

Abstract
Background
Neutrophils and Liver sinusoidal endothelial cells (LSECs) both contribute to sterile inflammatory injury during ischemia/reperfusion (I/R), a well-known liver surgical stress. Interleukin-33 (IL-33) has been shown to drive neutrophil infiltration during inflammatory responses through its receptor ST2. We recently reported that infiltrating neutrophils form neutrophil extracellular traps (NETs), which exacerbate sterile inflammatory injury in liver I/R. Here, we sought to determine the role of IL-33 in NET formation during liver sterile inflammation.
Methods
Evaluation of IL-33 forming NETs was investigated using partial liver I/R model to generate sterile injury in healthy WT, IL-33 and ST2 knockouts. Serum IL-33 and myeloperoxidase (MPO)-DNA levels were measured in both humans and mice after the first surgery. Liver damage was assessed. Mouse neutrophil depletion was performed by intraperitoneal injection of anti-Ly6G antibody before I/R.
Results
Patients undergoing liver resection show a significant increase in serum IL-33 compared to healthy volunteers. This coincided with higher serum - MPO-DNA complexes. NET formation was decreased in IL-33KO and ST2 KO mice compared with control after liver I/R. IL-33 or ST2 deficiency protected livers from I/R injury, whereas rIL-33 administration during I/R exacerbated hepatotoxicity and systemic inflammation. In vitro, IL-33 is released from LSECs to promote NET formation. IL-33 deficient LSECs failed to induce NETs. ST2 deficient neutrophils limited their capacity to form NETs in vitro and adoptive transfer of ST2 KO neutrophils to neutrophil-depleted WT mice significantly decreased NET formation.
Conclusions
Data establish that IL-33, mainly released from LSECs, causes excessive sterile inflammation after hepatic I/R by inducing NET formation. Therapeutic targeting IL-33/ST2 might extend novel strategies to minimize organ damage in various clinical settings that are associated with sterile inflammation.
Keywords: Liver sterile inflammation, ischemia/reperfusion injury, neutrophil extracellular trap, danger associated molecular pattern molecules, Liver sinusoidal endothelial cells, IL-33
Graphical abstract
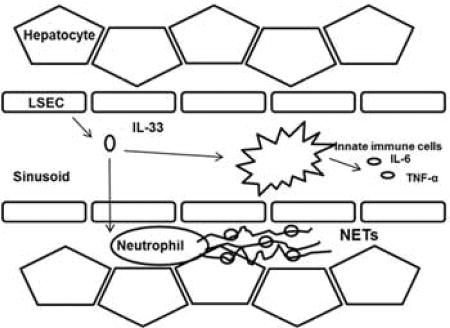
Introduction
During major liver resection or liver transplantation, the liver inevitably suffers direct mechanical and hypoxic injury with cellular necrosis. Further dysfunction and damage results from excessive activation of a sterile inflammatory response upon restoration of blood flow even in the absence of microbial pathogens [1, 2]. This process of ischemia and reperfusion (I/R) injury significantly contributes to the complications that follow these surgical procedures [3]. Both infiltrating neutrophils and liver sinusoidal endothelial cells (LSECs) have been implicated as critical cellular participants during liver I/R [4]. We have recently shown that in response to endogenous damage-associated molecular patterns (DAMP) molecules, infiltrating neutrophils form neutrophil extracellular traps (NETs) in the liver sinusoids during liver I/R. The NETs, in turn, were shown to exacerbate the inflammatory cascades augmenting liver injury [5].
IL-33 is a member of IL-1 family that resides in the nucleus, mainly found in barrier cells such as epithelial cells, keratinocytes, and LSECs [6, 7]. As a nuclear protein, IL-33 normally serves in the regulation of gene expression via binding to histones, histone methyltransferase, or NF-κB [6]. Like other DAMPs of nuclear origin, IL-33 is released into the extracellular space from severely injured or necrotic cells [7]. IL-33 binds to a cell surface receptor complex of IL-1 receptor-like 1 (IL1RL1) (also known as suppression of tumorgenicity 2, ST2) and IL-1 receptor accessory protein (IL1RAcP) [8]. Engagement of IL-33 with the transmembrane form of ST2 (ST2L) leads to activation of diverse intracellular signaling pathways, which are dependent on cell type and location [7]. In addition to IL-33/ST2 signaling at cell surfaces, the ST2 gene also encodes a soluble form of the protein (sST2) which lacks the transmembrane domain [9]. sST2 can function as a decoy receptor for IL-33, by interfering with the engagement of IL-33/ST2L at cell surfaces, thereby preventing activation of intracellular signaling pathways [10].
We have established that DAMPs facilitate NET formation during liver I/R leading to exaggerated sterile inflammatory liver injury [5]. IL-33 has previously been shown to play an important role in neutrophil infiltration, migration and activation [11–13]. However, the mechanism by which IL-33 acts as DAMP promoting sterile inflammatory liver injury is poorly understood. In the current study, we report a pathophysiological mechanism whereby IL-33 is released from LSECs after warm liver I/R to initiate a feed-forward mechanism involving ST2-dependent NET formation. These data establish IL-33/ST2 axis mediating NET formation in exacerbation of inflammation and hepatotoxicity during liver I/R.
Materials and Methods
Patient samples and data
We collected serum samples on postoperative day-1 from patients who underwent hepatectomy at the University of Pittsburgh. The majority of the patients (89%) in this study underwent liver resection for colorectal cancer metastases to the liver. All indications for surgery, and the characteristics of the background liver histology are listed in Supplementary Table 1. In particular, no patients had a diagnosis of chronic viral hepatitis or liver cirrhosis. We included sequential eligible patients, between the years 2010–2012, who were disease-free at the end of the operation and had a minimum one-year follow-up. Serum MPO-DNA complex levels for each sample were quantified and the fold-change was determined compared to healthy controls previously [14]. IL-33 serum levels were quantified and determined by ELISA (R&D Systems) All human materials used were obtained under an approved Institutional Review Board protocol. Written informed consent was received from all participants before inclusion.
Animals
Eight- to 12-week-old male wild-type (WT C57BL/6) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). IL-33 knockout (KO) mice were generated as described previously [15]. BALB/c ST2KO mice were originally obtained from A. McKenzie (Medical Research Council Laboratory of Molecular Biology, University of Cambridge, Cambridge, U.K.) [16] and provided by Dr. Heth R. Turnquist from the University of Pittsburgh (Pittsburgh, PA) [17]. Animal protocol approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh and the experiments were performed in adherence to National Institutes of Health guidelines for the use of laboratory animals.
Liver I/R
A nonlethal model of segmental (70%) hepatic warm ischemia and reperfusion was used [18]. Mice received ST2 Fc protein (100 µg per mouse; R&D Systems) intravenously 30 minutes before ischemia, as previously described [19]. Recombinant mouse IL-33 (carrier-free, 4, 8, or 10 µg per mouse; BioLegend) or PBS were injected intraperitoneally 1 h before or immediately after ischemia. Sham animals underwent anesthesia, laparotomy, and exposure of the portal triad without liver ischemia. Animals were sacrificed at predetermined time points (1 or 6 hours).
Neutrophil depletion, isolation, and adoptive transfer
Mouse neutrophils were isolated from bone marrow of tibias and femurs as described previously [5]. Neutrophil depletion was performed as described previously [20] with an intra-peritoneal injection of 500 µg anti-Ly6G antibody (1A8) (BioXCell) 24 and 2 hours before I/R. ST2 KO or WT freshly isolated neutrophils were injected into the spleens of WT mice just before I/R.
Quantification of NETs
To quantify NETs in cell culture supernatant or in mouse serum specifically, a capture ELISA myeloperoxidase (MPO) associated with DNA was performed as described [5]. Serum HMGB1, a marker of NETs [14], was quantified using an ELISA kit (IBL International). Serum nucleosome quantification was performed using the Cell Death kit (Roche).
Isolation, culture and treatment of hepatocytes, Nonparenchymal Cells and Kupffer cells
Hepatocytes were isolated, plated (3×106 cells/plate) and stimulated with hypoxia as previously described [21]. Supernatants from hypoxic or necrotic hepatocytes were harvested and used as conditioned media in subsequent co-culture assays. KCs were collected as previously described [22]. Mouse liver sinusoidal endothelial cell (LSEC) line was purchased from Cell Biologics or isolated from the liver Nonparenchymal cells (NPCs) using AutoMacs™ pro cell suspension. NPCs were stained with CD146 microbeads (Miltanyi) for 15 mins and resuspended in 5% RPMI-Hyclone Media.
ELISA
Serum IL-33, tumor necrosis factor α (TNF-α) and IL-6 levels in the mouse were detected by ELISA (R&D Systems) according to the manufacturer’s instructions. HMGB1 was quantified using ELISA kit (IBL International). Serum histones quantification was performed using Cell Death Kit (Roche).
Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis and Western Blotting
Western blot analysis for IL-33 (Nessy-1 Enzo Life Science); Cit-H3 (Abcam), ST2, HMGB1, histone H2A 2B, histone H3, histone H4 (Abcam); histone H1 (Santa Cruz); β-actin (Sigma); phospho-p38, p38, phospho-JNK, JNK, ERK, phospho-ERK, p65, and phospho-p65 (Cell Signaling), were performed. Protein extraction and Western blot analysis were performed following a standard protocol as described [23].
Real-Time Quantitative Live-Cell Analysis and visualization of NET formation
For Real-Time Quantitative Live-Cell Analysis, the NET formation was visualized and confirmed by labeling extracellular DNA with SYTOX Green (5 µM, Invitrogen) [24]. In brief, neutrophils were plated in a 12-well plate. NET formation was determined by plating the cells in the IncuCyte™ ZOOM Live-Cell Analysis System (Essen Bioscience, Ann Arbor, MI), which enables the automated noninvasive monitoring of live neutrophils in culture. Nine different locations per well were imaged every 10 minutes for NET formation.
Statistical Analysis
Results were expressed as the mean ± SE. Group comparisons were performed using ANOVA and Paired student’s t-test. For the human subjects’ data analysis, we compared the means of pre-operative and post-operative IL-33 serum levels using a paired student’s t representing the data graphically with their 95% confidence interval. The baseline characteristics of each group were compared using chi-square or Fisher’s exact tests for categorical variables. A p<0.05 was considered statistically significant and p<0.001 was considered highly significant.
Results
Serum IL-33 was significantly increased and correlated with NET formation in human patients underwent liver resection and up-regulated and released from LSECs after liver I/R in mice
Serum IL-33 levels have been shown to be significantly increased in patients with liver failure [25], viral hepatitis [26] and biliary atresia [27] compared with healthy controls. We measured IL-33 serum levels in a total of 41 patients, between the years 2010–2012, undergoing liver resection and 10 healthy age/gender-matched subjects. We compared preoperative serum IL-33 levels with healthy control and found no significant difference (median 2.169 pg/mL vs. median 1.716 pg/mL, p=0.24). Of note, when pre-operative IL-33 levels were analysed by liver background, no difference was observed based on liver histology that included various degrees of fatty liver, but no viral hepatitis or cirrhosis. Although recent data show that IL-33 levels may be increased at the tissue level in primary colorectal cancer [28] and in steatohepatitis [29]. In our cohort of human patients (Supplementary Table 1) this was not reflected in pre-operative IL-33 serum levels. A paired analysis of pre-operative and post-operative IL-33 levels was performed for 36 of 41 available serum pairs (Fig. 1A). Liver surgery resulted in a 5.1-fold increase in post-operative IL-33 compared with pre-operative IL-33 serum levels (Median 11 pg/mL vs. median 2.16 pg/ml, p<0.0001). When stratified for the extent of surgery, IL-33 levels after major resection were higher than after minor liver resection (Median 10.51 vs. 3.1 pg/ml, p<0.001) (Fig. 1B). Interestingly, there was a strong correlation between postoperative IL-33 levels and NET formation as measured by serum MPO-DNA complexes (a specific marker of NETs) (Spearman’s coefficient 0.7, p<0.001) (Fig. 1C).
Fig. 1. Sterile inflammatory injury causes increased secretion of serum IL-33 by LSECs accompanying NETs formation.

(A) Paired analysis of pre-operative and post-operative (n=36) IL-33 levels in patients (p<0.0001). (B) Serum IL-33 levels in patients undergoing major (n=32) or minor liver surgery (n=9) or in healthy controls (n=10). Higher serum IL-33 levels in liver resected patients (Median 3.1 pg/mL after minor and 10.51 pg/mL after major liver resection,) when compared with healthy control (Median 1.63 pg/mL). (C) Serum IL-33 and MPO-DNA levels showed increased correlation coefficient when measured postoperatively (Spearman’s coefficient 0.7, p<0.001). (D) Protein and serum levels of IL-33 were higher in liver ischemic tissue at 1h and 6h than sham mice; mean ± SEM (n=6–10). *,** P < 0.05 versus sham. (E) Immunofluorescent staining reveals an increase colocalization of LSECs and IL-33 staining after I/R
In vivo, following 60 min of warm ischemia, IL-33 protein expression in ischemic liver tissue was dramatically up-regulated as early as 1 h after reperfusion and then increased 6 h after reperfusion. Also, serum levels of IL-33 were significantly higher in mice with liver I/R at both 1 h and 6 h than in sham mice (Fig. 1D). Using immunofluorescent staining, we observed that LYVE-1 [30], a marker of LSECs, co-localized with IL-33, suggesting that LSECs are the major source of IL-33 after I/R (Fig. 1E).
IL33 promotes NETs formation during hepatic I/R
We recently demonstrated that NET formation occurs following liver I/R [5]. To determine whether IL-33 contributes to NET formation, we subjected IL-33 KO mice to liver I/R. Serum levels of circulating MPO-DNA complexes were significantly reduced in IL-33 KO mice compared to their WT counterparts (Fig. 2A). Serum levels of nucleosomes and HMGB1 after I/R, surrogate markers for NET formation, were also significantly less in IL-33 KO mice compared to WT controls (Supplementary Fig. 1A). Additionally, liver tissue from IL-33 KO mice had significantly lower levels of citrullinated histone H3 (cit-H3), another specific marker of NET formation, compared to WT mice undergoing hepatic I/R (Fig. 2A). IL-33 KO mice were protected from liver I/R injury with significantly lower levels of circulating sALT and lower extent of liver necrosis after hepatic I/R by histologic examination (Fig. 2B and Supplementary Fig. 1B). Of note, the number of neutrophils infiltration after hepatic I/R were significantly higher in the WT mice compared to IL-33KO (Supplementary Fig. 1E). Serum levels of TNF-α and IL-6 in IL-33 KO mice, two cytokines shown to be important in the pathogenesis of I/R injury [21], were also lower than in WT mice (Fig. 2C).
Fig. 2. IL-33 depletion leads to protective effects against inflammatory liver injury after I/R.

(A) Serum MPO-DNA levels and cit-H3 levels were measured in IL-33 KO mice after I/R. *P<0.05 versus WT. (B–C) Less serum ALT, quantification of necrotic area, and TNF-α and IL-6 levels were observed in IL-33 KO mice after I/R. *P<0.05 versus IL-33 WT. (D) Increased serum MPO-DNA levels and citH3 protein levels were observed in rIL-33 administered mice. *P<0.05 versus. PBS-treated mice. (E–F) Increased serum ALT, quantification of necrotic area, and TNF-α and IL-6 levels were observed in rIL-33 administered mice. *P<0.05 versus PBS-treated mice.
To confirm the functional role of IL-33 in NET formation after liver I/R, recombinant mouse IL-33 (rIL-33) was administered to mice immediately after I/R. rIL-33 at a dose of 10 µg injected intraperitoneally or intravenously caused no elevation of sALT in sham treated mice (data not shown). NET formation as measured by serum levels of MPO-DNA complexes, nucleosomes, and HMGB1, as well as tissue levels of cit-H3 in ischemic liver lobes were significantly increased in rIL-33-treated I/R mice compared to PBS-treated I/R mice (Fig. 2D and Supplementary Fig. 1C). Similarly, increased sALT levels (Fig. 2E), greater extent of necrosis (Fig. 2E and Supplementary Fig. 1D) in ischemic liver lobes as well as increased serum levels of TNF-α and IL-6, were observed in rIL-33 treated mice after I/R compared to PBS-treated mice. (Fig. 2F). These results suggest that IL-33 can further drive NET formation and concurrent tissue toxic inflammatory response following hepatic I/R.
Interestingly, Sakai et al. found seemingly contradictory results showing less hepatotoxicity if IL-33 was administered before I/R [31]. We confirmed these results and found significantly less NET formation and injury in mice with an injection of rIL-33 before I/R (Supplementary Fig. 2A–E). While rIL-33 administration exacerbated liver injury compared to controls after I/R, our data suggested that post-ischemic treatment of IL-33 functions to amplify the inflammatory cascade.
IL-33 modulates inflammatory signaling pathways after liver I/R
To further investigate how IL-33 might mediate the inflammatory response in liver I/R injury, we examined the mitogen-activated protein kinases (MAPK) and NF-κB signaling pathways. After 1 h of hepatic I/R, phosphorylation of c-Jun N-terminal kinase (JNK), p38, and extracellular signal-regulated kinase (ERK) increased compared to sham treated mice. In contrast, activation of these MAPK proteins was abrogated when IL-33 KO mice were subjected to I/R (Supplementary Fig. 3). Additionally, we observed an increase in NF-κB activation after 1 h of I/R in WT liver tissue by phosphorylation at serine 536 of the p65 subunit in WT mice, and this effect was significantly blunted in IL-33 KO mice (Supplementary Fig. 3). These results suggest that absence of IL-33 during I/R reduces activation of MAPKs and NF-κB in the liver, which is consistent with the findings that these mice have decreased inflammatory responses after I/R.
To determine the role of IL-33 in modulating the recruitment of innate immune cells in the liver following I/R, flow cytometry analysis with quantitative evaluation of DCs, neutrophils, inflammatory monocytes, and NK cells in ischemic liver tissue was performed. Liver I/R injury in WT mice led to significantly increased recruitment of DCs, neutrophils, inflammatory monocytes, and NK cells compared with sham WT mice (Supplementary Fig. 4). However, the ablation of IL-33 conferred a stable innate immune environment and numbers of DCs, neutrophils, inflammatory monocytes, and NK cells remained unchanged after liver I/R compared with sham KO mice. These data demonstrate that the absence of IL-33 down-regulates inflammatory immune responses by decreasing the influx of innate immune cells in the ischemic tissue after liver I/R.
IL-33 released from hypoxic LSECs stimulates neutrophils to form NETs in vitro
DAMPs are released from stressed cells during liver I/R to invoke an inflammatory response and subsequent liver damage [21, 23]. Consistent with other studies [19, 32], we also found IL-33 only localized in LSECs in the normal liver and overexpressed in both LSECs and hepatocytes after liver I/R (Fig. 1E). In vitro, we isolated murine LSECs or hepatocytes and exposed the cells in culture to hypoxia to mimic the ischemic microenvironment of I/R injury. IL-33 protein expression was significantly higher in LSECs than in hepatocytes at baseline and both were further up-regulated after stimulation with hypoxia (Supplementary Fig. 5A). Of note, we further isolated murine Hepatic Stellate Cells (HSC) and Kupffer cells and exposed them to hypoxia. Similarly, IL-33 protein expression was higher in LSECs than in HSC or Kupffer cells after stimulation with hypoxia (Supplementary Fig. 5B). Immunofluorescent staining of IL-33 in LSECs isolated from normoxic WT mice revealed strong nuclear staining whereas the LSECs exposed to hypoxia (1% oxygen) led to translocation of IL-33 into cytoplasm (Supplementary Fig. 6A).
Bone marrow derived neutrophils were incubated with the conditioned media from hypoxic or normoxic LSECs. Similar to the PMA-treated neutrophils (positive control), significantly higher levels of MPO-DNA complexes and cit-H3 levels were observed after stimulation of hypoxic LSECs media, compared to normal media stimulation (negative control) (Fig. 3A and Fig. 3B). Conversely, IL-33 neutralizing antibody treatment reduced MPO-DNA complex levels in hypoxic media-treated neutrophils. Similarly, NET formation was decreased after stimulation with conditioned media from hypoxic IL-33 KO LSECs compared with IL-33 WT LSECs. These results were confirmed by immunofluorescent imaging (Supplementary Fig. 6B).
Fig. 3. IL-33 released from hypoxic LSECs stimulates neutrophils to form NETs.

(A) Bone marrow derived neutrophils were cultured with conditioned media from hypoxic LSECs. Higher MPO-DNA levels were observed in the neutrophil media after stimulation of hypoxic media compared to levels in normoxic media. This effect was reduced significantly when treated with the anti-IL-33 neutralizing antibody (P<0.05). (B) Serum MPO-DNA levels demonstrate rIL-33 dose-dependent-increase when administered exogenously. When compared with rIL-33, neutrophils treated with LSECs hypoxia medium also demonstrated increase Cit-H3 protein levels. This was confirmed by immunofluorescence confocal microscopy Green: IL-33; red: actin; blue: DAPI (C). (D–E) NET formation was visualized in real-time by Incucyte® intravital confocal microscopy in response to rIL-33 indicating NET formation. This was quantified based on intra-vital video showing stimulation of rIL-33 significantly increased NET formation compared to PBS control and PMA.
We next determined whether IL-33 induces NET formation directly. Mouse bone marrow derived-neutrophils treated with different concentrations of rIL-33 demonstrated a dose-dependent increase in MPO-DNA complexes and cit-H3 levels (Fig. 3B and Supplementary Fig. 6C). Immunofluorescence confocal microscopy confirmed NET formation after stimulation of rIL-33 (Fig. 3C). For additional confirmation, we visualized NET formation by Incucyte® intravital confocal microscopy in real time in response to rIL-33 in vitro. Real-time video acquisition demonstrated a stable and robust SYTOX green release, indicating NET formation, beginning 1 hour after stimulation by rIL-33. Quantification of NET formation based on intra-vital video has also shown that stimulation of rIL-33 significantly increased NET formation compared with PBS control and almost as much as the PMA. (Fig. 3D and 3E and Supplementary video 1).
IL-33 induces NET formation through ST2 signaling
Although IL-33 has been shown to enhance neutrophil recruitment, whether neutrophils express IL-33 membrane receptor ST2L, remains controversial [11, 33]. Here, we confirm that neutrophils expressed the IL-33 receptor ST2L at baseline. Both mRNA and protein levels of ST2L increased dramatically after PMA or rIL-33 stimulation (Supplementary Fig. 6D and 6E), suggesting that neutrophils express ST2L that is up-regulated in response to rIL-33.
We next sought to determine whether IL33 induces NET formation by activating neutrophil ST2L. We measured MPO-DNA complexes in the media of ST2 KO neutrophils after stimulation of rIL-33, and found a significant decrease compared to WT neutrophils after stimulation (Fig. 4A). ST2 WT neutrophils stimulated with rIL-33 exhibited increased citrullination of histone H3 compared to untreated WT neutrophils. ST2 KO neutrophils failed to citrullinate histone H3 in response to rIL-33 (Fig. 4B). Furthermore, no NETs were observed in ST2 KO neutrophils after stimulation of exogenous rIL-33 and LSECs hypoxia media by immunofluorescence confocal microscopy (Fig. 4C and Supplementary Fig. 6F). Quantification of NETs by Incucyte® video revealed that rIL-33 stimulation failed to induce NETs in ST2 KO neutrophils (Fig. 4D). Collectively, these results suggest that IL-33 released from LSECs induced NET formation in naïve neutrophils in vitro through ST2L signaling pathways.
Fig. 4. NET formation is induced by IL-33 via ST2 signaling pathway.

(A) Quantification of MPO-DNA complexes in ST2 KO neutrophil media with rIL-33 treatment demonstrated a significant decrease (*P<0.05) compared to WT neutrophil media after stimulation. (B) ST2 WT neutrophils treated with rIL-33 exhibited increased cit-H3 compared to untreated neutrophils whereas ST2 KO neutrophils failed to citrullinate histone H3 in response to stimulation of rIL-33. (C) Confocal microscopy images revealed no NETs formation in ST2 KO neutrophils after rIL-33 stimulation. Green: IL-33; red: actin; blue: DAPI. (D) rIL-33 stimulation failed to induce NETs in ST2 KO neutrophils when quantified by Incucytes® intravital confocal microscopy.
NET formation after liver I/R in mice is dependent on IL33/ST2 signaling
To determine if IL-33 induces NET formation through ST2 in vivo, we subjected ST2 KO mice to liver I/R. We found that serum levels of circulating MPO-DNA complexes, nucleosomes and HMGB1, were significantly reduced in ST2 KO mice compared to their wildtype counterparts (Fig. 5A and Supplementary Fig. 7A). Of note, decreased number of neutrophils infiltration after hepatic I/R were observed in the ST2 KO mice compared to WT (Supplementary Fig. 7E). Similarly, less cit-H3 was observed in ischemic liver lobes in ST2 KO mice compared with WT mice after liver I/R (Fig. 5A). ST2 KO mice were found to be protected (Fig. 5B and Supplementary Fig. 7B) and had decreased serum levels of the inflammatory mediators, TNF-α and IL-6 response following liver I/R compared to their WT counterparts (Fig. 5C). Similar results of decreased hepatocellular injury and NET formation were also seen in sST2-treated mice compared to PBS-treated mice (Fig. 5D–F, Supplementary Fig. 7C and 7D).
Fig. 5. NETs formation in mice is dependent on IL-33/ST2 signaling pathway after liver Ischemia and reperfusion.

(A) Serum MPO-DNA levels and cit-H3 protein levels were measured in ST2 KO mice after I/R. *P<0.05 versus ST2 WT. (B–C) Decreased serum ALT, quantification of necrotic area, TNF-α and IL-6 levels were also observed in ST2 KO mice after I/R. *P<0.05 versus ST2 WT. (D) Decreased serum MPO-DNA complex levels and cit-H3 protein levels were observed. Data represents significant fold decrease in sST2 administered mice compare to PBS-treated mice. (E–F) Decreased serum ALT, quantification of necrotic area, TNF-α and IL-6 levels were observed in sST2 administered mice. *P<0.05 versus PBS-treated mice.
To further study whether NET formation during liver I/R is specifically dependent on ST2 on neutrophils in response to IL-33, we performed neutrophil depletion in WT mice by injection of anti-Ly6G monoclonal antibody. Neutrophils obtained from ST2 KO or WT mice were adoptively transferred into neutrophil-depleted WT mice that were then subjected liver I/R. We found that adoptive transfer of ST2 KO neutrophils resulted in significantly fewer circulating MPO-DNA complexes and lower tissue levels of citrullinated-histone H3 compared to WT mice receiving adoptive transfer of WT neutrophils (Fig. 6A). These results demonstrate that NET formation after liver I/R is a result of ST2 within circulating neutrophils. Moreover, liver damage, indicated by sALT levels, was significantly reduced in the mice receiving adoptive transfer of ST2 KO neutrophils and correlated with decreased NET formation (Fig. 6B).
Fig. 6. Adoptive transfer of ST2 KO neutrophils in WT neutrophil-depleted mice resulted in protective effects after liver I/R.

(A) Serum MPO-DNA levels and tissue cit-H3 protein levels were measured. Adoptive transfer of ST2 KO neutrophils resulted in significantly fewer circulating MPO-DNA and lower cit-H3 expression compared to WT mice receiving adoptive transfer of WT neutrophils. (B) Significantly reduced serum ALT levels indicated reduced liver injury in adoptively transferred ST2 KO neutrophils mice. (C) During liver I/R, DAMP IL-33 released from damaged LESCs, acts in a paracrine manner on ST2-expressing neutrophils to form NETs. Formation of NETs during I/R propagate hepatotoxicity and systemic inflammation.
Discussion
The cellular responses to I/R encompass a diverse network that includes cell injury and death due to innate and adaptive immune responses which ultimately culminate in end organ damage and dysfunction. It is known that the late phase of I/R is dependent on infiltrating immune cells and innate immune signaling [2]. In our present study, we describe an innate immune response to I/R by which the “dual-function” alarmin IL-33 initiates a feed-forward signaling pathway in neutrophils. Under liver I/R stress, IL-33 released from LSECs drives infiltrated neutrophils to form NETs through the IL-33/ST2 signaling pathway, consequently promoting hepatotoxicity and systemic inflammation.
Various types of cells can be stimulated to produce and release IL-33 [6], especially barrier cells such as keratinocytes, epithelial cells, fibroblasts, endothelial cells. Activated leukocytes, such as macrophages, dendritic cells and mast cells are also sources of IL-33. In the liver, LSECs, hepatocytes and hepatic stellate cells have been reported to express and release IL-33 in response to hepatic stress and injury [19, 34, 35]. We found that IL-33 is mainly expressed in LSECs in the normal liver and that expression of IL-33 in LSECs was drastically increased during liver I/R. Compared with hepatocytes and other cells LSECs serve as the main source of IL-33 after stimulation with hypoxia (Supplementary Fig. 5B).
The response to IL-33 signaling is determined by the cell types that express its receptor ST2. The main IL-33-responsive cell types are immune cells, including CD4+ T cells, CD8+ T cells, Treg cells, dendritic cells, NK cells, NKT cells, mast cells, eosinophils, and macrophages [7]. In context, these cell types shape an organ and tissue-specific response within their respective microenvironment. However, little is known about the role of IL33/ST2 signaling in neutrophils. Whether neutrophils express ST2 is controversial [11, 13, 33]. In our findings, we show that mouse bone marrow-derived neutrophils express ST2 and that expression is markedly increased by PMA or IL-33. Our present study provides here, to our knowledge, the first evidence that IL-33 directly induces neutrophils through ST2 to form NETs and that this can be blocked by gene depletion of ST2 on neutrophils. Our human data shows consistent results that elevated IL-33 serum levels in patients undergoing liver resection strongly correlates with postoperative NET production. We recently demonstrated that HMGB1 and histones from damaged hepatocytes, serve as DAMPs, elicit NET formation during liver sterile inflammation which exacerbates organ damage and inflammatory response [5]. Here, we show that another nuclear protein released into the extracellular space after cellular insult, IL-33 has similar DAMP functions and can initiate NET formation during liver I/R. Additionally, we found that damaged LSECs serves as the major resource of IL-33 during liver I/R. Whether damaged or stressed LSECs actively or passively released IL-33 in response to liver I/R injury remains unclear and needs further investigation.
IL-33 has been previously demonstrated as hepato-protective with IL-33 pre-treatment or anti-ST2 neutralizing antibody treatment in a mouse model of warm hepatic I/R [31]. However, over-expression of sST2 before I/R has been shown to reduce injury, indicating the pathophysiologic role of IL-33/ST2 signaling in warm I/R [36]. While we confirmed this protective effect with pre-treatment of IL-33 in liver I/R, we observed that IL-33 promotes NET formation, causing liver damage and systemic inflammation when administered after ischemia. The discordance of these findings seems to be a result of using different time points of IL-33 treatment. Pre-treatment of IL-33 thus might confer a pre-conditioning effect on the liver, a phenomenon that has been well-documented [37]. Additionally, pre-treatment with IL-33 alters the immune cell phenotype of ST2-expressing cells in the liver - including DC’s, NK-T cells, and regulatory T cells [38] – which might well reduce the subsequent innate immune response after ischemic injury. Also, pre-treatment of IL-33 might lead to sST2 secretion as a decoy receptor of IL-33, against its further damage effects. This should be an active area of future investigation into the seeming paradoxical function of IL-33 in liver injury.
Interestingly, recent studies have also demonstrated controversial roles of IL-33 in the model of Concanavalin-A (Con-A) induced-acute hepatitis [15] [39] [40]. Furthermore, IL-33/ST2 signaling has been shown to have a protective role in other diseases [41, 42]. While these studies shed light into the potential mechanism of IL-33, it is unknown whether these mechanisms may be operant in the same manner during I/R injury. IL-33 role may depend on inflammation site and stimulant types or timing in relation to the hepatotoxic insult.
In conclusion, we demonstrate a novel pathologic mechanism whereby warm hepatic I/R injury causes up-regulation and release of the DAMP molecule IL-33 primarily from LSECs, which acts in a paracrine manner on ST2-expressing neutrophils to form NETs, to propagate hepatotoxicity and systemic inflammation (Fig. 6C). These findings confirm the notion that endogenous DAMP signaling drives sterile inflammatory responses after I/R injury. Furthermore, measures to disrupt the IL-33/ST2 signaling axis - such as administration of IL-33 decoy receptor, sST2 or neutralization of IL-33 - during I/R would protect against hepatic inflammatory injury and confer a potential therapeutic intervention for sterile inflammation occurring liver surgeries.
Supplementary Material
Highlights.
Patients underwent major liver resection showed a significant increase in serum IL-33 levels
Evaluation of IL-33 forming NETs via ST2 signaling pathway is investigated in liver I/R model
LSECs released IL-33 resulted in sterile inflammation after hepatic I/R by inducing NET formation
Lay summary.
Liver ischemia and reperfusion injury results in the formation of neutrophil extracellular traps inevitably contribute to organ damage in liver surgeries. Here we show, IL-33 is released from liver sinusoidal endothelial cells to promote NET formation during liver I/R, in turn, exacerbate inflammatory cascades augmenting sterile inflammation.
Acknowledgments
We thank Xinghuang Liao, Nicole Hays, Junda Chen, Hongji Zhang and Kimberly Ferrero for technical assistance in preparing the manuscript. We thank Jannat Malik for linguistic revision.
Financial Support: This work was supported by Howard Hughes Medical Institute Physician-Scientist Award (A.T.), R01-GM95566 (A.T.), R00-HL097155, R01HL122489 and 1R21AI121981 (H.T), and National Natural Science Foundation of China Grant No. 81470902 (H.H.).
Abbreviations
- DAMP
Damage-associated molecular pattern
- PAMP
Pathogen-associated molecular pattern
- LSEC
Liver sinusoidal endothelial cell
- PRR
pattern recognition receptor
- TLR
Toll-Like Receptor
- I/R
Ischemia/Reperfusion
- HMGB1
High Mobility Group Box 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: Authors disclose no conflicts
Authors contributions:
To design and study conception: HY, HWC, AT, HH
To acquisition of data: HY, HWC, ST, DJVAW, PL, BRR, DBS, HRT, HH
To analysis and interpretation: HY, HWC, ST, DJVAW, PL, BRR, VS, DBS, HRT, AT, HH
Participation in drafting: HY, HH
Participation in revising: HY, HWC, ST, HRT, AT, HH
All authors approved the final version.
Reference List
- 1.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143:1158–1172. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Lu L, Zhou H, Ni M, Wang X, Busuttil R, Kupiec-Weglinski J, et al. Innate Immune Regulations and Liver Ischemia-Reperfusion Injury. Transplantation. 2016;100:2601–2610. doi: 10.1097/TP.0000000000001411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec-Weglinski JW. Ischaemia-reperfusion injury in liver transplantation--from bench to bedside. Nature reviews Gastroenterology & hepatology. 2013;10:79–89. doi: 10.1038/nrgastro.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peralta C, Jimenez-Castro MB, Gracia-Sancho J. Hepatic ischemia and reperfusion injury: effects on the liver sinusoidal milieu. Journal of hepatology. 2013;59:1094–1106. doi: 10.1016/j.jhep.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 5.Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. DAMPs-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. 2015 doi: 10.1002/hep.27841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin NT, Martin MU. Interleukin 33 is a guardian of barriers and a local alarmin. Nature immunology. 2016;17:122–131. doi: 10.1038/ni.3370. [DOI] [PubMed] [Google Scholar]
- 7.Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nature reviews Immunology. 2016;16:676–689. doi: 10.1038/nri.2016.95. [DOI] [PubMed] [Google Scholar]
- 8.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 9.Bergers G, Reikerstorfer A, Braselmann S, Graninger P, Busslinger M. Alternative promoter usage of the Fos-responsive gene Fit-1 generates mRNA isoforms coding for either secreted or membrane-bound proteins related to the IL-1 receptor. The EMBO journal. 1994;13:1176–1188. doi: 10.1002/j.1460-2075.1994.tb06367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kakkar R, Lee RT. The IL-33/ST2 pathway: therapeutic target and novel biomarker. NatRevDrug Discov. 2008;7:827–840. doi: 10.1038/nrd2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alves-Filho JC, Sonego F, Souto FO, Freitas A, Verri WA, Jr, Auxiliadora-Martins M, et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. NatMed. 2010;16:708–712. doi: 10.1038/nm.2156. [DOI] [PubMed] [Google Scholar]
- 12.Hueber AJ, Alves-Filho JC, Asquith DL, Michels C, Millar NL, Reilly JH, et al. IL-33 induces skin inflammation with mast cell and neutrophil activation. EurJImmunol. 2011;41:2229–2237. doi: 10.1002/eji.201041360. [DOI] [PubMed] [Google Scholar]
- 13.Verri WA, Jr, Souto FO, Vieira SM, Almeida SC, Fukada SY, Xu D, et al. IL-33 induces neutrophil migration in rheumatoid arthritis and is a target of anti-TNF therapy. AnnRheumDis. 2010;69:1697–1703. doi: 10.1136/ard.2009.122655. [DOI] [PubMed] [Google Scholar]
- 14.Tohme S, Yazadani HO, Al-Khafaji AB, Chidi AP, Lougharn P, Mowen K, et al. Neutrophil Extracellular Traps Promote the Development and Progression of Liver Metastases after Surgical Stress. Cancer Res. 2016 doi: 10.1158/0008-5472.CAN-15-1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, Ishii A, et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. ProcNatlAcadSciUSA. 2010;107:18581–18586. doi: 10.1073/pnas.1003059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Townsend MJ, Fallon PG, Matthews DJ, Jolin HE, McKenzie AN. T1/ST2-deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. J Exp Med. 2000;191:1069–1076. doi: 10.1084/jem.191.6.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matta BM, Lott JM, Mathews LR, Liu Q, Rosborough BR, Blazar BR, et al. IL-33 is an unconventional Alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2+ regulatory T cells. J Immunol. 2014;193:4010–4020. doi: 10.4049/jimmunol.1400481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsung A, Stang MT, Ikeda A, Critchlow ND, Izuishi K, Nakao A, et al. The transcription factor interferon regulatory factor-1 mediates liver damage during ischemia-reperfusion injury. AmJPhysiol GastrointestLiver Physiol. 2006;290:G1261–G1268. doi: 10.1152/ajpgi.00460.2005. [DOI] [PubMed] [Google Scholar]
- 19.Marvie P, Lisbonne M, L'helgoualc'h A, Rauch M, Turlin B, Preisser L, et al. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. JCell MolMed. 2010;14:1726–1739. doi: 10.1111/j.1582-4934.2009.00801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, Dematteo RP. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology. 2009 doi: 10.1002/hep.23365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, et al. Endogenous histones function as alarmins in sterile inflammatory liver injury through toll-like receptor 9. Hepatology. 2011 doi: 10.1002/hep.24501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang H, Chen HW, Evankovich J, Yan W, Rosborough BR, Nace GW, et al. Histones Activate the NLRP3 Inflammasome in Kupffer Cells during Sterile Inflammatory Liver Injury. Journal of immunology. 2013;191:2665–2679. doi: 10.4049/jimmunol.1202733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. JExpMed. 2005;201:1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:2817–2822. doi: 10.1073/pnas.1414055112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roth GA, Zimmermann M, Lubsczyk BA, Pilz J, Faybik P, Hetz H, et al. Up-regulation of interleukin 33 and soluble ST2 serum levels in liver failure. JSurgRes. 2010;163:e79–e83. doi: 10.1016/j.jss.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 26.Cacopardo B, Rita Pinzone M, Palermo F, Nunnari G. Changes in serum Interleukin-33 concentration before and after treatment with pegylated interferon alfa-2a plus ribavirin in patients with chronic hepatitis C genotype 1b infection. Hepat Mon. 2012;12:e7611. doi: 10.5812/hepatmon.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Razumilava N, Gores GJ, Walters S, Mizuochi T, Mourya R, et al. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. The Journal of clinical investigation. 2014;124:3241–3251. doi: 10.1172/JCI73742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Davis C, Shah S, Hughes D, Ryan JC, Altomare D, et al. IL-33 promotes growth and liver metastasis of colorectal cancer in mice by remodeling the tumor microenvironment and inducing angiogenesis. Mol Carcinog. 2017;56:272–287. doi: 10.1002/mc.22491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vasseur P, Dion S, Filliol A, Genet V, Lucas-Clerc C, Jean-Philippe G, et al. Endogenous IL-33 has no effect on the progression of fibrosis during experimental steatohepatitis. Oncotarget. 2017 doi: 10.18632/oncotarget.18335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strauss O, Phillips A, Ruggiero K, Bartlett A, Dunbar PR. Immunofluorescence identifies distinct subsets of endothelial cells in the human liver. Sci Rep. 2017;7:44356. doi: 10.1038/srep44356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakai N, Van Sweringen HL, Quillin RC, Schuster R, Blanchard J, Burns JM, et al. Interleukin-33 Is hepatoprotective during liver ischemia/reperfusion in mice. Hepatology. 2012;56:1468–1478. doi: 10.1002/hep.25768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel 'alarmin'? PloS one. 2008;3:e3331. doi: 10.1371/journal.pone.0003331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. The Journal of allergy and clinical immunology. 2008;121:1484–1490. doi: 10.1016/j.jaci.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arshad MI, Piquet-Pellorce C, L'helgoualc'h A, Rauch M, Patrat-Delon S, Ezan F, et al. TRAIL but not FasL and TNFalpha, regulates IL-33 expression in murine hepatocytes during acute hepatitis. Hepatology. 2012 doi: 10.1002/hep.25893. [DOI] [PubMed] [Google Scholar]
- 35.McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, et al. Interleukin-33- dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. 2013;39:357–371. doi: 10.1016/j.immuni.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin H, Huang BJ, Yang H, Huang YF, Xiong P, Zheng F, et al. Pretreatment with soluble ST2 reduces warm hepatic ischemia/reperfusion injury. BiochemBiophysResCommun. 2006;351:940–946. doi: 10.1016/j.bbrc.2006.10.166. [DOI] [PubMed] [Google Scholar]
- 37.Montalvo-Jave EE, Pina E, Montalvo-Arenas C, Urrutia R, Benavente-Chenhalls L, Pena-Sanchez J, et al. Role of ischemic preconditioning in liver surgery and hepatic transplantation. Journal of gastrointestinal surgery : official journal of the Society for Surgery of the Alimentary Tract. 2009;13:2074–2083. doi: 10.1007/s11605-009-0878-7. [DOI] [PubMed] [Google Scholar]
- 38.Turnquist HR, Zhao Z, Rosborough BR, Liu Q, Castellaneta A, Isse K, et al. IL-33 Expands Suppressive CD11b+ Gr-1int and Regulatory T Cells, including ST2L+ Foxp3+ Cells, and Mediates Regulatory T Cell- Dependent Promotion of Cardiac Allograft Survival. JImmunol. 2011;187:4598–4610. doi: 10.4049/jimmunol.1100519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Volarevic V, Mitrovic M, Milovanovic M, Zelen I, Nikolic I, Mitrovic S, et al. Protective role of IL-33/ST2 axis in Con A-induced hepatitis. JHepatol. 2011 doi: 10.1016/j.jhep.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 40.Arshad MI, Rauch M, L'helgoualc'h A, Julia V, Leite-de-Moraes MC, Lucas-Clerc C, et al. NKT cells are required to induce high IL-33 expression in hepatocytes during ConA-induced acute hepatitis. EurJImmunol. 2011;41:2341–2348. doi: 10.1002/eji.201041332. [DOI] [PubMed] [Google Scholar]
- 41.Chen WY, Hong J, Gannon J, Kakkar R, Lee RT. Myocardial pressure overload induces systemic inflammation through endothelial cell IL-33. Proceedings of the National Academy of Sciences of the United States of America. 2015 doi: 10.1073/pnas.1424236112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu AK, Hung KW, Yuen MY, Zhou X, Mak DS, Chan IC, et al. IL-33 ameliorates Alzheimer's disease-like pathology and cognitive decline. Proceedings of the National Academy of Sciences of the United States of America. 2016 doi: 10.1073/pnas.1604032113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.