

Abstract
The heavy metal nickel is a known carcinogen, and occupational exposure to nickel compounds has been implicated in human lung and nasal cancers. Unlike many other environmental carcinogens, however, nickel does not directly induce DNA mutagenesis, and the mechanism of nickel-related carcinogenesis remains incompletely understood. Cellular nickel exposure leads to signaling pathway activation, transcriptional changes and epigenetic remodeling, processes also impacted by hypoxia, which itself promotes tumor growth without causing direct DNA damage. One of the mechanisms by which hypoxia contributes to tumor growth is the generation of genomic instability via down-regulation of high-fidelity DNA repair pathways. Here, we find that nickel exposure similarly leads to down-regulation of DNA repair proteins involved in homology-dependent DNA double-strand break repair (HDR) and mismatch repair (MMR) in tumorigenic and non-tumorigenic human lung cells. Functionally, nickel induces a defect in HDR capacity, as determined by plasmid-based host cell reactivation assays, persistence of ionizing radiation-induced DNA double-strand breaks and cellular hypersensitivity to ionizing radiation. Mechanistically, we find that nickel, in contrast to the metalloid arsenic, acutely induces transcriptional repression of HDR and MMR genes as part of a global transcriptional pattern similar to that seen with hypoxia. Finally, we find that exposure to low-dose nickel reduces the activity of the MLH1 promoter, but only arsenic leads to long-term MLH1 promoter silencing. Together, our data elucidate novel mechanisms of heavy metal carcinogenesis and contribute to our understanding of the influence of the microenvironment on the regulation of DNA repair pathways.
Carcinogenic nickel leads to transcriptional repression of the DNA repair pathways of homologous recombination and mismatch repair in tumorigenic and non-tumorigenic human lung cells in a manner analogous to hypoxic stress, potentially contributing to nickel-induced carcinogenesis.
Introduction
Nickel and certain other metals, including arsenic, chromium and cadmium, are established human carcinogens (1). Exposure to nickel primarily occurs via inhalation in industrial workers mining, processing and producing nickel-containing products, though exposure in the general population can also occur via oral consumption of contaminated water or skin contact with consumer products (2). Occupational exposure to nickel is a significant risk factor for cancers of the respiratory system, with epidemiologic studies demonstrating 3-fold and 18-fold increases in the rates of lung and sinonasal cancers, respectively, in nickel-exposed workers (3–5). In animal studies, inhalation of nickel has also been shown to cause lung carcinomas, and injection with nickel particles leads to the growth of sarcomas and liver tumors (1). Nickel exists as water-soluble and water-insoluble compounds, which both can enter cells via ion transporters and phagocytosis, respectively. In general, the carcinogenicity of nickel compounds correlates with the accumulation of intracellular nickel ions (Ni2+), which are therefore thought to be the active molecule (2).
Despite its carcinogenic effects, nickel does not form DNA adducts and demonstrates very low or no mutagenicity in most mutational assays. Instead, the carcinogenicity of nickel has been largely attributed to its effects on epigenetic modifications and gene expression through the inhibition of iron- and 2-oxoglutarate-dependent dioxygenases, including hypoxia-inducible factor (HIF) prolyl hydroxylases and Jumonji-domain-containing histone demethylases (reviewed in (6–8)). Nickel ions deplete intracellular iron by blocking membrane ion transporters and can also displace iron from the active site of dioxygenase enzymes, inhibiting their catalytic activity (9,10). Importantly, iron- and 2-oxoglutarate-dependent dioxygenases also require molecular oxygen to catalyze their oxidation reactions, so hypoxia similarly leads to inhibition of their activity. Thus, many of the transcriptional and epigenetic changes induced by nickel exposure are related to those induced by hypoxic stress, which similarly promotes cancer without direct mutagenesis.
HIF prolyl hydroxylases act on the HIF α-subunits, modifying specific proline residues, which allows their recognition by the von Hippel–Lindau (VHL) protein subunit of an E3 ubiquitin ligase complex with subsequent polyubiquitination and proteasomal degradation. Inhibition of the HIF prolyl hydroxylases leads to stabilization of HIF α-subunits, their dimerization with a constitutively expressed β-subunit, and transcriptional co-regulation of hypoxia-response genes. Therefore, nickel, like hypoxia, induces the expression of genes involved in glucose metabolism, angiogenesis, and cell growth (11,12).
The Jumonji-domain-containing histone demethylases remove methyl groups from lysine and arginine residues in histone tails, which can promote either transcription activation or repression depending on the specific residue. In vitro nickel treatment has been shown to lead to global cellular increases in methylation at histone H3 lysines 4, 9 and 36 (H3K4, H3K9 and H3K36) and decreases in histone lysine acetylation (13–15). In vivo, differences in global histone modifications and DNA methylation have been reported in nickel-exposed workers compared to unexposed controls (16–18). Nickel, likely, also induces gene-specific epigenetic changes, as activating histone marks such as H3K4 di- and tri-methylation (H3K4me2/3) and H3K9 acetylation (H3K9ac) and repressive histone marks such as H3K9 di-and tri-methylation (H3K9me2/3) localize to different regions within the nucleus (15). Nickel-induced epigenetic changes have also been shown to silence a gpt transgene and regulate expression of the endogenous Spry2 gene (13,19). Hypoxia similarly leads to increased H3K4me2/3 and H3K9me2/3 and decreased H3K9ac, with gene-specific modifications correlating with transcriptional regulation (19–21).
Several transcriptional and epigenetic targets of nickel have been discovered that may contribute to carcinogenesis, including Spry2 and the hypoxia-response genes that promote angiogenesis, metabolic reprogramming and tumor growth. An additional mechanism by which hypoxia contributes to tumorigenesis is the repression of cellular DNA repair pathways (reviewed in (22)). Specifically, hypoxic stress leads to transcriptional down-regulation of the homology-dependent DNA double-strand break repair (HDR) proteins, BRCA1, RAD51 and FANCD2 and the mismatch repair (MMR) proteins MLH1 and MSH2 and can generate stable silencing of the BRCA1 and MLH1 promoters (23–28). Other high-fidelity pathways of DNA repair including nucleotide excision repair (NER) and base excision repair (BER) are also reduced under hypoxic conditions by transcriptional or translational mechanisms, while the error-prone DNA double-strand break repair pathway of non-homologous end joining (NHEJ) is not a regular target of hypoxic stress (23,29,30). Hypoxia-induced DNA repair deficiencies generate genomic instability, which leads to more aggressive cancer behavior.
The metalloid arsenic, implicated in human lung, skin, liver, bladder and prostate cancer, has functional similarities to nickel and potentially hypoxia (reviewed in (6–8)). Like nickel and hypoxia, arsenic has very low mutagenic activity despite its high transforming potential, and it induces global and gene-specific changes in histone modifications and DNA methylation, leading to altered gene expression patterns that are believed to drive tumorigenesis (15,31,32). For example, arsenic exposure has been shown to lead to promoter hypermethylation of p53, p16INK4a and RASSF1A tumor suppressor genes and, conversely, to transcriptional up-regulation of the FOXM1, CDC6, CDC25A and Cyclin D1 oncogenes (33–35). Regarding the hypoxia-response pathway, arsenic has been shown to lead to up-regulation of HIF-1 and VEGF, likely through the induction of reactive oxygen species and activation of PI3K/AKT and MAPK/ERK signaling pathways (36–41). However, arsenic-induced VEGF expression has been shown to be independent of HIF-1 and arsenic-stabilized HIF-1 may not be completely functional, as it was unable to activate transcription of reporter constructs (38,41–43), suggesting that nickel and arsenic may overlap with hypoxia in different ways.
Given the similar transcriptional and epigenetic changes induced by nickel, arsenic and hypoxia, we hypothesized that they may regulate DNA repair pathways in a similar manner. Of note, several connections between nickel or other metals and DNA repair have been reported. Most notably, nickel and arsenic can inhibit the repair of UV-induced and oxidative base DNA damage by the NER and BER pathways, possibly through direct interaction with zinc finger repair enzymes such as XPA and PARP (44–50). Nickel can also directly inhibit the DNA alkylation repair enzymes ABH2 and ABH3 by replacing iron at the catalytic site (7,51). Recently, arsenic was shown to directly inhibit a RING finger E3 ubiquitin ligase leading to reduced HR and NHEJ repair (52). At the gene expression level, silencing of the MGMT promoter has been demonstrated in nickel-transformed cells and could contribute to reduced repair of O6-methylguanine lesions (53). In vivo, DNA repair genes were found to be overrepresented among differentially expressed genes in nickel refinery workers compared to controls, with 29 under-expressed and 2 over-expressed DNA repair genes in the high nickel exposure group (54). Nickel-exposed workers have also been shown to have lower levels of the BER glycosylase OGG1 with a correlative increase in oxidative base lesions (55). Finally, arsenic and chromium have been shown to epigenetically silence the MLH1 promoter in cell culture and in exposed human subjects (56–59). Thus, nickel and other metals are clearly implicated in DNA repair, but whether nickel leads to coordinated transcriptional and epigenetic down-regulation of DNA repair via hypoxia-like pathways remains unknown.
In this study, we have investigated the regulation of DNA repair by nickel in tumorigenic and non-tumorigenic human lung cells. We found that cellular exposure to nickel leads to reduced expression of the HDR proteins, BRCA1, RAD51, and FANCD2, and the MMR protein MLH1 in a dose-dependent manner, without affecting the expression of NHEJ proteins. This down-regulation was associated with impaired DNA double-strand break repair in a plasmid-based host cell reactivation assay specific for HDR, as well as persistent unrepaired chromosomal DNA double-strand breaks and reduced clonogenic survival following ionizing radiation. Using targeted expression analysis and global microarray analysis, we demonstrated that nickel-induced repression of HDR and MMR proteins occurs at the mRNA level through transcriptional changes that are more closely related to hypoxia than to other metals. Finally, we investigated whether nickel can lead to MLH1 promoter silencing, and found short-term inactivation, but not stable silencing, in our assay system.
Materials and methods
Cell culture
A549 and HeLa cells were grown in high-glucose DMEM supplemented with 10% FBS (Invitrogen). MCF7 cells were grown in RPMI Medium 1640 supplemented with 10% FBS. RKO cells containing the MLH1 promoter reporter construct were grown in MEM supplemented with 10% FBS and 0.8 mg/mL G418 (American Bio). BEAS-2B cells were grown in Airway Epithelial Cell Basal Medium (ATCC) supplemented with Bronchial Epithelial Cell Growth Kit (ATCC). BEAS-2B cells were authenticated by short tandem repeat profiling at the Yale DNA Analysis Facility and comparison to the published profile. All other cell lines were obtained from ATCC.
Chemicals and hypoxia
Nickel(II) chloride (Sigma) was dissolved in H2O at 250 mM prior to each treatment and used at final concentrations of 0–500 μM. Sodium (meta)arsenite (Sigma) was dissolved in H2O at 100 mM and used at final concentrations of 0–10 μM. For exposure to hypoxia, cells were placed in a Galaxy R Series 170 R CO2 incubator equipped with an electrochemical oxygen sensor, and the oxygen level was reduced to 1% by the controlled addition of nitrogen via a two-stage pressure regulator and an inline pressure regulator.
Western blot analysis
Frozen cell pellets were lysed in AZ lysis buffer (50 mM Tris, 250 mM NaCl, 1% Igepal, 0.1% SDS, 5 mM EDTA, 10 mM Na2P2O7, 19 mM NaF) supplemented with Protease Inhibitor Cocktail (Roche) on ice for 20 min. Cellular debris was cleared by centrifugation and lysate protein concentration was quantified using the DC Protein Assay (Bio-Rad). Equal amounts of protein were subjected to SDS-PAGE in Mini-PROTEAN TGX gradient gels (Bio-Rad) and then transferred to nitrocellulose membrane. Antibodies used for western blot analysis are provided in the Supplementary Methods.
HR and NHEJ assays
These assays were performed as previously described (60). Details are provided in the Supplemental Methods.
Irradiation
Irradiation was performed using an X-RAD 320 Biological Irradiator (Precision X-Ray Inc.) set at 320 kV, 12.5 mA, 2 mm Al filter, 20 × 20 cm collimator and 50.0 cm SSD (source to sample distance) with dose rate 2.4 Gy/min. Cells in sterile plastic 6-well culture dishes were irradiated at room temperature without removal of media for 2.1 min to achieve a total dose of 5 Gy.
Comet assays
Cells were pretreated with NiCl2 for 45 h and irradiated in 6-well format. Cells were harvested 24 h post-irradiation and resuspended in LM Agarose (Trevigen). Neutral single-cell gel electrophoresis was conducted using the CometAssay Electrophoresis System (Trevigen) according to the manufacturer’s protocol. Data were collected with an EVOS FL microscope (Advance Microscopy Group) and analyzed with CometScore software (TriTek Corporation). Statistical analyses by Mann–Whitney test were performed using GraphPad Prism Version 6.0a for MAC OS X (GraphPad Software).
Clonogenic survival assays
Cells were pretreated with NiCl2 for 45 h and irradiated in 6-well format. Immediately following irradiation, cells were reseeded in triplicate at 100–5000 cells/well in 6-well plates. Cells were cultured for 9 days until colonies formed, replacing culture media every 3 days. Cells were permeabilized with 0.9% saline solution and stained with crystal violet in 80% methanol. Colonies with >50 cells were counted manually. Statistical analyses by unpaired t-test were performed using GraphPad Prism Version 6.0a for MAC OS X.
Reverse transcription quantitative PCR (RT-qPCR)
Total RNA was prepared using the RNeasy Mini Kit (Qiagen). The optional on-column DNase digestion was performed with the RNase-Free DNase Set (Qiagen) to eliminate genomic DNA. Complementary DNA (cDNA) was synthesized using 750 ng RNA in the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The resulting cDNA was diluted 1:5 and used in triplicate PCRs containing TaqMan Gene Expression Assay premixed primers and probes for BRCA1, FANCD2, RAD51, MLH1, NHEJ1, XRCC4, LIG4, and 18S rRNA and TaqMan Fast Universal PCR Master Mix (Applied Biosystems). A StepOnePlus Real-Time PCR System (Thermo Fisher Scientific) was used to measure fluorescence intensity in real-time and to calculate cycle thresholds. Ct values were normalized to 18S rRNA and relative expression was calculated using the –ΔΔCt method. Statistical analyses by unpaired t-test were performed using GraphPad Prism Version 6.0a for MAC OS X.
Expression microarray analysis
Total RNA from BEAS-2B cells treated with 100 μM NiCl2, 5 μM NaAsO2, or 1% O2 and from untreated control cells was prepared using the RNeasy Mini Kit in 3 independent experiments. The optional on-column DNase digestion was performed with the RNase-Free DNase Set to eliminate genomic DNA. Gene expression profiling was performed at the Yale Center for Genome Analysis using a Human HT-12 v4 BeadChip (Illumina). Data was analyzed by normalization to control samples and identification of genes with mRNA transcripts increased >1.8-fold or decreased <0.5-fold. Expression values with coefficients of variance between replicates of >0.5 were excluded from analysis.
MLH1 promoter silencing assays
The MLH1 promoter reporter construct was originally generated by cloning 5 kb of the MLH1 promoter into a modified pDsRed2-N1 vector (Clontech) in which the CMV promoter was removed and blasticidin resistance (BSR) and thymidine kinase (TK) genes were inserted in-frame with DsRed2 (61). RKO cells stably transfected with the MLH1 promoter reporter have been previously described (27). BEAS-2B and A549 cells were transfected with the MLH1 promoter reporter using FuGENE 6 Transfection Reagent (Promega) and Lipofectamine 3000 Reagent (Thermo Fisher Scientific), respectively. Stable transfection and functional expression of the reporter were selected for with 0.4 mg/mL G418 and 4 μg/mL blasticidin (Invivogen) or 1 mg/mL G418 and 10 μg/mL blasticidin, in BEAS-2B and A549 cells respectively. Integration of the MLH1 promoter plasmid was confirmed via PCR with MLH1 promoter and plasmid-specific primers. To measure silencing of the MLH1 promoter reporter, cells were seeded in 10-cm dishes at 105 and 5 × 105 cells/dish in triplicate under selection with G418 and 10 μg/mL ganciclovir (Invivogen). Plating efficiency was determined by seeding cells in 10-cm dishes at 500 cells/dish in triplicate under selection with G418 only. Cells were cultured for 10 to 14 days until colonies formed, replacing culture media and selection agents every 3 to 4 days. Cells were then permeabilized with 0.9% saline solution and stained with crystal violet in 80% methanol, and colonies with >50 cells were counted manually. Silencing frequency was determined by normalizing the surviving fraction with ganciclovir selection to plating efficiency for each condition. Statistical analyses by unpaired t-test were performed using GraphPad Prism Version 6.0a for MAC OS X.
Results
Nickel exposure leads to reduced expression and activity of HDR, but not NHEJ, proteins
To investigate the effects of nickel exposure on DNA repair, we began by analyzing the expression of DNA repair proteins in a panel of human cancer cell lines following treatment with nickel(II) chloride (NiCl2). Lung carcinoma A549, cervical carcinoma HeLa, and breast carcinoma MCF7 cells, were treated with 0 to 500 μM NiCl2 for 48 h and protein expression was analyzed by western blotting. We observed a dose-dependent decrease in expression of the HDR proteins, BRCA1, FANCD2 and RAD51, as well as the MMR protein MLH1 in all three cell lines (Figure 1A and Supplementary Figure S1). In contrast, there was no significant change in the protein levels of the NHEJ proteins DNA-PKcs, KU80, XRCC4 or LIG4 with nickel treatment (Figure 1A and Supplementary Figure S1). Because nickel exposure is most closely tied to human lung cancer, we wanted to determine the effect of nickel exposure in the setting of normal human lung cells. We therefore performed western blotting for DNA repair proteins in non-tumorigenic BEAS-2B cells, which are an immortalized cell line derived from normal human bronchial epithelium, after treatment with 0 to 250 μM NiCl2 for 48 h. As in the human cancer cell lines, we observed a dose-dependent decrease in expression of BRCA1, FANCD2, RAD51 and MLH1 expression, but no significant changes in NHEJ protein expression (Figure 1B, lanes 1–3, and Supplementary Figure S1). For comparison, we also analyzed expression of these proteins following culture in 1% O2 conditions for 48 h, and similarly found reduced expression of BRCA1, FANCD2, RAD51 and MLH1, but not of NHEJ proteins (Figure 1B, lane 4).
Figure 1.

Nickel treatment induces down-regulation of HR, but not NHEJ, protein expression and activity in human cancer cell lines and non-tumorigenic human lung cells. (A) Western blot analysis of HR and NHEJ proteins in human cancer cell lines treated with 0, 100, 250 or 500 μM NiCl2 for 48 h. Quantitation of band density is presented in Supplementary Figure S1 and uncropped blots are available in Supplementary Figures S7 and S8. (B) Western blot analysis of HR and NHEJ proteins in BEAS-2B cells treated with 0, 100 or 250 μM NiCl2 or hypoxia (1% O2) for 48 h. Quantitation of band density is presented in Supplementary Figure S1 and uncropped blots are available in Supplementary Figure S9. (C) Relative HR activity determined by reactivation of the HR luciferase plasmid reporter in A549 and BEAS-2B cells pretreated with 250 μM NiCl2 for 48 h or BEAS-2B cells pretreated with 1% O2 for 48 h. HR repair is normalized to mock-treated control cells. (D) Relative NHEJ activity determined by reactivation of the NHEJ luciferase plasmid reporter in A549 and BEAS-2B treated as in panel C. NHEJ repair is normalized to mock-treated control cells. In panels C and D, statistical analyses by one-sample t-test were performed comparing relative repair values to a hypothetical value of 1. Columns, mean of 3 independent experiments; error bars, SEM; **significant at P < 0.01; *significant at P < 0.05; # significant at P < 0.1; ns, not significant.
We next wanted to determine whether nickel-induced repression of HDR protein expression leads to a functional impairment in homologous recombination (HR). To answer this question, we utilized plasmid-based luciferase reactivation double-strand break repair assays specific for HR or NHEJ (60). The HR reporter plasmid contains a CMV promoter-driven firefly luciferase gene with an inactivating I-SceI restriction enzyme site and a downstream promoter-less copy of wild type firefly luciferase (Figure 1C). Digestion of this plasmid with I-SceI induces a double-strand DNA break within the CMV promoter-driven luciferase gene, which can be repaired by HR using the downstream luciferase gene as a template to allow reactivation of luciferase expression. Following pretreatment with 0 or 250 μM NiCl2 for 48 h, A549 and BEAS-2B cells were transfected with I-SceI-digested HR reporter plasmid and assayed for reactivation of luciferase activity 48 h post-transfection. We found that nickel exposure led to an approximately 50% reduction in HR repair in both A549 and BEAS-2B cells (Figure 1C). For comparison, we also pretreated BEAS-2B cells with hypoxia at 1% O2 for 48 h, and found a similar 50% impairment in HR capacity. To determine the specificity of HR repression by nickel, we used an NHEJ reporter plasmid consisting of a CMV promoter-driven wild type firefly luciferase gene with a HindIII restriction enzyme site located between the promoter and the firefly luciferase gene (Figure 1D). Digestion of this plasmid with HindIII induces a double-strand DNA break separating the promoter from the luciferase gene, which can be repaired by NHEJ in the absence of a homologous template. In A549 and BEAS-2B cells, we found that pretreatment with 250 μM NiCl2 for 48 h did not reduce NHEJ activity and actually stimulated NHEJ in A549 cells (Figure 1D). Hypoxia pretreatment similarly did not repress NHEJ in BEAS-2B cells.
Nickel inhibits repair of IR-induced DNA double-strand breaks in tumorigenic and non-tumorigenic lung cells
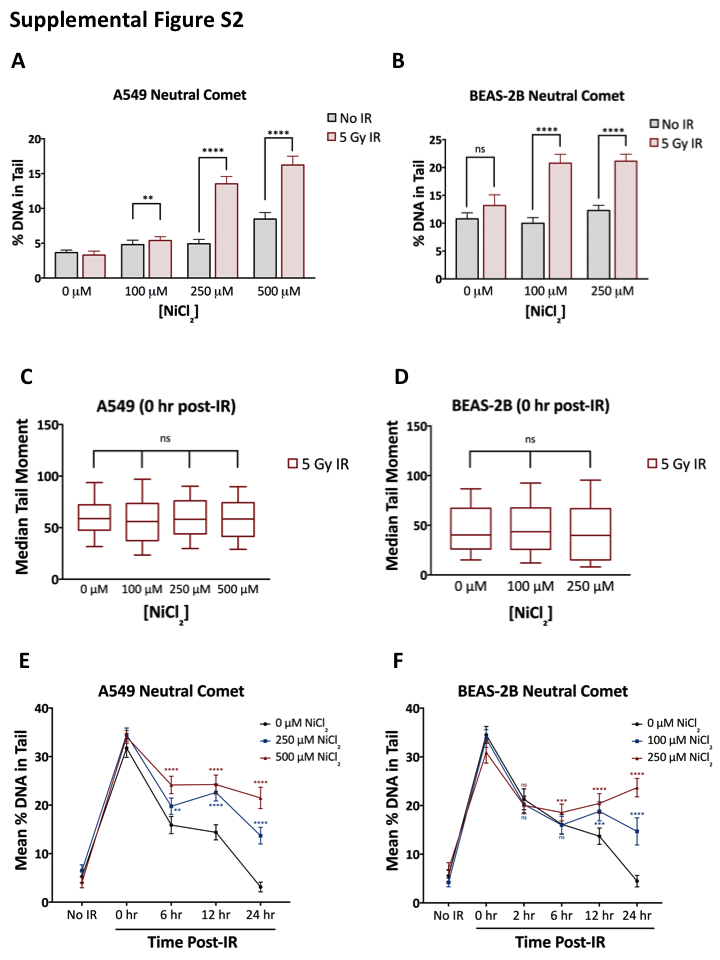
Based on our finding that nickel reduces cellular HDR capacity, we hypothesized that nickel treatment would induce cellular sensitivity to DNA double-strand breaks induced by ionizing radiation (IR). To test this, we measured the persistence of IR-induced DNA double-strand breaks in nickel-treated A549 and BEAS-2B cells using the neutral comet assay. A549 and BEAS-2B cells were pretreated with 0 to 500 μM NiCl2 for 45 h, irradiated with 5 Gy IR, and assayed for DNA double-strand breaks 24 h post-IR. In both cell lines, we observed that, at 0 μM NiCl2, there was no increase in DNA double-strand breaks, as measured by the median comet tail moment or % DNA in the comet tail, in irradiated cells indicating that cells effectively repaired the DNA breaks within 24 h (Figure 2A–D and Supplementary Figure S2A and B). In contrast, treatment with NiCl2 in both cell lines led to dose-dependent increases in the DNA double-strand breaks persisting 24 h post-IR in irradiated cells (Figure 2A–D and Supplementary Figure S2A and B). At 250 μM and 500 μM NiCl2 in BEAS-2B and A549 cells, respectively, we observed small increases in the median comet tail moment in non-irradiated cells (Figure 2A–D), indicating that, at high doses, nickel-induced repression of HR may limit repair of spontaneous DNA double-strand breaks within cells. Of note, there was no increase in two markers of apoptosis, activated Caspase-3 and cleaved PARP1, in nickel-treated and/or irradiated cells 24 h post-irradiation, indicating that the increased levels of DNA double-strand breaks detected in the comet assay at this time-point are not simply reflecting apoptotic DNA breakage (Supplementary Figure S3A and B).
Figure 2.

Nickel exposure compromises repair of IR-induced DNA double-strand breaks and induces cellular sensitivity to IR. Neutral comet assay in A549 (A) or BEAS-2B (B) cells pretreated with 0, 100, 250 or 500 μM NiCl2 for 45 h and irradiated with 0 or 5 Gy, assayed 24 h post-irradiation. Median comet tail moments from analysis of ≥100 comets/sample are presented as box-and-whisker plots. Statistical analyses were by Mann–Whitney test. Boxes, lower and upper quartiles; middle line, median; whiskers, 10th to 90th percentiles; ****significant at P < 0.0001; **significant at P < 0.01; ns, not significant. Representative images of comets observed in A549 (C) and BEAS-2B (D) cells treated and assayed as in panels A and B. Clonogenic survival in A549 (E) and BEAS-2B (F) cells exposed to ionizing radiation following pretreatment with NiCl2 for 45 h. Clonogenic survival was normalized to survival at 0 Gy for each dose of NiCl2. Statistical analyses by unpaired t-test were performed with comparison to 0 μM NiCl2 at each radiation dose. Points, mean of six replicates; error bars, SEM; ***significant at P < 0.001; **significant at P < 0.01; *significant at P < 0.05; ns, not significant.
The repair of IR-induced DNA double-strand breaks has been shown to occur with biphasic kinetics, with NHEJ responsible for fast repair of the majority of breaks occurring in cells throughout the cell cycle and HDR responsible for slow repair of a smaller subset of complex or heterochromatin-associated breaks occurring in S/G2-phase cells (62,63). Therefore, inhibition of NHEJ blocks repair of IR-induced breaks at early time-points whereas inhibition of HDR leads to elevated persistent breaks only at later time-points. To further explore which repair pathways are affected by nickel, we used the neutral comet assay to analyze the repair of IR-induced DNA double-strand breaks over time. In both A549 and BEAS-2B cells, we found that nickel pretreatment had no significant effect on the level of DNA double-strand breaks induced immediately following irradiation, indicating that nickel did not enhance radiation-induced DNA damage (Supplementary Figure S2C and D). Moreover, we observed that nickel pretreatment had little or no effect on repair at early time-points (2 h and 6 h post-IR) as treated and control cells demonstrated similar reductions in the number of DNA double-strand breaks over time (Supplementary Figure S2E and F). However, at later time-points (12 h and 24 h post-IR), control cells demonstrated further reduction in the number of DNA double-strand breaks whereas nickel-treated cells demonstrated failure to repair remaining breaks in a dose-dependent manner (Supplementary Figure S2E and F). Altogether, these results strongly support our conclusion that nickel is inhibiting the HDR rather than the NHEJ pathway of DNA double-strand break repair.
Reduced capacity to repair IR-induced DNA double-strand breaks can lead to reduced cellular survival post-IR. We therefore performed clonogenic survival assays in A549 and BEAS-2B cells pretreated with 0 to 500 μM NiCl2 and then irradiated with 0 to 5 Gy IR. In A549 cells, we observed significant radiosensitization at 250 and 500 μM NiCl2 (Figure 2E). Nickel treatment in non-irradiated cells did not significantly affect clonogenic survival in A549 cells (Supplementary Figure S4A). Similarly, we observed reduced clonogenic survival of irradiated BEAS-2B cells pretreated with 100 or 250 μM NiCl2 (Figure 2F). Treatment with 250 μM NiCl2, by itself, did reduce BEAS-2B clonogenic survival about 30%, but 100 μM NiCl2 did not (Supplementary Figure S4B). Thus, nickel radiosensitizes human lung cells at doses that do not directly affect clonogenic survival and acts synergistically at higher doses.
Nickel-induced repression of HDR occurs via transcriptional reprogramming in similarity to hypoxic stress
We next wanted to determine the mechanism underlying the repression of HDR protein expression and activity induced by nickel exposure. Based on the known transcriptional changes induced by nickel and their similarity to hypoxia, we hypothesized that transcriptional and epigenetic changes induced by hypoxia-like signaling pathways might be responsible. We therefore began by measuring mRNA levels of DNA repair genes in A549 cells exposed to 0 to 500 μM NiCl2 for 48 h. We found that nickel induced dose-dependent decreases in BRCA1, FANCD2, RAD51 and MLH1 mRNA levels but little or no decrease in expression of several NHEJ genes (Figure 3A). In nickel-treated BEAS-2B cells, we similarly found significant decreases in FANCD2 and MLH1 mRNA levels, non-significant trends towards reduced expression of BRCA1 and RAD51 mRNA, and no change in NHEJ gene mRNA levels with nickel treatment (Figure 3B). We also obtained comparable results in nickel-treated MCF7 cells and similar but less significant results in nickel-treated HeLa cells (Supplementary Figure S5A and B). These changes in DNA repair gene mRNA levels are in concordance with our western blot analysis (Figure 1A and B) and indicate that nickel, like hypoxia, reduces HR and MMR via transcriptional control.
Figure 3.

Nickel exposure induces acute transcriptional changes similar to hypoxic exposure and leads to reduced mRNA levels of HR, but not NHEJ genes. (A) qRT-PCR analysis of HR and NHEJ gene expression in A549 cells after treatment with 0, 250 or 500 μM NiCl2 for 48 h. (B) qRT-PCR analysis of HR and NHEJ gene expression in BEAS-2B cells after treatment with 0, 100 or 250 μM NiCl2 for 48 h. (C) qRT-PCR analysis of HR gene expression in A549 cells after treatment with 0, 1 or 10 μM NaAsO2 for 48 h. (D) qRT-PCR analysis of HR gene expression in BEAS-2B cells after treatment with 0, 1 or 5 μM NaAsO2 for 48 h. In panels A-D, mRNA expression levels were normalized to 18S rRNA expression and presented relative to control samples. Statistical analyses by unpaired t-test were performed with comparison to control samples. Columns, mean of three replicates; bars, SEM; ***significant at P < 0.001; **significant at P < 0.01; *significant at P < 0.05; #significant at P < 0.1; ns, not significant. (E) Venn diagrams showing genes down-regulated <0.5-fold or up-regulated >1.8-fold in BEAS-2B cells treated in triplicate with 100 μM NiCl2, 5 μM NaAsO2 or 1% O2 for 48 h, based on mRNA expression profiling using an Illumina Human HT-12 v4 Expression BeadChip. (F) Heat map demonstrating hierarchal clustering of differentially expressed genes in BEAS-2B cells treated and analyzed by expression profiling as in Panel E.
Arsenic is also thought to induce carcinogenesis in part through transcriptional reprogramming (6–8,32); however, we have observed different effects on DNA repair gene expression as compared to nickel and hypoxia. Treatment with up to 5 or 10 μM sodium arsenite (NaAsO2) for 48 h had no effect on BRCA1, FANCD2 or MLH1 mRNA expression in A549 or BEAS-2B cells (Figure 3C and D). At the protein level, arsenic exposure induced variable changes in DNA repair protein levels across different cell lines, but with no discernable significant trends (data not shown). Interestingly, in BEAS-2B cells treated with 100 μM NiCl2 or 5 μM NaAsO2, doses which induce expression of the Metallothionein 1 and 2 mRNA transcripts, we observed an increase in VEGF mRNA expression comparable to hypoxia with nickel but not arsenic treatment (Supplementary Figure S5C). We therefore suspected that the acute transcriptional changes induced by nickel may be more closely related to hypoxia than other metals or metalloids.
To further characterize the connection between hypoxia- and nickel-induced transcriptional down-regulation of DNA repair, we compared global transcriptional changes via gene expression microarray after hypoxia, nickel, or for comparison, arsenic exposure. BEAS-2B cells were treated with 100 μM NiCl2, 5 μM NaAsO2 or 1% O2 for 48 h, and mRNA expression profiles were analyzed using an Illumina Human HT-12 v4 Expression BeadChip. To compare expression changes induced by nickel, arsenic and hypoxia, we analyzed the overlap between the most highly up-regulated and down-regulated genes in each sample. Using cutoffs of >1.8-fold up-regulation and <0.5-fold down-regulation, we found many more common genes between the nickel and hypoxia samples (5 up-regulated, 11 down-regulated) than between the arsenic and hypoxia samples (2 up-regulated, 1 down-regulated) or between the nickel and arsenic samples (0 up-regulated, 1 down-regulated) (Figure 3E). Using hierarchal clustering analysis, we similarly found closer clustering of gene expression patterns with nickel and hypoxia treatment compared to arsenic treatment (Figure 3F).
Nickel and arsenic repress MLH1 promoter activity, but only arsenic generates stable promoter silencing
Chronic exposure to hypoxia and has been shown to lead to stable epigenetic silencing of certain DNA repair gene promoters, including the BRCA1 and MLH1 promoters (26,27). Arsenic and chromium can also lead to epigenetic inactivation of MLH1 (57,59). To investigate whether nickel can generate stable silencing of DNA repair genes, we utilized RKO cells harboring a chromosomally-integrated MLH1 promoter reporter construct which has previously been utilized to study hypoxia-induced gene silencing (27). This construct contains the MLH1 promoter upstream of the thymidine kinase (TK) and blasticidin resistance (BSR) genes, allowing selection of cells with an active MLH1 promoter using blasticidin and cells with a silenced MLH1 promoter using ganciclovir (Figure 4A).
Figure 4.

Nickel acutely increases MLH1 promoter inactivation while arsenic induces long-term MLH1 promoter silencing. (A) Schematic of the MLH1 promoter reporter silencing construct. The active MLH1 promoter confers ganciclovir sensitivity and blasticidin resistance while MLH1 promoter silencing leads to ganciclovir resistance and blasticidin sensitivity. TK, thymidine kinase gene; BSR, blasticidin resistance gene; Neo, neomycin resistance gene; GCV, ganciclovir, Blast, blasticidin. (B) MLH1 promoter silencing frequency in RKO cells after 1 week of treatment with 100 μM NiCl2, 0.5 μM NaAsO2, or hypoxia (1% O2), compared to untreated control cells. (C) MLH1 promoter silencing frequency in RKO cells over 3 weeks of treatment with 100 μM NiCl2, 0.5 μM NaAsO2 or hypoxia (1% O2), compared to untreated control cells. (D) MLH1 promoter silencing frequency in BEAS-2B cells after 1 week of treatment with 100 μM NiCl2, 1 μM NaAsO2 or hypoxia (1% O2), compared to untreated control cells. (E) MLH1 promoter silencing frequency in BEAS-2B cells over 3 weeks of treatment with 100 μM NiCl2, 1 μM NaAsO2 or hypoxia (1% O2), compared to untreated control cells. In panels B–E, statistical analyses by unpaired t-test were performed with comparison to untreated control cells at each time-point. Columns and points, mean of three replicates; error bars, SEM; ***significant at P < 0.001; **significant at P < 0.01; *significant at P < 0.05; #significant at P < 0.1; ns, not significant.
To test the potential epigenetic effects of long-term low-dose metal exposure, we treated the RKO cells containing the MLH1 promoter reporter continuously with 100 μM NiCl2 or 0.5 μM NaAsO2, which were the maximum doses that we determined to have no significant effect on cell growth with long-term treatment. At weekly intervals, we determined MLH1 promoter silencing by measuring the frequency of ganciclovir resistance in clonogenic assays. After one week of treatment, we observed an approximately 2-fold increase in MLH1 promoter inactivation with both nickel and arsenic (Figure 4B). However, upon longer treatment, enhanced MLH1 promoter silencing persisted only in the arsenic-treated cells (Figure 4C). For comparison with published data, we grew the RKO cells under hypoxic conditions (1%) in parallel and observed increased MLH1 promoter silencing in hypoxic cells as previously reported (27) (Figure 4C).
We next generated BEAS-2B and A549 cells with stable integration of the MLH1 promoter reporter construct, in order to test our results in human lung cells. These BEAS-2B and A549 cells were likewise treated with 100 μM NiCl2, 1 μM NaAsO2 or 1% oxygen, and MLH1 promoter silencing was measured at weekly intervals. We found that after one week of treatment, nickel, arsenic, and hypoxia all increased MLH1 promoter inactivation (Figure 4D and Supplementary Figure S6A), but again only arsenic and hypoxia led to long-term stable MLH1 promoter silencing (Figure 4E and Supplementary Figure S6B).
Discussion
In this study, we have identified and characterized a novel mechanism of DNA repair regulation by the carcinogenic heavy metal nickel. In short, nickel exposure in human tumorigenic and non-tumorigenic lung cells leads to transcriptional down-regulation of the HDR proteins BRCA1, RAD51 and FANCD2 and the MMR protein MLH1, without down-regulation of NHEJ factors. This DNA repair factor repression appears functionally significant as nickel treatment reduces repair of DNA double-strand breaks by HDR, but not NHEJ, and acts as a radiosensitizer by preventing efficient repair of IR-induced DNA double-strand breaks. Finally, nickel initially led to reduced activity of the MLH1 promoter, but did not generate stable silencing as we and others have observed with hypoxia and arsenic. Together, our findings support a model in which nickel inhibits high-fidelity DNA repair pathways through acute hypoxia-mimetic transcriptional pathways, potentially contributing to nickel-induced carcinogenesis.
The gene expression and functional changes in DNA repair that we observe in nickel-treated cells have similarities to those induced by hypoxic stress. Specifically, hypoxia represses the high-fidelity HDR and MMR pathways but not the error-prone NHEJ pathway, through multiple transcriptional and epigenetic mechanisms (23–28). Given that nickel and hypoxia are known to have convergent effects on the iron- and 2-oxoglutarate-dependent dioxygenases leading to activation of HIF-dependent transcription and inhibition of histone demethylation, nickel likely regulates HDR and MMR expression through transcriptional and epigenetic mechanisms analogous to hypoxia. Arsenic can also lead to stabilization of HIF-1, but it does not activate the same hypoxia-induced transcriptional response (42,43), and we found that arsenic did not lead to immediate changes in DNA repair gene transcription or protein expression. Interestingly, other studies have found that, although most heavy metals induce significant changes in transcription, the effects of different metals are largely independent (64). In our microarray analysis in BEAS-2B cells, we also found greater overlap between the transcriptional effects of nickel and hypoxia than between nickel and arsenic. These results support our hypothesis that nickel acutely down-regulates HDR and MMR but not NHEJ via transcriptional changes that are possibly more similar to hypoxia than other metals.
Although nickel-induced transcriptional down-regulation of DNA repair resembles the effect of hypoxia, we did not find comparable DNA repair gene silencing effects. In our assay using the MLH1 promoter reporter, we found that nickel initially increased the fraction of cells with MLH1 promoter silencing, but more prolonged treatment with nickel did not generate stable silencing. In contrast, we did observe an increase in stable silencing with hypoxia or arsenic treatment, consistent with previous studies (27,58,59). We hypothesize that this difference may be due to the potential of these treatments to induce DNA methylation. Nickel, arsenic, and hypoxia can all increase global DNA methylation and induce silencing at specific gene promoters. However, nickel is unique in that its effects on DNA methylation seem to be limited to regions adjacent to heterochromatin, suggesting that it may act by inducing ‘heterochromatin spreading’ rather than de novo DNA hypermethylation (65,66). The integration site of the MLH1 promoter reporter construct in our assay may have limited our ability to detect nickel-induced gene silencing. Thus, by including arsenic in this study, we have been able to elucidate some of the similarities and differences between the effects of nickel and hypoxia on DNA repair.
Previous studies have suggested that nickel can regulate certain types of DNA repair, either through direct enzyme inhibition or through regulation of gene expression. Specifically, nickel has been shown to inhibit nucleotide and base excision repair, likely through direct inhibition of zinc finger or Jumonji domain-containing repair enzymes such as XPA, PARP and ABH2/3, thereby sensitizing cells to UV irradiation and alkylating agents (44–51). Nickel exposure has also been associated with down-regulation of a number of DNA repair genes including MGMT, OGG1, and other DNA repair genes identified in an in vivo study conducted by Arita et al. (53–55). Down-regulation of the hypoxia-regulated HDR and MMR genes that we observed in our study illustrates an additional way in which nickel may regulate DNA repair. Of the DNA repair genes that we investigated, three were present in the group of 29 DNA repair genes that Arita et al. found to be reduced in peripheral blood mononuclear cells of nickel refinery workers (54). These three genes, NHEJ1, XRCC5 and PRKDC, are all involved in NHEJ. We did not observe decreased expression of NHEJ1 mRNA expression or decreased protein expression of KU80 or DNA-PKcs (the protein products of XRCC5 and PRKDC), possibly due to the different cell types under study or the differences in duration of nickel exposure. In addition, it is possible that the effects of nickel exposure in vivo, through interactions with other environmental exposures or complex endogenous factors, may differ from the pure effects of nickel on cells in vitro. Further studies will be necessary to determine whether nickel-induced repression of HDR and MMR genes occurs in a functionally important manner in vivo.
Nickel does not induce mutagenesis in most assays that detect direct DNA damage, yet it has been shown to induce chromosomal aberrations when cells are treated for longer periods of time (2). In addition, nickel can potentiate the effects of DNA damaging agents such as cisplatin and mitomycin C (45). Cells with reduced repair of DNA double-strand breaks exhibit higher levels of baseline genomic instability and sensitivity to DNA damaging agents. Thus, our findings of nickel-induced down-regulation of HDR are consistent with the time course of nickel-induced genetic lesions and the co-carcinogenic effect of nickel with DNA damaging agents, and together with previous literature, suggest that nickel can impact cellular DNA repair on multiple levels, ranging from direct enzyme inhibition to modulation of DNA repair factor expression. Moreover, our study extends the reach of nickel-mediated DNA repair regulation to the HDR and MMR pathways. One previous study investigated the effect of nickel on the repair of IR-induced DNA breaks but used only short-term nickel exposure and found inhibition of DNA double-strand break repair only with extremely high toxic dose of nickel (67). More recently, nickel was shown to sensitize yeast cells to ionizing radiation, consistent with our results in human cells (68). To our knowledge, our data are the first to demonstrate inhibition of DNA double-strand break repair and radiosensitization in human cells with non-toxic doses of nickel.
In conclusion, our results reveal a potential connection between nickel and hypoxia-mediated down-regulation of cellular HDR and MMR DNA repair pathways. Nickel-induced inhibition of HDR and MMR is particularly relevant as a potential source of genomic instability that could contribute to nickel-related lung and nasal cancers. Altogether, our study contributes to the molecular understanding of heavy metal carcinogenesis, which will ultimately aid in the development of preventative and curative cancer therapies.
Supplementary Material
Supplementary data are available at Carcinogenesis online.
Funding
Research reported in this publication was supported by NIH grant R01ES005775 to P.M.G., NIH Medical Scientist Program Training Grant T32GM007205, and NIH National Institute of General Medical Sciences award T32GM007223.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Peter Koo for generously providing reagents, and we thank Yuhong Lu for technical assistance.
Conflict of Interest Statement: None declared.
Abbreviations
- HDR
homology-dependent DNA double-strand break repair
- MMR
mismatch repair
- NER
nucleotide excision repair
- NHEJ
non-homologous end joining
References
- 1. National Toxicology P. (2011) Nickel compounds and metallic nickel. Rep. Carcinog., 12, 280–283. [PubMed] [Google Scholar]
- 2. Lu H., et al. (2005) Carcinogenic effect of nickel compounds. Mol. Cell. Biochem., 279, 45–67. [DOI] [PubMed] [Google Scholar]
- 3. Andersen A., et al. (1996) Exposure to nickel compounds and smoking in relation to incidence of lung and nasal cancer among nickel refinery workers. Occup. Environ. Med., 53, 708–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Navarro Silvera S.A., et al. (2007) Trace elements and cancer risk: a review of the epidemiologic evidence. Cancer Causes Control, 18, 7–27. [DOI] [PubMed] [Google Scholar]
- 5. Binazzi A., et al. (2015) Occupational exposure and sinonasal cancer: a systematic review and meta-analysis. BMC Cancer, 15, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Salnikow K., et al. (2008) Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium. Chem. Res. Toxicol., 21, 28–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chervona Y., et al. (2012) Carcinogenic metals and the epigenome: understanding the effect of nickel, arsenic, and chromium. Metallomics, 4, 619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chervona Y., et al. (2012) The control of histone methylation and gene expression by oxidative stress, hypoxia, and metals. Free Radic. Biol. Med., 53, 1041–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davidson T.L., et al. (2006) Soluble nickel inhibits HIF-prolyl-hydroxylases creating persistent hypoxic signaling in A549 cells. Mol. Carcinog., 45, 479–489. [DOI] [PubMed] [Google Scholar]
- 10. Chen H., et al. (2009) Iron- and 2-oxoglutarate-dependent dioxygenases: an emerging group of molecular targets for nickel toxicity and carcinogenicity. Biometals, 22, 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Salnikow K., et al. (2000) Carcinogenic nickel induces genes involved with hypoxic stress. Cancer Res., 60, 38–41. [PubMed] [Google Scholar]
- 12. Salnikow K., et al. (2003) GeneChip analysis of signaling pathways effected by nickel. J. Environ. Monit., 5, 206–209. [DOI] [PubMed] [Google Scholar]
- 13. Chen H., et al. (2006) Nickel ions increase histone H3 lysine 9 dimethylation and induce transgene silencing. Mol. Cell. Biol., 26, 3728–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ke Q., et al. (2006) Alterations of histone modifications and transgene silencing by nickel chloride. Carcinogenesis, 27, 1481–1488. [DOI] [PubMed] [Google Scholar]
- 15. Zhou X., et al. (2009) Effects of nickel, chromate, and arsenite on histone 3 lysine methylation. Toxicol. Appl. Pharmacol., 236, 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arita A., et al. (2012) Global levels of histone modifications in peripheral blood mononuclear cells of subjects with exposure to nickel. Environ. Health Perspect., 120, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arita A., et al. (2012) The effect of exposure to carcinogenic metals on histone tail modifications and gene expression in human subjects. J. Trace Elem. Med. Biol., 26, 174–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ma L., et al. (2015) Histone Methylation in Nickel-Smelting Industrial Workers. PLoS One, 10, e0140339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen H., et al. (2010) Hypoxia and nickel inhibit histone demethylase JMJD1A and repress Spry2 expression in human bronchial epithelial BEAS-2B cells. Carcinogenesis, 31, 2136–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson A.B., et al. (2008) Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutat. Res., 640, 174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tausendschön M., et al. (2011) Hypoxia causes epigenetic gene regulation in macrophages by attenuating Jumonji histone demethylase activity. Cytokine, 53, 256–262. [DOI] [PubMed] [Google Scholar]
- 22. Scanlon S.E., et al. (2015) Multifaceted control of DNA repair pathways by the hypoxic tumor microenvironment. DNA Repair (Amst)., 32, 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bindra R.S., et al. (2005) Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res., 65, 11597–11604. [DOI] [PubMed] [Google Scholar]
- 24. Bindra R.S., et al. (2007) Co-repression of mismatch repair gene expression by hypoxia in cancer cells: role of the Myc/Max network. Cancer Lett., 252, 93–103. [DOI] [PubMed] [Google Scholar]
- 25. Bindra R.S., et al. (2007) Repression of RAD51 gene expression by E2F4/p130 complexes in hypoxia. Oncogene, 26, 2048–2057. [DOI] [PubMed] [Google Scholar]
- 26. Lu Y., et al. (2011) Hypoxia-induced epigenetic regulation and silencing of the BRCA1 promoter. Mol. Cell. Biol., 31, 3339–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu Y., et al. (2014) Silencing of the DNA mismatch repair gene MLH1 induced by hypoxic stress in a pathway dependent on the histone demethylase LSD1. Cell Rep., 8, 501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scanlon S.E., et al. (2014) Hypoxic stress facilitates acute activation and chronic downregulation of fanconi anemia proteins. Mol. Cancer Res., 12, 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rezvani H.R., et al. (2010) Hypoxia-inducible factor-1alpha regulates the expression of nucleotide excision repair proteins in keratinocytes. Nucleic Acids Res., 38, 797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chan N., et al. (2014) Hypoxia provokes base excision repair changes and a repair-deficient, mutator phenotype in colorectal cancer cells. Mol. Cancer Res., 12, 1407–1415. [DOI] [PubMed] [Google Scholar]
- 31. Zhou X., et al. (2008) Arsenite alters global histone H3 methylation. Carcinogenesis, 29, 1831–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Riedmann C., et al. (2015) Inorganic arsenic-induced cellular transformation is coupled with genome wide changes in chromatin structure, transcriptome and splicing patterns. BMC Genomics, 16, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mass M.J., et al. (1997) Arsenic alters cytosine methylation patterns of the promoter of the tumor suppressor gene p53 in human lung cells: a model for a mechanism of carcinogenesis. Mutat. Res., 386, 263–277. [DOI] [PubMed] [Google Scholar]
- 34. Cui X., et al. (2006) Chronic oral exposure to inorganic arsenate interferes with methylation status of p16INK4a and RASSF1A and induces lung cancer in A/J mice. Toxicol. Sci., 91, 372–381. [DOI] [PubMed] [Google Scholar]
- 35. Liu Y., et al. (2014) Aberrant overexpression of FOXM1 transcription factor plays a critical role in lung carcinogenesis induced by low doses of arsenic. Mol. Carcinog., 53, 380–391. [DOI] [PubMed] [Google Scholar]
- 36. Duyndam M.C., et al. (2001) Induction of vascular endothelial growth factor expression and hypoxia-inducible factor 1alpha protein by the oxidative stressor arsenite. J. Biol. Chem., 276, 48066–48076. [DOI] [PubMed] [Google Scholar]
- 37. Gao N., et al. (2004) Arsenite induces HIF-1alpha and VEGF through PI3K, Akt and reactive oxygen species in DU145 human prostate carcinoma cells. Mol. Cell. Biochem., 255, 33–45. [DOI] [PubMed] [Google Scholar]
- 38. Soucy N.V., et al. (2004) Signaling pathways for arsenic-stimulated vascular endothelial growth factor-a expression in primary vascular smooth muscle cells. Chem. Res. Toxicol., 17, 555–563. [DOI] [PubMed] [Google Scholar]
- 39. Liu L.Z., et al. (2011) Role and mechanism of arsenic in regulating angiogenesis. PLoS One, 6, e20858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang F., et al. (2013) Arsenic induces the expressions of angiogenesis-related factors through PI3K and MAPK pathways in SV-HUC-1 human uroepithelial cells. Toxicol. Lett., 222, 303–311. [DOI] [PubMed] [Google Scholar]
- 41. Watcharasit P., et al. (2014) β-catenin involvement in arsenite-induced VEGF expression in neuroblastoma SH-SY5Y cells. Environ. Toxicol., 29, 672–678. [DOI] [PubMed] [Google Scholar]
- 42. Duyndam M.C., et al. (2003) Evidence for a role of p38 kinase in hypoxia-inducible factor 1-independent induction of vascular endothelial growth factor expression by sodium arsenite. J. Biol. Chem., 278, 6885–6895. [DOI] [PubMed] [Google Scholar]
- 43. Li Q., et al. (2006) Effects of 12 metal ions on iron regulatory protein 1 (IRP-1) and hypoxia-inducible factor-1 alpha (HIF-1alpha) and HIF-regulated genes. Toxicol. Appl. Pharmacol., 213, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hartwig A., et al. (1994) Nickel(II) interferes with the incision step in nucleotide excision repair in mammalian cells. Cancer Res., 54, 4045–4051. [PubMed] [Google Scholar]
- 45. Krueger I., et al. (1999) Nickel(II) increases the sensitivity of V79 Chinese hamster cells towards cisplatin and transplatin by interference with distinct steps of DNA repair. Carcinogenesis, 20, 1177–1184. [DOI] [PubMed] [Google Scholar]
- 46. Hartwig A., et al. (2002) Interference by toxic metal ions with DNA repair processes and cell cycle control: molecular mechanisms. Environ. Health Perspect., 110 (suppl 5), 797–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hu W., et al. (2004) Nickel (II) enhances benzo[a]pyrene diol epoxide-induced mutagenesis through inhibition of nucleotide excision repair in human cells: a possible mechanism for nickel (II)-induced carcinogenesis. Carcinogenesis, 25, 455–462. [DOI] [PubMed] [Google Scholar]
- 48. Woźniak K., et al. (2004) Nickel impairs the repair of UV- and MNNG-damaged DNA. Cell. Mol. Biol. Lett., 9, 83–94. [PubMed] [Google Scholar]
- 49. Ding W., et al. (2009) Inhibition of poly(ADP-ribose) polymerase-1 by arsenite interferes with repair of oxidative DNA damage. J. Biol. Chem., 284, 6809–6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hu J., et al. (2016) Metal binding mediated conformational change of XPA protein:a potential cytotoxic mechanism of nickel in the nucleotide excision repair. J. Mol. Model., 22, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen H., et al. (2010) Nickel ions inhibit histone demethylase JMJD1A and DNA repair enzyme ABH2 by replacing the ferrous iron in the catalytic centers. J. Biol. Chem., 285, 7374–7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang F., et al. (2014) Arsenite binds to the RING finger domains of RNF20-RNF40 histone E3 ubiquitin ligase and inhibits DNA double-strand break repair. J. Am. Chem. Soc., 136, 12884–12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ji W., et al. (2008) Epigenetic silencing of O6-methylguanine DNA methyltransferase gene in NiS-transformed cells. Carcinogenesis, 29, 1267–1275. [DOI] [PubMed] [Google Scholar]
- 54. Arita A., et al. (2013) Gene expression profiles in peripheral blood mononuclear cells of Chinese nickel refinery workers with high exposures to nickel and control subjects. Cancer Epidemiol. Biomarkers Prev., 22, 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu S., et al. (2015) Dynamic changes in DNA damage and repair biomarkers with employment length among nickel smelting workers. Biomed. Environ. Sci., 28, 679–682. [DOI] [PubMed] [Google Scholar]
- 56. Takahashi Y., et al. (2005) Microsatellite instability and protein expression of the DNA mismatch repair gene, hMLH1, of lung cancer in chromate-exposed workers. Mol. Carcinog., 42, 150–158. [DOI] [PubMed] [Google Scholar]
- 57. Sun H., et al. (2009) Modulation of histone methylation and MLH1 gene silencing by hexavalent chromium. Toxicol. Appl. Pharmacol., 237, 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hossain M.B., et al. (2012) Environmental arsenic exposure and DNA methylation of the tumor suppressor gene p16 and the DNA repair gene MLH1: effect of arsenic metabolism and genotype. Metallomics, 4, 1167–1175. [DOI] [PubMed] [Google Scholar]
- 59. Treas J., et al. (2013) Chronic exposure to arsenic, estrogen, and their combination causes increased growth and transformation in human prostate epithelial cells potentially by hypermethylation-mediated silencing of MLH1. Prostate, 73, 1660–1672. [DOI] [PubMed] [Google Scholar]
- 60. Czochor J.R., et al. (2016) miR-155 overexpression promotes genomic instability by reducing high-fidelity polymerase delta expression and activating error-prone DSB repair. Mol. Cancer Res., 14, 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fang M., et al. (2014) The BRAF oncoprotein functions through the transcriptional repressor MAFG to mediate the CpG Island Methylator phenotype. Mol. Cell, 55, 904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Beucher A., et al. (2009) ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J., 28, 3413–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shibata A., et al. (2011) Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J., 30, 1079–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Andrew A.S., et al. (2003) Genomic and proteomic profiling of responses to toxic metals in human lung cells. Environ. Health Perspect., 111, 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Costa M., et al. (2005) Nickel carcinogenesis: epigenetics and hypoxia signaling. Mutat. Res., 592, 79–88. [DOI] [PubMed] [Google Scholar]
- 66. Jose C.C., et al. (2014) Epigenetic dysregulation by nickel through repressive chromatin domain disruption. Proc. Natl. Acad. Sci. USA, 111, 14631–14636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Takahashi S., et al. (2000) Inhibition of repair of radiation-induced DNA double-strand breaks by nickel and arsenite. Radiat. Res., 154, 686–691. [DOI] [PubMed] [Google Scholar]
- 68. Song S., et al. (2008) Radiosensitization of yeast cells by inhibition of histone h4 acetylation. Radiat. Res., 170, 618–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.