 19. Schuber F., et al. (2004)Structure and enzymology of ADP-ribosyl cyclases: conserved enzymes that produce multiple calcium mobilizing metabolites. Curr. Mol. Med., 4, 249–261. [DOI] [PubMed] [Google Scholar]
- 20. Howard M., et al. (1993)Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science, 262, 1056–1059. [DOI] [PubMed] [Google Scholar]
- 21. Aarhus R., et al. (1995)ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J. Biol. Chem., 270, 30327–30333. [DOI] [PubMed] [Google Scholar]
- 22. Lund F.E., et al. (1999)CD38 signaling in B lymphocytes is controlled by its ectodomain but occurs independently of enzymatically generated ADP-ribose or cyclic ADP-ribose. J. Immunol., 162, 2693–2702. [PubMed] [Google Scholar]
- 23. Nishina H., et al. (1994)Cell surface antigen CD38 identified as ecto-enzyme of NAD glycohydrolase has hyaluronate-binding activity. Biochem. Biophys. Res. Commun., 203, 1318–1323. [DOI] [PubMed] [Google Scholar]
- 24. Newman P.J. (1999)Switched at birth: a new family for PECAM-1. J. Clin. Invest., 103, 5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Malavasi F., et al. (2008)Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev., 88, 841–886. [DOI] [PubMed] [Google Scholar]
- 26. Deaglio S., et al. (2003)CD38 is a signaling molecule in B-cell chronic lymphocytic leukemia cells. Blood, 102, 2146–2155. [DOI] [PubMed] [Google Scholar]
- 27. Lin P., et al. (2004)Flow cytometric immunophenotypic analysis of 306 cases of multiple myeloma. Am. J. Clin. Pathol., 121, 482–488. [DOI] [PubMed] [Google Scholar]
- 28. Laubach J.P., et al. (2014)Daratumumab granted breakthrough drug status. Expert Opin. Investig. Drugs, 23, 445–452. [DOI] [PubMed] [Google Scholar]
- 29. Drach J., et al. (1994)Retinoic acid-induced expression of CD38 antigen in myeloid cells is mediated through retinoic acid receptor-alpha. Cancer Res., 54, 1746–1752. [PubMed] [Google Scholar]
- 30. Gao Y., et al. (2007)Retinoic acid-induced CD38 antigen promotes leukemia cells attachment and interferon-gamma/interleukin-1beta-dependent apoptosis of endothelial cells: implications in the etiology of retinoic acid syndrome. Leuk. Res., 31, 455–463. [DOI] [PubMed] [Google Scholar]
- 31. Malavasi F., et al. (2011)CD38 and chronic lymphocytic leukemia: a decade later. Blood, 118, 3470–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vaisitti T., et al. (2015)The enzymatic activities of CD38 enhance CLL growth and trafficking: implications for therapeutic targeting. Leukemia, 29, 356–368. [DOI] [PubMed] [Google Scholar]
- 33. Santidrian A.F., et al. (2013)Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J. Clin. Invest., 123, 1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cockayne D.A., et al. (1998)Mice deficient for the ecto-nicotinamide adenine dinucleotide glycohydrolase CD38 exhibit altered humoral immune responses. Blood, 92, 1324–1333. [PubMed] [Google Scholar]
- 35. Avilés-Salas A., et al. (2017)Reproducibility of the EGFR immunohistochemistry scores for tumor samples from patients with advanced non-small cell lung cancer. Oncol. Lett., 13, 912–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Young G.S., et al. (2006)Decreased cADPR and increased NAD+ in the Cd38-/- mouse. Biochem. Biophys. Res. Commun., 346, 188–192. [DOI] [PubMed] [Google Scholar]
- 37. Rhodes D.R., et al. (2004)ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia, 6, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Burger J.A., et al. (2006)CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood, 107, 1761–1767. [DOI] [PubMed] [Google Scholar]
- 39. Deaglio S., et al. (2007)CD38 and ZAP-70 are functionally linked and mark CLL cells with high migratory potential. Blood, 110, 4012–4021. [DOI] [PubMed] [Google Scholar]
- 40. Vaisitti T., et al. (2010)CD38 increases CXCL12-mediated signals and homing of chronic lymphocytic leukemia cells. Leukemia, 24, 958–969. [DOI] [PubMed] [Google Scholar]
- 41. Levy A., et al. (2012)CD38 deficiency in the tumor microenvironment attenuates glioma progression and modulates features of tumor-associated microglia/macrophages. Neuro. Oncol., 14, 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liao S., et al. (2017)CD38 enhances the proliferation and inhibits the apoptosis of cervical cancer cells by affecting the mitochondria functions. Mol Carcinog., 56, 2245–2257. [DOI] [PubMed] [Google Scholar]
- 43. Deshpande D.A., et al. (2017)CD38 in the pathogenesis of allergic airway disease: Potential therapeutic targets. Pharmacol. Ther., 172, 116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shi G., et al. (2007)Identification of an alternative G{alpha}q-dependent chemokine receptor signal transduction pathway in dendritic cells and granulocytes. J. Exp. Med., 204, 2705–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Partida-Sánchez S., et al. (2001)Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat. Med., 7, 1209–1216. [DOI] [PubMed] [Google Scholar]
- 46. Cambronne X.A., et al. (2016)Biosensor reveals multiple sources for mitochondrial NAD⁺. Science, 352, 1474–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu J., et al. (2017)Cytosolic interaction of type III human CD38 with CIB1 modulates cellular cyclic ADP-ribose levels. Proc Natl Acad Sci USA, 114, 8283–8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Camacho-Pereira J., et al. (2016)CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab., 23, 1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chalkiadaki A., et al. (2015)The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer, 15, 608–624. [DOI] [PubMed] [Google Scholar]
- 50. van de Donk N.W., et al. (2016)Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol Rev, 270, 95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Damle R.N., et al. (1999)Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood, 94, 1840–1847. [PubMed] [Google Scholar]
- 52. Dürig J., et al. (2002)CD38 expression is an important prognostic marker in chronic lymphocytic leukaemia. Leukemia, 16, 30–35. [DOI] [PubMed] [Google Scholar]
- 53. Chevallier P., et al. (2002)CD38 expression and secondary 17p deletion are important prognostic factors in chronic lymphocytic leukaemia. Br. J. Haematol., 116, 142–150. [DOI] [PubMed] [Google Scholar]
- 54. Wang L., et al. (2015)CD38 expression predicts poor prognosis and might be a potential therapy target in extranodal NK/T cell lymphoma, nasal type. Ann. Hematol., 94, 1381–1388. [DOI] [PubMed] [Google Scholar]
- 55. Karakasheva T.A., et al. (2015)CD38-expressing myeloid-derived suppressor cells promote tumor growth in a murine model of esophageal cancer. Cancer Res., 75, 4074–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Patton D.T., et al. (2011)The PI3K p110δ regulates expression of CD38 on regulatory T cells. PLoS One, 6, e17359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ganesan A.P., et al. (2013)Tumor-infiltrating regulatory T cells inhibit endogenous cytotoxic T cell responses to lung adenocarcinoma. J. Immunol., 191, 2009–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McKeage K. (2016)Daratumumab: first global approval. Drugs, 76, 275–281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.