

Abstract
Background
Allergic asthma is a heterogeneous chronic inflammatory disease of the airways with a massive infiltration of eosinophils or neutrophils mediated by allergen-specific TH2 and TH17 cells, respectively. Therefore successful treatment of allergic asthma will require suppression of both TH2 and TH17 cells.
Objective
We sought to investigate the role of the TH17 cell pathway in regulating TH2 cell responses in allergic asthma.
Methods
Allergic asthma was induced by intranasal challenge with proteinase allergens in C57BL/6, Il17a−/−Il17f−/−, and retinoic acid receptor–related orphan receptor γt (RORγt)gfp/gfp mice. A pharmacologic RORγt inhibitor was used to evaluate its preventive and therapeutic effects in allergic asthma. Characteristics of allergic airway inflammation were analyzed by using flow cytometry, histology, quantitative real-time PCR, and ELISA. Mixed bone marrow chimeric mice, fate mapping analysis, short hairpin RNA transduction, and in vitro T-cell differentiation were used for mechanistic studies.
Results
Mice deficient in IL-17A and IL-17F, as well as RORγt, exhibited a significant reduction not only in TH17 cell responses but also in TH2 cell responses in an animal model of allergic asthma. Similarly, mice treated with an RORγt inhibitor had significantly diminished TH17 and TH2 cell responses, leading to reduced neutrophil and eosinophil numbers in the airway. RORγt-deficient T cells were intrinsically defective in differentiating into TH2 cells and expressed increased levels of B-cell lymphoma 6 (Bcl6). Bcl6 knockdown resulted in a remarkable restoration of TH2 cell differentiation in RORγt- deficient T cells. Blockade of RORγt also significantly hampered the differentiation of human TH2 and TH17 cells from naive CD4+ T cells.
Conclusion
RORγt in T cells is required for optimal TH2 cell differentiation by suppressing Bcl6 expression; this finding suggests that targeting RORγt might be a promising approach for the treatment of allergic asthma by concomitantly suppressing TH17 and TH2 cell responses in the airway.
Keywords: Allergic asthma, TH17 cell, TH2 cell, retinoic acid receptor–related orphan receptor γt, B-cell lymphoma 6
Graphical Abstract
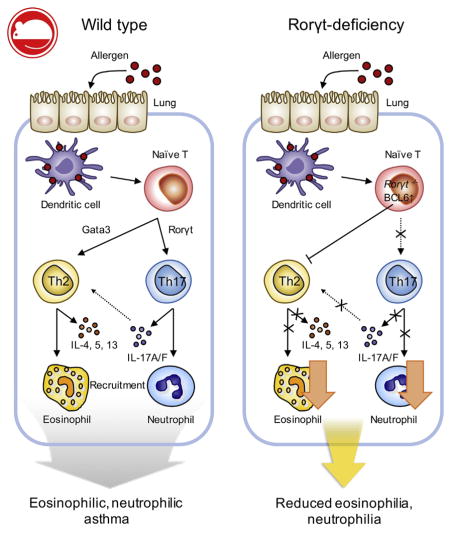
Asthma is a heterogeneous disease of the lung and airway characterized by distinct symptoms, such as airway hyperresponsiveness, mucus production, infiltration of inflammatory granulocytes, and shortness of breath, which could be life-threatening.1,2 The prevalence of this disease has markedly increased over the past several decades, and it has become one of the major global health problems, affecting approximately 300 million persons worldwide.1
Asthma can be categorized into allergic and nonallergic asthma based on the types of triggering stimuli.3 Allergic asthma is a common form of asthma caused by sensitization against allergens, such as pollen, house dust mites, fur dander from pets, or fungi, whereas nonallergic asthma is caused by irritants, such as tobacco smoke, ozone, diesel exhaust particles, or airborne virus.1,4
Allergic asthma has been considered to be mediated by TH2 cells; however, recent studies uncovered the involvement of TH17 cells as an additional critical contributor in the pathogenesis of allergic asthma in animal models and human subjects.5 TH2 cells mediate eosinophilic asthma by secreting type 2 cytokines, such as IL-4, IL-5, IL-9, and IL-13. These cytokines induce B-cell isotype switching to IgE (IL-4), recruit eosinophils (IL-5) and mast cells (IL-9 and IL-13) into the lung and airway, and induce goblet cell hyperplasia and tissue remodeling (IL-13), leading to airway hyperresponsiveness.2,6 On the other hand, TH17 cells have been regarded as a critical mediator of steroid-resistant neutrophilic asthma.7–9 Increased IL-17A levels were observed in the lungs of patients with asthma, and these were positively correlated with neutrophilic inflammation, increased airway hyperresponsiveness, and a steroid-resistant type of severe asthma.10 A mixed TH2 and TH17 cell response in the airways has been associated with the severity of allergic asthma.9,11 IL-17A stimulates airway epithelial cells to secrete the chemokines CXCL1 and CXCL8, which in turn recruit neutrophils.12 In addition, IL-17A causes airway remodeling through upregulation of α-smooth muscle actin in fibroblasts.13 Hence TH2 and TH17 cells exert nonredundant pathogenic roles during the development of allergic asthma by inducing eosinophilic and neutrophilic inflammation, respectively.
Because of their critical contributions to the development of allergic asthma, a number of experimental and clinical studies have addressed whether blockade of either the TH2 or TH17 cell pathway ameliorates clinical symptoms of the disease. In particular, multiple clinical trials have demonstrated that antibodies against TH2 cell cytokines (IL-4, IL-5, and IL-13) and their receptors in patients with moderate-to-severe asthma had limited clinical benefits. For instance, antibodies to IL-5 or its receptor are generally effective in reducing eosinophilia, whereas they have little to no effect on airway hyperresponsiveness.14 Antibodies to IL-13 or its receptor lead to reduced airway hyperresponsiveness, with little effect in eosinophilia.15,16 More recently, a human mAb to IL-17RA did not improve asthmatic symptoms in patients with moderate-to-severe asthma.17
In this regard Choy et al18 recently reported that antibodies to TH2 cell cytokines enhance TH17 cell responses and neutrophilia, whereas anti–IL-17A enhances TH2 cell responses and eosinophilia in animal models of allergic asthma. This study indicates that pulmonary TH2 and TH17 cell responses are mutually regulated and suggests that blockade of the TH cell pathway alone leads to exaggeration of the other pathway. Supporting this notion, GATA-3 and IL-13 are shown to inhibit TH17 cell differentiation.19,20 Hence combined blockade of both TH2 and TH17 cell pathways might be considered to achieve therapeutic benefits in controlling asthma without adverse effects.
In the present study we aimed to investigate the role of the TH17 pathway in regulating TH2 cell responses in allergic airway inflammation by using an animal model of proteinase-induced allergic asthma. We also aimed to investigate the clinical relevance of blocking the TH17 pathway by analyzing T cells from patients with allergic asthma.
METHODS
Ethics statements
Animal experiments, including induction of allergic lung inflammation in Il17a−/−Il17f−/− double-deficient mice and subsequent analysis, fate mapping, and adoptive transfer of in vitro–differentiated TH17 cells, were done with protocols approved by Institutional Animal Care and Use Committees of the MD Anderson Cancer Center. All remaining animal experiments were reviewed and approved by Seoul National University Institutional Animal Care and Use Committee (IACUC nos. SNU-140602-2-7 and SNU-140217-6-8). Collection of human blood samples from healthy volunteers and subsequent experimental procedures were reviewed and approved by the Seoul National University Institutional Review Board. The protocol approval number is 1608/001-006. Collection of blood samples from patients with allergic asthma and related experimental procedures were reviewed and approved by Seoul National University Bundang Hospital Institutional Review Board (no. B-1603/340-310). Written informed consent was obtained from all human subjects before their involvement.
Animals
C57BL/6 mice were purchased from Orient Bio (Seongnam, Korea). Rorγtgfp/gfp, B6.SJL, and Tcrb−/− mice were purchased from the Jackson Laboratory (Bar Harbor, Me). Il17a−/−Il17f−/− double-deficient mice, IL-17F fate reporter mice, Il17fCreR26ReYFP mice, and IL-17F reporter mice (Il17frfp) were described previously.21–23 All mice were maintained in a specific pathogen-free facility at Seoul National University or MD Anderson Cancer Center. Mice aged 6 to 12 weeks were used.
Reagents
Ursolic acid (UA), busulfan (Sigma-Aldrich, St Louis, Mo) and SR2211 (Calbiochem, Nottingham, United Kingdom) were dissolved in dimethyl sulfoxide (DMSO). For in vitro use, UA and SR2211 were further diluted with PBS (GenDEPOT, Barker, Tex). In vitro cell cultures of lymphoid cells were performed in RPMI 1640 supplemented with 10% FBS (Gen-DEPOT), 2 mmol/L L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, 55 μmol/L 2-mercaptoethanol, and 10 μg/mL gentamicin. For PLAT-E cell cultures, Dulbecco modified Eagle medium supplemented with 10% FBS, 1 μg/mL puromycin, and 10 μg/mL blasticidin was used. All cell-culture reagents, except FBS, were the products of Gibco (Carlsbad, Calif).
Animal models of allergic asthma
To induce allergic lung inflammation, we adopted an animal model of proteinase-induced allergic asthma induced by repeated intranasal challenge with fungal proteinase allergens.24 In brief, mice were anesthetized with isoflurane (Terrell; Piramal, Bethlehem, Pa) and challenged intranasally with 7 μg of proteinase from Aspergillus oryzae (PAO; Sigma-Aldrich) plus 20 μg of ovalbumin (OVA; Grade V from Sigma-Aldrich) in 50 μL of PBS every other day for 4 times (days 0, 2, 4, and 6). For the therapeutic model, mice were challenged on days 0, 2, 4, 6, and 12. In experiments with UA, mice were injected intraperitoneally with 150 mg/kg UA dissolved in DMSO or DMSO alone as a vehicle control. In some experiments UA was injected on days 0, 2, 4, and 6 in preventive format and days 6, 8, 10, and 12 in therapeutic format as described in Fig E1, A and C, in this article’s Online Repository at www.jacionline.org. For IFN-γ neutralization, anti-mouse IFN-γ (XMG1.2; Bio X Cell, West Lebanon, NH) or rat IgG1 (HRPN; Bio X Cell) as an isotype control were injected intraperitoneally (200 μg per mouse per injection) on days 0, 2, 4, and 6. Twenty-four hours after the last challenge, mice were killed with CO2 for further analysis.
For chronic asthma models, we injected mice with intranasal PAO/OVA and intraperitoneal UA on days 0, 2, 4, 11, and 13 (see Fig E2, A, in this article’s Online Repository at www.jacionline.org). Alternatively, we intraperitoneally immunized mice with an OVA-alum mixture (100 μg of OVA, Imject Alum; Thermo Scientific, Waltham, Mass) on days 0 and 14 and injected them with intranasal OVA (50 μg) and intraperitoneal UA on days 25, 26, and 27 (see Fig E2, E). Twenty-four hours after the last challenge, mice were killed with CO2 for further analysis.
Analysis of bronchoalveolar lavage fluid
A 20-gauge IV catheter (BD Biosciences, San Jose, Calif) was inserted into the trachea and flushed twice with 500 and 800 μL of cold PBS to collect bronchoalveolar lavage (BAL) fluid. Cytokine levels were determined by means of ELISAwith first-flushed 500 μL of BAL fluid. Ten thousand cells in BAL fluid were attached on the slide by Cytopro Centrifuge (Wescor; Logan, Utah) and stained with Diff-Quik staining kits (Sysmex, Kobe, Japan), according to the manufacturer’s protocol. Absolute numbers of macrophages, eosinophils, neutrophils, and lymphocyte in BAL fluid were calculated based on total cell counts.
Analysis of lymphoid cells in the lung
The left lobe of the lung was used for histologic analysis. The rest of the lobes were placed in 3 mL of RPMI 1640 containing 0.5 mg/mL collagenase (Type IV; Gibco), 2 mg/mL Dispase (Gibco), and 25 U/ml DNase I (Bio Basic, Markham, Ontario, Canada). Lungs were first dissociated with the gentle-MACS Dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany) and then digested for 30 minutes at 37°C with continuous agitation, followed by a second dissociation with the gentleMACS Dissociator. Cells were filtered through 100-μm nylon mesh and washed with cold PBS containing 1.5% FBS. Lymphoid cells were further isolated by using lymphocyte separation medium (MP Biomedicals, Santa Ana, Calif), according to the manufacturer’s instructions.
Histology
The left lobe of the lung was fixed in neutral buffered 10% formalin solution (Sigma) for at least 24 hours. Formalin-fixed samples were sent to the Pathology Center at Seoul National University College of Medicine for hematoxylin and eosin and periodic acid–Schiff staining.
Lymph node restimulation
The mediastinal lymph nodes were minced physically in cold PBS containing 1.5% FBS and filtered through a 100-μm nylon mesh. Lymphoid cells (1 × 106/mL) were cultured in the presence of 0, 10, and 50 μg/mL OVA for 3 days. Culture supernatants were used for cytokine ELISA.
In vitro murine TH cell differentiation
CD4+ cells from the spleen and peripheral lymph nodes were positively selected with CD4 microbeads (L3T4; Miltenyi Biotec). Subsequently, naive CD4+ T cells were sorted as CD4+CD25−CD62LhighCD44low cells with the FACSAria III cell sorter (BD Biosciences) and stimulated with plate-coated anti-CD3 (1 μg/mL, 145-2C11: Bio X Cell) and soluble anti-CD28 (2 μg/mL, 37.51; Bio X Cell) for 4 days. For TH2 differentiation, IL-2 (10 ng/mL; eBioscience, San Diego, Calif) and IL-4 (10 ng/mL; PeproTech, Rocky Hills, NJ) were added. For IFN-γ neutralization experiment, anti–IFN-γ (5 μg/mL, XMG1.2; Bio X Cell) was added also.
Quantitative real-time PCR
Total RNA was prepared with TRIzol Reagents (Invitrogen, Carlsbad, Calif). cDNA was then synthesized with Oligo(dT) primers and reverse transcriptase included in the RevertAid cDNA synthesis kit (Thermo Fisher Scientific), and gene expression levels were examined with the Applied Biosystems 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, Calif) by using iTaq SYBR Green Supermix (Bio-Rad Laboratories, Hercules, Calif). Data were normalized to β-actin (Actb) for reference. The following primers were used: Rorγt, 5′-CCGCTGAGAGGGCTTCAC-3′ and 5′-TGCAGGAGTAGGCCACATTACA-3′; B-cell lymphoma 6 (Bcl6), 5′-CACACCCGTCCATCATTGAA-3′ and 5′-TGTCCTCACGGTGCCTTT TT-3′; interferon regulatory factor 4 (Irf4), 5′-CACCAAAGCACAG AGTCACCT-3′ and 5′-TCCTCTGGATGGCTCCAGATG-3′; Gata3, 5′-AG AACCGGCCCCTTATGAA-3′ and 5′-AGTTCGCGCAGGATGTCC-3′; Il4, 5′-AGATCACGGCATTTTGAACG-3′ and 5′-TTTGGCACATCCA TCTCCG-3′; Il5, 5′-CGCTCACCGAGCTCTGTTG-3′ and 5′-CCAATG CATAGCTGGTGATTTTT-3′; Il13, 5′-GCTTATTGAGGAGCTGAGCA ACA-3′ and 5′-GGCCAGGTCCACACTCCATA-3′; and Actb, 5′-TGGA ATCCTGTGGCATCCATGAAAC-3′and 5′-TAAAACGCAGCTCAGTAAC AGTCCG-3′.
Mixed bone marrow chimeric mice
A day before bone marrow transfer, Tcrb−/− mice were injected with 35 mg/kg busulfan (Sigma-Aldrich) to ablate bone marrow cells from recipient mice. Bone marrow cells of B6.SJL (CD45.1+/+) and Rorγtgfp/gfp mice were obtained from femurs and tibia by flushing with cold PBS. These bone marrow cells were mixed at a 1:1 ratio and transferred into busulfan-treated Tcrb−/− mice through the tail vein (1.5 × 107 cells per mouse). Six to 8 weeks later, the reconstituted mice were intranasally challenged with PAO/OVA every other day for a total of 6 times. Twenty-four hours after the last challenge, mice were killed by means of CO2 inhalation for analysis.
BCL6 knockdown in naive T cells by means of retroviral infection
Retroviral supernatants were made by transfecting PLAT-E cell lines with either negative control LMP vector or 2 kinds of LMP-shBcl6. Transfection was performed with FuGene HD transfection reagent (Promega, Madison, Wis), according to the manufacturer’s protocol. Naive T cells were sorted from splenocytes and stimulated with plate-bound αCD3 and soluble αCD28. For TH2 differentiation, IL-2, IL-4, and αIFN-γ were added. After 24 hours, cells were infected with retroviral supernatants containing 8 μg/mL Polybrene by means of spin infection (1000g for 60 minutes at 30°C). After spin infection, cells were washed with PBS and again placed on the αCD3-coated plate with the same initial TH2 differentiating condition. After another 72 hours, cells were analyzed for cytokine production by using flow cytometry.
Flow cytometry and antibodies
For intracellular cytokine analysis, cells were stimulated with phorbol 12-myristate 13-acetate (100 ng/mL; Sigma-Aldrich) and ionomycin (1 μmol/L; Sigma-Aldrich) plus Brefeldin A and monensin (both from eBioscience) before staining. IC fixation buffer and permeabilization buffer from eBioscience were used for intracellular cytokine staining, and the Foxp3 staining kit (eBioscience) was used for BCL6 staining, according to the manufacturer’s instructions. Samples were analyzed with the FACSVerse flow cytometer (BD Biosciences), and acquired data were analyzed with FlowJo software (TreeStar, Ashland, Ore). The following antibodies were used for flow cytometric analysis or cell sorting: Alexa Fluor 488–conjugated antibodies to mouse CD62L (MEL-14) and IFN-γ (XMG1.2); fluorescein isothiocyanate–conjugated antibody to human CD45RA (HI100); phycoerythrin-conjugated rat IgG1 (eBRG1; eBioscience); antibodies to mouse CD25 (PC61), IL-17A (TC11-18H10.1), and mouse/human BCL6 (IG191E/A8); peridinin-chlorophyll-protein complex/Cy5.5–conjugated antibodies to mouse CD4 (GK1.5), CD45.1 (A20), and IFN-γ; phycoerythrin/Cy7-conjugated antibodies to mouse IL-13 (eBio13A; eBioscience), mouse/human CD44 (IM7), human CD4 (OKT4), and CD45RO (UCHL1); allophycocyanin (APC)–conjugated antibodies to mouse/human IL-5 (TRFK5), human CD25 (BC96), and IL-4 (8D4-8; eBioscience); Alexa Fluor 647–conjugated antibody to mouse IL-4 (11B11); APC/Cy7-conjugated antibodies to mouse CD4 (GK1.5) and human CD3ε (OKT3); and Pacific blue–conjugated antibodies to mouse CD45.2 (104) and human CD4 (RPA-T4). In some experiments the frequency of IL-4– and/or IL-5–producing TH2 cells was determined by staining cells with Alexa Fluor 647– and APC-conjugated antibodies before analyzing them with a same channel in the flow cytometer. IL-4– and/or IL-5–positive cells were designated as IL-4/5+ cells.
ELISA
Cytokine levels in cultured supernatants or BAL fluid were measured by using ELISA kits, according to the manufacturer’s protocol. For mouse IFN-γ, IL-4, and IL-5, ELISA kits were from R&D Systems (Minneapolis, Minn) or BioLegend (San Diego, Calif). For mouse IL-17A, ELISA kits were from BioLegend. For mouse IL-13 and human IL-13 and IL-17A, ELISA kits were from eBioscience.
In vitro human naive CD4+ T-cell differentiation and effector memory CD4+ T-cell restimulation
Human peripheral blood was obtained from healthy volunteers who are not taking any medications. PBMCs were prepared by using lymphocyte separation medium (MP Biomedicals), according to the manufacturer’s instructions. Subsequently, total CD4+ T cells were isolated by means of negative selection with CD14 microbeads (Miltenyi Biotec), followed by positive selection with CD4 microbeads (Miltenyi Biotec). Naive and effector memory CD4+ T cells were further sorted as CD3+CD4+ CD45RA+CD45RO−CD25− and CD3+CD4+CD45RA−CD45RO+ cells, respectively, by using the FACSAria III cell sorter (BD Biosciences). For naive T-cell differentiation, naive T cells undergoing fluorescence-activated cell sorting (FACS) were stimulated with plate-coated anti-CD3 (10 μg/mL, OKT3; BioLegend) and soluble anti-CD28 (2 μg/mL, CD28.2; BioLegend) for 4 or 7 days. For TH2 polarization, IL-2 (10 ng/mL), IL-4 (10 ng/mL), and anti–IFN-γ (5 μg/mL, B27; BioLegend) were added. For TH17 polarization, IL-2 (10 ng/mL), IL-6 (20 ng/mL), TGF-β (10 ng/mL), IL-23 (20 ng/mL), IL-21 (20 ng/mL), IL-1β (20 ng/mL), anti–IFN-γ (5 μg/mL), and anti–IL-4 (5 μg/mL, MP4-25D2; BioLegend) were added. TGF-β was from PeproTech, and the rest of the cytokines were from eBioscience. Effector memory CD4+ T cells were stimulated with plate-coated anti-CD3 (10 μg/mL) for 48 hours in the presence of UA or DMSO.
Human PBMC isolation and in vitro stimulation
Human peripheral blood was obtained from allergic asthmatic patients who meet the following criteria. First, asthma was diagnosed based on a positive bronchodilator response or methacholine provocation test result. Second, positivity of the Dermatophagoides farinae skin response was determined with skin prick tests by using the cutoff level of both a wheal size greater than 4 mm and an allergen/histamine wheal size ratio of 1 or greater. Third, systemic corticosteroids were not administrated in the most recent month. PBMCs were prepared by using Lymphocyte Separation Medium (MP Biomedicals), according to the manufacturer’s instruction. Cells (2 × 105) were stimulated with 50 μg/mL HDM extract (D farinae; Greer Laboratories, Lenoir, NC) in the presence of 2 μmol/L UA or DMSO as a vehicle control for 7 days.
Fate mapping study
IL-17F fate reporter mice (Il17fCreR26ReYFP) were sensitized by means of an intraperitoneal injection of OVA-alum on days 0 and 17 and challenged with intranasal OVA on days 27, 28, and 29. Lung mononuclear cells were prepared by using collagenase treatment with manual disruption. Lymphoid cells
Adoptive transfer of in vitro–differentiated TH17 cells
Naive CD4+CD25−CD62LhighCD44low T cells were isolated from Il17frfp mice crossed with OT-II mice (Il17frfp×OT-II mice). Naive CD4 T cells were activated with OVA peptide and irradiated antigen-presenting cells under TH17 conditions for 4 days, and red fluorescent protein (RFP)+ cells were sorted by using FACS. B6.SJL congenic (CD45.1+) recipient mice were sensitized by means of an intraperitoneal injection of OVA-alum on days 0 and 17 and challenged with intranasal OVA on days 27, 28, and 29. RFP+ cells (1 × 106) were transferred to recipient mice on day 16. Asthma-induced B6.SJL congenic mice were assessed for IL-17A, IL-13, IL-4, and IFN-γ expression by using intracellular staining.
Statistics
Data were graphed and analyzed with Prism 6 software (GraphPad Software, La Jolla, Calif). Statistical significance between 2 groups was determined by using the 2-tailed Student t test. For comparison of more than 3 groups, 1-way ANOVA and Tukey tests were used. P values of less than .05 were considered statistically significant.
RESULTS
Il17a−/−Il17f−/− double-knockout mice exhibit reduced TH2 cell responses to protease allergens in the airway
To investigate the role of TH17 cells in allergic asthma, we used Il17a−/−Il17f−/−(designated as double-knockout [dKO]) mice.23 Wild-type and dKO mice were intranasally injected with mixtures of fungal PAO/OVA as a model allergen. Intranasal PAO/OVA is known to induce allergen-specific TH2 cells, as well as TH17 cells, in the airway, offering an ideal model to study the potential mutual regulation between the 2 TH cell subsets.24–26 As expected, wild-type mice expressed increased levels of IL-17A and IL-17F producers among CD4+ T cells in the lungs, which were absent in dKO mice (Fig 1, A–C). Of note, frequencies and numbers of TH2 cells were also significantly diminished in the lungs of dKO mice, and frequencies and numbers of TH1 cells were increased compared with those in wild-type mice (Fig 1, A–C.) IL-4 and IL-5 levels in BAL fluid from dKO mice were also significantly lower than those in wild-type mice (Fig 1, D). A similar decrease in the frequency of TH2 cells was also observed in CD4+ T cells from BAL fluid (see Fig E3 in this article’s Online Repository at www.jacionline.org). Thus combined deficiency of IL-17A and IL-17F resulted in a significant reduction of the TH2 cell population in the airways in an animal model of allergic asthma.
FIG. 1.

Il17a−/−Il17f−/− dKO mice exhibit reduced TH2 cell responses against intranasal allergens. A, Representative FACS plot of CD4+ T cells from the lung. FSC, Forward scatter. B and C, Percentages (Fig 1, B) and absolute numbers (Fig 1, C) of CD4+ T-cell subsets. D, Levels of IFN-γ, IL-4, and IL-5 in BAL fluid. The graph shows means ± SEMs. *P < .05, **P < .01, and ***P < .001. WT, Wild-type. were further isolated by using lymphocyte separation medium. CD4+ yellow fluorescent protein (YFP)+ IL-17A+ cells represent stable TH17 cells, whereas CD4+YFP+IL-17A− cells represent exTH17 cells.
A distinct TH2-TH17 cell population expressing both IL-17A and IL-4/IL-5 was observed in wild-type mice but not dKO mice (Fig 1, A). This observation raised the hypothesis that the reduced TH2 cell responses in dKO mice might be due to the lack of these TH2-TH17 cells originating from TH17 cells and that IL-17A− TH2 cells are partially deviated from TH17 cells because we also observed a diminished frequency of IL-17A− TH2 cells (10.03 ± 0.56 [wild-type] vs 6.93 ± 1.02 [dKO], P = .04). To test this hypothesis and define the origin of the TH2-TH17 cell population, we first used a fate mapping system with Il17fCreR26ReYFP mice,21 which enables us to track cells that activated IL-17F expression regardless of their present expression of this cytokine. We found that few enhanced YFP+ cells (exTH17 cells) produced TH2 cytokines, indicating that TH17 cells did not become TH2 cytokine–producing cells in the airway (see Fig E4, A, in this article’s Online Repository at www.jacionline.org). Next, we adoptively transferred in vitro–generated OVA-specific TH17 cells purified from Il17frfp×OT-II mice22 into congenic mice before challenging the recipient with intranasal OVA. Donor TH17 cells appeared to be stable and did not produce TH2 cytokines (see Fig E4, B and C). These results together indicate that TH2-TH17 cells found in the airways of asthmatic mice are not derived from TH17 cells and suggest that the reduced TH2 cell responses in dKO mice are not due to the lack of TH17 cells.
Retinoic acid receptor–related orphan receptor γt–deficient mice exhibit reduced TH2 cell responses to allergens in the airway
Because the lack of IL-17A and IL-17F showed reduced TH2 cell responses in an animal model of allergic asthma, we hypothesized that TH17 cell responses play a crucial role in generation of TH2 cell responses. To further explore the role of TH17 cell responses on TH2 cells in allergic asthma, we sought to determine the role of retinoic acid receptor–related orphan receptor γt (RORγt), a signature transcriptional factor responsible for the TH17 cell program, on TH2 cell responses during the development of allergic asthma. We administered PAO/OVA intranasally in wild-type and Rorγtgfp/gfp (RORγt-deficient) mice. As expected,27 RORγt-deficient mice showed a profound reduction in the frequency and number of TH17 cells in the lung compared with wild-type mice (Fig 2, A–C). Interestingly, RORγt-deficient mice also exhibited significantly diminished TH2 cell populations, both in frequency and number, whereas those of TH1 cells were increased compared with values in wild-type mice (Fig 2, AC). Consistently, amounts of IL-4, IL-5, and IL-17A were all significantly lower in BAL fluid of RORγt-deficient mice compared with that of wild-type mice (Fig 2, D). Both eosinophil and neutrophil numbers in BAL fluid were also significantly reduced in the former group (Fig 2, E). These results demonstrate that, in addition to diminished TH17 cell responses and neutrophilia, RORγt-deficient mice did not mount optimal TH2 cell responses and eosinophilia in this animal model of allergic asthma.
FIG. 2.

RORγt-deficient mice exhibit reduced TH2 and TH17 responses against intranasal allergens. A, Representative FACS plot of lymphocytes from the lung. B and C, Percentages (Fig 2, B) and absolute numbers (Fig 2, C) of lung CD4+ T-cell subsets. D, Levels of IFN-γ, IL-4, IL-5, and IL-17A in BAL fluid. E, Absolute numbers of total cells, eosinophils (eo), macrophages (mac), lymphocytes (lym), and neutrophils (neu) in BAL fluid. Data are representative of 3 independent experiments. The graph shows means ± SEMs. *P < .05, **P < .01, and ***P < .001. ns, Not significant.
An RORγt inhibitor suppresses both TH2 and TH17 cell responses in allergic asthma
Diminished pulmonary TH2 cell responses in RORγt-deficient mice prompted us to hypothesize that targeting RORγt might be effective in suppressing both TH2 cell– and TH17 cell–mediated inflammation in the airway. To test this possibility, we used the RORγt inhibitor UA, which has been shown to selectively inhibit RORγt in vitro and in vivo during TH17 cell differentiation.28 Groups of C57BL/6 mice were intranasally administered PAO/OVA and additionally given UA or DMSO as a vehicle (see Fig E1, A). Consistent with the results observed in RORγt-deficient mice (Fig 2), mice treated with UA had significantly reduced frequencies and numbers of TH2 and TH17 cells while having increased TH1 cell frequencies and numbers in BAL fluid (Fig 3, A and B). A similar tendency was also observed in CD4+T cells in the lung (data not shown). When lymphoid cells from the mediastinal lymph nodes were restimulated with OVA, CD4+ T cells from UA-treated mice produced significantly lower levels of IL-4, IL-5, and IL-17 while producing significantly higher levels of IFN-γ compared with those from vehicle-treated mice (see Fig E1, B). In addition, mice treated with UA exhibited significantly reduced eosinophil and neutrophil numbers in BAL fluid compared with those in vehicle-treated mice (Fig 3, C). Moreover, the former group had significantly reduced infiltration of inflammatory cells around the airways with reduced mucus production than seen in the latter group (Fig 3, D). Hence treatment with UA prevented development of TH17 and TH2 cell responses and blocked allergic inflammation in an animal model of allergic asthma.
FIG. 3.

The RORγt inhibitor UA suppresses both TH2- and TH17-mediated allergic inflammation in the air-ways. A–D, Results from preventive model. E–H, Results from therapeutic model. Frequencies (Fig 3, A and E) and absolute numbers (Fig 3, B and F) of BAL fluid CD4+ T-cell subsets. Fig 3, C and G, Absolute numbers of total cells, macrophages (mac), eosinophils (eo), neutrophils (neu), and lymphocytes (lym) in BAL fluid. Fig 3, D and H, Histologic analysis of the lung. Data are representative of 3 independent experiments. H&E, Hematoxylin and eosin; PAS, periodic acid–Schiff. The graph shows means ± SEMs. *P < .05, **P < .01, and ***P < .001. ns, Not significant.
We next investigated whether the RORγt inhibitor can ameliorate allergic T-cell responses in asthmatic mice. Mice were intranasally administered PAO/OVA to establish allergic airway inflammation before being injected with UA or vehicle (see Fig E1, C). Compared with mice treated with vehicle, mice treated with UA had a moderate but significant reduction in the frequencies and numbers of TH17 and TH2 cells in BAL fluid, whereas those of TH1 cells were increased (Fig 3, E and F). A similar tendency was also observed in OVA-restimulated lymphoid cells of the mediastinal lymph nodes (see Fig E1, D). Consistent with reduced TH2 and TH17 responses in UA-treated mice, eosinophil and neutrophil numbers in BAL fluid were all remarkably lower (Fig 3, G). Reduction of infiltrated inflammatory cell numbers and mucus production in the UA-treated mice were also observed (Fig 3, H).
To further determine whether RORγt blockade ameliorates allergic TH2 and TH17 cell responses in more chronic settings, we used 2 additional allergic asthma models induced by repeated intranasal PAO/OVA challenges or by systemic sensitization with OVA and alum before subsequent challenges with intranasal OVA (see Fig E2, A and E). In both models we observed a significant reduction in the numbers of both TH17 and TH2 cells in the BAL fluid of UA-treated mice compared with those in vehicle-treated mice (see Fig E2, B and C, and E2, F and G, respectively). Consistently, eosinophil and neutrophil numbers in BAL fluid were profoundly lower in the former than in the latter (see Fig E2, D and H). Collectively, these results demonstrate that treatment with the RORγt inhibitor UA significantly ameliorated both eosinophilic and neutrophilic inflammation in the airways in animal models of allergic asthma associated with reduced TH2 and TH17 cell numbers.
Diminished TH2 cell responses by RORγt blockade are not due to increased IFN-γ levels
RORγt-deficient T cells and T cells from UA-treated mice produced higher levels of IFN-γ compared with their respective control T cells. Because IFN-γ is a strong inhibitor of TH2 cell differentiation,29 it is possible to surmise that the increased IFN-γ levels led to diminished TH2 cell responses in UA-treated mice. To address this possibility, we examined whether neutralizing IFN-γ restores TH2 cell responses in the UA-treated mice. Compared with control IgG treatment, we observed that anti–IFN-γ treatment significantly decreased the frequency of IFN-γ producers among CD4+T cells in the UA-treated mice, indicating the IFN-γ–neutralizing effect of the antibody (Fig 4, A). By contrast, we observed comparable frequencies and numbers of TH2 and TH17 cells between the control IgG– and anti–IFN-γ–treated groups in the UA-treated mice (Fig 4, A and B). Consistently, differential cell counts in BAL fluid also showed that mice treated with UA exhibited reduced eosinophilia and neutrophilia regardless of anti–IFN-γ treatment (Fig 4, C). Thus the reduction of TH2 cell responses by the RORγt inhibitor is independent of IFN-γ.
FIG. 4.
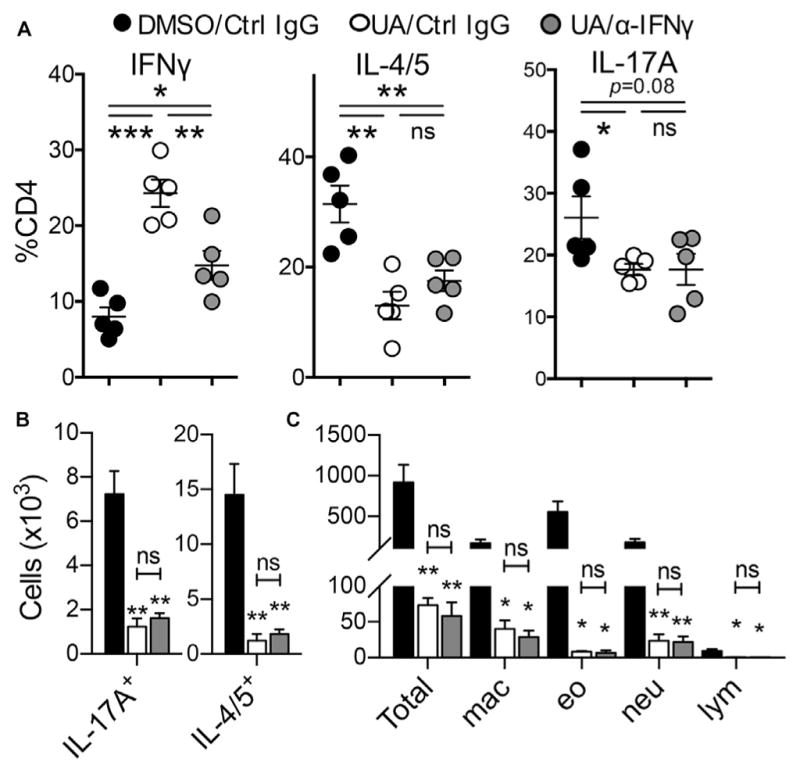
Reduction of TH2 cell responses by UA treatment is IFN-γ independent. A and B, Frequency (Fig 4, A) and absolute number (Fig 4, B) of BAL fluid CD4+ T-cell subsets. C, Absolute numbers of total cells, macrophages (mac), eosinophils (eo), neutrophils (neu), and lymphocytes (lym) in BAL fluid. Data are representative of 3 independent experiments. The graph shows means ± SEMs. *P < .05, **P < .01, and ***P < .001. ns, Not significant.
T cell–intrinsic function of RORγt in TH2 cell differentiation
The observed diminished pulmonary TH2 cell responses in RORγt-deficient mice and UA and αIFN-γ–treated mice led us to hypothesize that RORγt regulates TH2 cell differentiation in a T cell–intrinsic manner. To determine whether RORγt plays any role in pulmonary TH2 cell responses in a T cell–intrinsic manner in vivo, we generated mixed bone marrow chimeric mice by transferring a 1:1 mixture of wild-type (CD45.1+) and RORγt-deficient (CD45.2+) bone marrow cells into bone marrow–ablated Tcrb−/− mice (Fig 5, A). After reconstitution, the chimeric mice were intranasally challenged with PAO/OVA. Compared with wild-type CD4+ T cells, RORγt-deficient CD4+ T cells exhibited significantly reduced TH2 and TH17 cell responses in the lung and BAL fluid while exhibiting increased TH1 cell responses (Fig 5, B and C). Hence RORγt in T cells is required for optimal differentiation of TH2 cells in the airway in vivo.
FIG. 5.

T cell–intrinsic role of RORγt in TH2 cell differentiation. A–C, Results from bone marrow chimeric mice. Fig 5, A, Schematic outline of mixed bone marrow chimeric mouse generation. Fig 5, B, Representative FACS plot of lung CD4+ T cells. Fig 5, C, Frequency of lung CD4+ T-cell subsets. D–F, Differentiation of TH2 cells from naive RORγt-deficient CD4+ T cells. Fig 5, D, Expression of the indicated cytokines in CD4+ T cells after differentiation. Fig 5, E, Levels of IL-13 and IL-5 in supernatants. Fig 5, F, mRNA expression of indicated genes examined by using quantitative real-time PCR. G and H, Analysis of T cells from steady-state Rorγt+/gfp or Rorγtgfp/gfp mice. Fig 5, G, Splenocytes were analyzed for their BCL6 expression by using flow cytometry. Fig 5, H, Sorted CD44hiCD62L−CD4+ T cells were stimulated with plate-coated αCD3, and levels of the indicated cytokines were measured. Data are representative of at least 2 independent experiments. The graph shows means ± SEMs. WT, Wild-type. *P < .05, **P < .01, and ***P < .001. nd, Not detected.
To further investigate the T cell–intrinsic role of RORγt in differentiation of TH2 cells, we used an in vitro TH2 cell differentiation system in which naive CD4+ T cells from Rorγtgfp/gfp mice were stimulated with plate-coated αCD3 in the presence of soluble αCD28, IL-2, and IL-4. Of note, RORγt-deficient T cells showed a significantly lower frequency of IL-4/IL-5–producing TH2 cells than wild-type T cells (Fig 5, D). The production of IL-13 and IL-5 in the cultured supernatants was also significantly decreased in RORγt-deficient T cells (Fig 5, E). In addition, this inefficient differentiation of RORγt-deficient T cells into TH2 cells was independent of IFN-γ because anti–IFN-γ treatment had little effect on the production of IL-13 and IL-5 (data not shown).
To further examine the role of RORγt in the TH2 cell program, we analyzed mRNA transcript levels known to be associated with TH cell differentiation. Consistent with the protein data, we observed a significant reduction in levels of Il4, Il5, and Il13 transcripts in RORγt-deficient T cells compared with those of wild-type T cells (Fig 5, F). Levels of Gata3 and Irf4 were also slightly reduced in RORγt-deficient T cells. Interestingly, we observed that Bcl6 expression was significantly higher in RORγt-deficient T cells than wild-type T cells. These data demonstrate that RORγt-deficient T cells express lower levels of TH2 cell signature genes but express a higher level of Bcl6 than wild-type T cells under TH2 cell differentiation conditions.
Because we observed an increased expression of Bcl6 transcript in the RORγt-deficient T cells under TH2-skewing conditions in vitro, we investigated whether RORγt-deficient T cells expressed increased BCL6 in vivo. To this end, we compared BCL6 expression in CD44hiCD62L−CD4+ T cells between Rorγt+/gfp and Rorγtgfp/gfp mice. Compared with heterozygotes, RORγt-deficient mice had an increased frequency of BCL6-expressing cells among effector/memory CD4+ T cell population (Fig 5, G). When the CD44hiCD62L−CD4+ T cells were subjected to plate-coated anti-CD3 stimulation, RORγt-deficient T cells produced significantly less IL-4, IL-13, and IL-17A than wild-type T cells, whereas IFN-γ levels were comparable (Fig 5, H). Collectively, RORγt deficiency resulted in increased expression of BCL6 in effector memory T cells associated with diminished TH2 and TH17 cytokine production at steady state in vivo.
Downregulation of Bcl6 restores TH2 cell differentiation in RORγt-deficient T cells
Because BCL6 is known to suppress the differentiation of naive T cells into TH2 cells,30 we hypothesized that the increased Bcl6 expression accounts for the decreased TH2 cell differentiation in RORγt-deficient T cells. To test this hypothesis, we used 2 retro-viral short hairpin RNA constructs targeting Bcl6 and found that they significantly diminished the expression of BCL6 (Fig 6, A). As depicted in Fig 6, B, knockdown of BCL6 in RORγt-deficient T cells resulted in a remarkable increase in the frequency of IL-13–producing TH2 cells during TH2 cell differentiation in vitro. Wild-type or BCL6-deficient naive CD4+ T cells were subjected to TH2 cell–skewing conditions to directly investigate the role of BCL6 in TH2 cell differentiation. BCL6-deficient naive CD4+ T cells exhibited an increase in the frequency of IL-4/IL-5– and IL-13–producing cells compared with that of wild-type CD4+ T cells (Fig 6, C). Consistently, amounts of IL-5 and IL-13 in supernatants were significantly greater in the BCL6-deficient than wild-type T cells, whereas IL-4 production was comparable (Fig 6, D). Thus downregulation of BCL6 restores the decreased TH2 differentiation in RORγt-deficient T cells. These results strongly support the notion that RORγt facilitates TH2 cell differentiation by inhibiting BCL6 expression.
FIG. 6.

BCL6 negatively regulates TH2 cell differentiation in RORγt-deficient T cells. A, Expression of BCL6 in T cells after transduction with the indicated short hairpin RNA. B, Frequency of IL-13–producing T cells. GFP, Green fluorescent protein. C and D, Naive CD4+ T cells from wild-type (WT) or Bcl6f/fCD4-Cre mice were cultured in TH2-skweing conditions. Fig 6, C, Expression of the indicated cytokines were analyzed by using flow cytometry. Fig 6, D, Levels of IL-4, IL-5, and IL-13 in supernatants. Data are representative of at least 2 independent experiments. The graph shows means ± SEMs. *P < .05, **P < .01, and ***P < .001.
Effects of RORγt inhibition in human TH2 cell differentiation
Finally, we sought to determine the effects of RORγt inhibition on the differentiation of human TH2 cells from naive CD4+ T cells. Naive CD4+ T cells from healthy donors were stimulated under TH17 or TH2 cell differentiation conditions in the presence or absence of UA. As expected,28 RORγt inhibition by UA almost completely suppressed IL-17A production under TH17-skewing conditions (Fig 7, A). To check the effects of RORγt inhibition on restimulation of effector/memory TH17 cells, CD3+CD4+CD45RA−CD45RO+ T cells from the same healthy volunteers were stimulated with plate-bound CD3 antibody. IL-17A production was significantly suppressed by the addition of UA in culture (Fig 7, A). In addition, we also observed that addition of UA into the TH2-skewing culture significantly inhibited the production of IL-13 compared with vehicle control (Fig 7, B). Consistently, the frequency of IL-4+ cells was significantly reduced in UA-treated T cells compared with that in vehicle-treated T cells (Fig 7, C). However, we observed no detectable levels of IL-13 from the supernatants of anti-CD3–stimulated effector/memory CD4+ T cells (Fig 7, B). Similarly, another RORγt inhibitor, SR2211, was found to inhibit the differentiation of human TH2 and TH17 cells in vitro (see Fig E5 in this article’s Online Repository at www.jacionline.org).31
FIG. 7.

Blockade of RORγt negatively affects differentiation of human TH2 cells. A–C, Human naive CD4+ T cells from healthy volunteers were cultured in TH17- or TH2-skewing conditions, and effector memory CD4+ T (TEM) cells were stimulated with plate-coated αCD3. Fig 7, A, Levels of IL-17A in culture media from TH17 or TEM conditions. Fig 7, B, Levels of IL-13 in culture media from TH2 or TEM conditions. Fig 7, C, Percentage changes in IL-4 producers in UA-treated CD4+ T cells. Each symbol represents an individual donor. D, PBMCs from human allergic asthmatic patients were stimulated with HDM extract for 7 days. Levels of IL-17A and IL-13 are shown. Each symbol represents an individual patient. Data are representative of at least 3 independent experiments. The graph shows means ± SEMs. **P < .01 and ***P < .001.
We next sought to explore whether inhibition of RORγt suppresses allergen-specific effector memory TH2 and TH17 cells in asthmatic patients. Allergic asthmatic patients were selected based on their response against the house dust mite allergen D farinae in skin prick tests. PBMCs from the patients were simulated with 50 μg/mL D farinae extract in the presence of DMSO or UA before IL-17A and IL-13 levels were measured. As shown in Fig 7, D, addition of UA significantly reduced production of IL-17A from D farinae–specific CD4+ T cells. By contrast, no evident difference was observed in IL-13 levels between the DMSO- and UA-treated cells, indicating that RORγt inhibition has little role in suppressing allergen-specific effector/memory TH2 cells. Together, these results demonstrate that RORγt inhibition resulted in suppression of both TH2 and TH17 cell differentiation from naive T cells while exerting little to no suppressing effector/memory TH2 cell responses in human subjects.
DISCUSSION
Despite the nonredundant pathogenic roles of TH2 and TH17 cells in allergic airway inflammation, it has been unclear how the simultaneous inhibition of these 2 TH cell subsets can be achieved. Our findings in the present study are that (1) Il17a−/−Il17f −/− dKO mice showed reduction of TH17 and TH2 cell responses on proteinase allergen challenge, (2) RORγt-deficient mice exhibited significantly diminished TH2 and TH17 cells in the airway in response to proteinase allergen, (3) pharmacologic inhibition of RORγt also inhibited TH2 and TH17 cell responses in the airway in wild-type mice in an IFN-γ–independent and T cell–intrinsic manner, (4) inhibition of TH2 cell responses in RORγt-deficient T cells was restored by knockdown of BCL6, and (5) pharmacologic inhibition of RORγt also inhibited human TH2 and TH17 cell differentiation. Overall, our findings indicate that RORγt blockade can simultaneously suppress TH2 and TH17 cell responses during allergic asthma.
Among diverse animal models of allergic asthma, we used a PAO/OVA-induced asthma model because this model exhibits both TH2 and TH17 cell–mediated allergic airway inflammation, offering an ideal model to study any mutual regulation between TH2 and TH17 cell responses.25 In addition, proteinase allergens used in this model contains very low amounts of endotoxin, minimizing allergen-independent inflammation, which likely interferes with interpretation of TH2 and TH17 cell–mediated responses.32 RORγt-deficient mice lack lymph nodes33; therefore the reduced induction of TH2 cell responses in RORγt-deficient mice could be due to the lack of airway-draining lymph nodes where antigen-induced T-cell differentiation occurs rather than a role of RORγt in TH2 cell differentiation. To address this issue, we examined TH2 cell responses in RORγt-deficient T cells in the mixed chimeric Tcrb−/− mice given a 1:1 mixture of bone marrow cells from wild-type and RORγt-deficient mice, which have normal secondary lymphoid organs, including airway draining lymph nodes, and found that RORγt-deficient T cells still exhibited a profound reduction in TH2 cell responses on intranasal allergen challenges. Therefore we concluded that RORγt in T cells is required for optimal TH2 cell responses in vivo.
Because TH2 and TH17 cell responses play critical and nonredundant roles in the pathogenesis of allergic asthma, there have been a number of attempts to block either pathway in animal models, as well as in human subjects.15 However, blocking either the TH2 or TH17 pathway with anti–IL-13 or anti–IL-17 alone has been shown not to be sufficient for the treatment of allergic asthma because such treatment resulted in upregulation of the other pathway.18 Similarly, a recent study by Park et al34 demonstrated that coadministration of anti–IL-17 and anti–IL-13 reduced both TH2 and TH17 cell responses in the lung in an animal model of antigen-induced pulmonary arterial remodeling, whereas anti–IL-17 or anti–IL-13 alone resulted in upregulation of TH2 or TH17 downstream effector genes, respectively. Collectively, these reports imply that simultaneous targeting of both TH2 and TH17 cell responses would be a promising strategy for the treatment of allergic asthma, whereas targeting either pathway alone might be ineffective because of activation of the other pathway.
In this regard our previous study examined whether blockade of signal transducer and activator of transcription 3 (STAT3) inhibits both TH2 and TH17 cell responses in the airway because the activation of STAT3 is indispensable for TH17 cell differentiation35,36 and STAT3 has been shown recently to be required for optimal TH2 cell differentiation.37 Although STAT3-deficient T cells exhibited reduced TH2 and TH17 cell differentiation in the bronchial lymph nodes, they showed more robust TH2 cell responses in the airway compared with STAT3-sufficient T cells, suggesting that STAT3 blockade might increase TH2 cell responses in the airway.26 Unlike STAT3-deficient T cells, we observed a consistent reduction of TH2 cell responses in airways of mice with RORγt–deficient T cells, as well as in mice treated with an RORγt inhibitor. Based on these observations, we propose that blockade of RORγt would offer a promising approach for the treatment of allergic asthma in human subjects.
What is the underlying mechanism for the diminished TH2 cell responses caused by RORγt blockade? IFN-γ from TH1 cell responses can suppress TH2 and TH17 cell responses.38,39 However, increased TH1 cell responses are not likely the direct the mechanism of diminished TH2 cell responses because neutralization of IFN-γ did not restore TH2 cell responses in RORγt-deficient T cells in vitro, as well as in UA-treated mice in vivo. Of note, we observed that the level of Bcl6 transcript was increased in RORγt-deficient T cells stimulated under TH2-skewing conditions and that the expression of BCL6 in CD44hiCD4+ T cells was increased in RORγt-deficient mice at steady state. The antagonism between BCL6 and RORγt or GATA-3 is well documented. BCL6 functions as a transcriptional repressor antagonizing the function of RORγt, as well as the expression of Gata3, leading to inhibition of TH17 and TH2 cell differentiation, respectively.40,41 In addition, BCL6-deficient mice have more robust TH2 cell–mediated inflammatory responses than wild-type mice,42 and IL-13 is the most downregulated molecule by BCL6.43 Importantly, we found that knockdown of BCL6 significantly restored the frequency of IL-13 producers in RORγt-deficient T cells under TH2-skewing conditions, indicating that the increased BCL6 expression in the absence of RORγt played a critical role in the observed diminished TH2 cell responses.
Thus we propose that RORγt blockade inhibits TH2 cell responses, at least in part by inducing the expression of BCL6 in T cells. At this moment, it is not clear how RORγt regulates BCL6, and further studies are needed to identify the underlying molecular mechanisms. To rule out any possible nonredundant and compensatory functions between IL-17A and IL-17F,44–47 we used Il17a−/−Il17f−/− dKO mice in our allergic asthma model and found a significantly reduced TH2 cell response in the airways of dKO mice in vivo. Il17a−/−Il17f−/− dKO mice still have intact RORγt, suggesting that the observed diminished TH2 cell responses in dKO mice are not likely associated with reduced RORγt expression. Thus we speculate that the mechanism governing the reduced TH2 cell responses in dKO mice differs from that induced by RORγt deficiency. It is possible that IL-17A and IL-17F stimulate the production of soluble factors, such as complement,48 that could facilitate TH2 cell responses from nonimmune cells in the lung. Hence we propose that both RORγt and TH17 cytokines contribute to the generation of TH2 cell responses in the airway, presumably through distinct mechanisms. BCL6 is essential for the generation of follicular helper T cells,49 as well as follicular regulatory T cells.50,51 Hence one can speculate that RORγt can also play an important role in controlling germinal center reactions by modulating the expression of BCL6 in follicular helper T cells, follicular regulatory T cells, or both. Further studies will be needed to dissect the role of RORγt in germinal center reactions.
Importantly, we observed that pharmacologic inhibition of RORγt by UA also decreased significantly the differentiation of human TH2 and TH17 cells. UA is known to inhibit activation of STAT3 and nuclear factor κB in addition to RORγt; however, it specifically inhibits RORγt but not STAT3 or nuclear factor κB concentrations of less than 2 μmol/L, the concentration we used in the present study.28 We also observed that another RORγt inhibitor, SR2211, also inhibited human TH2 cell differentiation. Together with the results obtained with RORγt-deficient T cells, these findings strongly support the notion that RORγt blockade suppresses the TH2 cell differentiation program in mice and human subjects. In addition, the same treatment also suppressed production of allergen-induced IL-17A from the PBMCs of patients with allergic asthma. Production of TH2 cell cytokines from the PBMCs of patients with allergic asthma was not affected by treatment with the RORγt inhibitor. Clinical longitudinal studies reported that monosensitized children with atopic asthma become polysensitized over time, indicating that new allergen-specific TH2 or TH17 cells continuously develops over the course of time.52,53 Hence, although inhibition of RORγt is less efficient in controlling established memory TH2 responses, it could eventually be beneficial for treatment of established allergic asthma by suppressing newly developing TH2 and TH17 cells. Supporting this notion, we observed that treatment with the RORγt inhibitor reduced all aspects of allergic asthma in our therapeutic model.
In summary, our study has unveiled a crucial role of RORγt in differentiation of TH2 cells in allergic asthma. Based on our findings, we propose that targeting RORγt would be beneficial for the treatment of TH2 cell–mediated eosinophilic asthma, as well as TH17 cell–mediated neutrophilic asthma. Further immunologic and pharmacologic studies will be needed to identify effective RORγt inhibitors with few side effects before considering therapeutic use for allergic asthma in human subjects.
Supplementary Material
Key messages.
RORγt in T cells is necessary for optimal TH2 cell responses during allergic asthma.
Inhibition of TH2 cell responses in RORγt-deficient T cells depends on BCL6.
Targeting RORγt might be a promising strategy for the treatment of allergic asthma through concomitant suppression of TH2 and TH17 cells in the airway.
Acknowledgments
Supported by research grants 2017R1A2B3007392 (to Y.C.) from the National Research Foundation of Korea and HI14C2282 (to Y.C.) from the Korean Health Technology R&D Project, Ministry of Health & Welfare; the Cancer Prevention and Research Institute of Texas (RP130078; to S.H.C.); and the Department of Defense (W81XWH-16-1-0100; to S.H.C.). H.N. is a recipient of the Global Ph.D. Fellowship Program through NRF (2015H1A2A1030805) funded by the Ministry of Education of Korea.
We thank Drs Kyu-Won Kim and Sung-Jin Bae (Seoul National University) for their support in flow cytometric analysis and the entire Chung laboratory for suggestion and discussion. We thank Ms Da-Sol Kuen for proofreading the manuscript.
Abbreviations used
- APC
Allophycocyanin
- BAL
Bronchoalveolar lavage
- BCL6
B-cell lymphoma 6
- dKO
Double knockout
- DMSO
Dimethyl sulfoxide
- FACS
Fluorescence-activated cell sorting
- OVA
Ovalbumin
- PAO
Proteinase from Aspergillus oryzae
- RFP
Red fluorescent protein
- RORγt
Retinoic acid receptor–related orphan receptor γt
- STAT3
Signal transducer and activator of transcription 3
- UA
Ursolic acid
- YFP
Yellow fluorescent protein
Footnotes
Disclosure of potential conflict of interest: S. H. Chang received grants from the Cancer Prevention and Research Institute of Texas (RP130078) and Department of Defense (W81XWH-16-1-0100) for this work. Y. Chung received grants from National Research Foundation of Korea (2017R1A2B3007392) and Ministry of Health and Welfare of Korea (HI14C2282) for this work. The rest of the authors declare that they have no relevant conflicts of interest.
References
- 1.Kim HY, DeKruyff RH, Umetsu DT. The many paths to asthma: phenotype shaped by innate and adaptive immunity. Nat Immunol. 2010;11:577–84. doi: 10.1038/ni.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holgate ST. Innate and adaptive immune responses in asthma. Nat Med. 2012;18:673–83. doi: 10.1038/nm.2731. [DOI] [PubMed] [Google Scholar]
- 3.Romanet-Manent S, Charpin D, Magnan A, Lanteaume A, Vervloet D, Group EC. Allergic vs nonallergic asthma: what makes the difference? Allergy. 2002;57:607–13. doi: 10.1034/j.1398-9995.2002.23504.x. [DOI] [PubMed] [Google Scholar]
- 4.Asher I, Baena-Cagnani C, Boner A, Canonica GW, Chuchalin A, Custovic A, et al. World Allergy Organization guidelines for prevention of allergy and allergic asthma. Int Arch Allergy Immunol. 2004;135:83–92. doi: 10.1159/000080524. [DOI] [PubMed] [Google Scholar]
- 5.Yu S, Kim HY, Chang YJ, DeKruyff RH, Umetsu DT. Innate lymphoid cells and asthma. J Allergy Clin Immunol. 2014;133:943–51. doi: 10.1016/j.jaci.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 6.Corry DB, Kheradmand F. Biology and therapeutic potential of the interleukin-4/interleukin-13 signaling pathway in asthma. Am J Respir Med. 2002;1:185–93. doi: 10.1007/BF03256608. [DOI] [PubMed] [Google Scholar]
- 7.Newcomb DC, Peebles RS., Jr Th17-mediated inflammation in asthma. Curr Opin Immunol. 2013;25:755–60. doi: 10.1016/j.coi.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cosmi L, Liotta F, Maggi E, Romagnani S, Annunziato F. Th17 cells: new players in asthma pathogenesis. Allergy. 2011;66:989–98. doi: 10.1111/j.1398-9995.2011.02576.x. [DOI] [PubMed] [Google Scholar]
- 9.Al-Ramli W, Prefontaine D, Chouiali F, Martin JG, Olivenstein R, Lemiere C, et al. T(H)17-associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol. 2009;123:1185–7. doi: 10.1016/j.jaci.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 10.Alcorn JF, Crowe CR, Kolls JK. TH17 cells in asthma and COPD. Annu Rev Physiol. 2010;72:495–516. doi: 10.1146/annurev-physiol-021909-135926. [DOI] [PubMed] [Google Scholar]
- 11.Irvin C, Zafar I, Good J, Rollins D, Christianson C, Gorska MM, et al. Increased frequency of dual-positive TH2/TH17 cells in bronchoalveolar lavage fluid characterizes a population of patients with severe asthma. J Allergy Clin Immunol. 2014;134:1175–86. e7. doi: 10.1016/j.jaci.2014.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roussel L, Houle F, Chan C, Yao Y, Berube J, Olivenstein R, et al. IL-17 promotes p38 MAPK-dependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. J Immunol. 2010;184:4531–7. doi: 10.4049/jimmunol.0903162. [DOI] [PubMed] [Google Scholar]
- 13.Bellini A, Marini MA, Bianchetti L, Barczyk M, Schmidt M, Mattoli S. Interleukin (IL)-4, IL-13, and IL-17A differentially affect the profibrotic and proinflammatory functions of fibrocytes from asthmatic patients. Mucosal Immunol. 2012;5:140–9. doi: 10.1038/mi.2011.60. [DOI] [PubMed] [Google Scholar]
- 14.Leckie MJ, ten Brinke A, Khan J, Diamant Z, O’Connor BJ, Walls CM, et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyperresponsiveness, and the late asthmatic response. Lancet. 2000;356:2144–8. doi: 10.1016/s0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- 15.Chung KF. Targeting the interleukin pathway in the treatment of asthma. Lancet. 2015;386:1086–96. doi: 10.1016/S0140-6736(15)00157-9. [DOI] [PubMed] [Google Scholar]
- 16.Scheerens H, Arron JR, Zheng Y, Putnam WS, Erickson RW, Choy DF, et al. The effects of lebrikizumab in patients with mild asthma following whole lung allergen challenge. Clin Exp Allergy. 2014;44:38–46. doi: 10.1111/cea.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Busse WW, Holgate S, Kerwin E, Chon Y, Feng J, Lin J, et al. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med. 2013;188:1294–302. doi: 10.1164/rccm.201212-2318OC. [DOI] [PubMed] [Google Scholar]
- 18.Choy DF, Hart KM, Borthwick LA, Shikotra A, Nagarkar DR, Siddiqui S, et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med. 2015;7:301ra129. doi: 10.1126/scitranslmed.aab3142. [DOI] [PubMed] [Google Scholar]
- 19.van Hamburg JP, de Bruijn MJ, Ribeiro de Almeida C, van Zwam M, van Meurs M, de Haas E, et al. Enforced expression of GATA3 allows differentiation of IL-17-producing cells, but constrains Th17-mediated pathology. Eur J Immunol. 2008;38:2573–86. doi: 10.1002/eji.200737840. [DOI] [PubMed] [Google Scholar]
- 20.Newcomb DC, Zhou W, Moore ML, Goleniewska K, Hershey GK, Kolls JK, et al. A functional IL-13 receptor is expressed on polarized murine CD4+ Th17 cells and IL-13 signaling attenuates Th17 cytokine production. J Immunol. 2009;182:5317–21. doi: 10.4049/jimmunol.0803868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ichiyama K, Gonzalez-Martin A, Kim BS, Jin HY, Jin W, Xu W, et al. The microRNA-183-96-182 cluster promotes T helper 17 cell pathogenicity by negatively regulating transcription factor Foxo1 expression. Immunity. 2016;44:1284–98. doi: 10.1016/j.immuni.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang J, Yuan Q, Zhu H, Yin L, Hong S, Dong Z, et al. IL-17C/IL-17RE augments T cell function in autoimmune hepatitis. J Immunol. 2017;198:669–80. doi: 10.4049/jimmunol.1600977. [DOI] [PubMed] [Google Scholar]
- 24.Kheradmand F, Kiss A, Xu J, Lee SH, Kolattukudy PE, Corry DB. A protease-activated pathway underlying Th cell type 2 activation and allergic lung disease. J Immunol. 2002;169:5904–11. doi: 10.4049/jimmunol.169.10.5904. [DOI] [PubMed] [Google Scholar]
- 25.Lim H, Kim YU, Yun K, Drouin SM, Chung Y. Distinct regulation of Th2 and Th17 responses to allergens by pulmonary antigen presenting cells in vivo. Immunol Lett. 2013;156:140–8. doi: 10.1016/j.imlet.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Lim H, Cho M, Choi G, Na H, Chung Y. Dynamic control of Th2 cell responses by STAT3 during allergic lung inflammation in mice. Int Immunopharmacol. 2015;28:846–53. doi: 10.1016/j.intimp.2015.03.051. [DOI] [PubMed] [Google Scholar]
- 27.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 28.Xu T, Wang X, Zhong B, Nurieva RI, Ding S, Dong C. Ursolic acid suppresses interleukin-17 (IL-17) production by selectively antagonizing the function of RORgamma t protein. J Biol Chem. 2011;286:22707–10. doi: 10.1074/jbc.C111.250407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oriss TB, McCarthy SA, Morel BF, Campana MA, Morel PA. Crossregulation between T helper cell (Th)1 and Th2: inhibition of Th2 proliferation by IFN-gamma involves interference with IL-1. J Immunol. 1997;158:3666–72. [PubMed] [Google Scholar]
- 30.Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity. 2009;31:457–68. doi: 10.1016/j.immuni.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 31.Kumar N, Lyda B, Chang MR, Lauer JL, Solt LA, Burris TP, et al. Identification of SR2211: a potent synthetic RORgamma-selective modulator. ACS Chem Biol. 2012;7:672–7. doi: 10.1021/cb200496y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Millien VO, Lu W, Shaw J, Yuan X, Mak G, Roberts L, et al. Cleavage of fibrinogen by proteinases elicits allergic responses through Toll-like receptor 4. Science. 2013;341:792–6. doi: 10.1126/science.1240342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. 2004;5:64–73. doi: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- 34.Park SH, Chen WC, Esmaeil N, Lucas B, Marsh LM, Reibman J, et al. Interleukin 13- and interleukin 17A-induced pulmonary hypertension phenotype due to inhalation of antigen and fine particles from air pollution. Pulm Circ. 2014;4:654–68. doi: 10.1086/678511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–63. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 36.Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, et al. Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179:4313–7. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- 37.Stritesky GL, Muthukrishnan R, Sehra S, Goswami R, Pham D, Travers J, et al. The transcription factor STAT3 is required for T helper 2 cell development. Immunity. 2011;34:39–49. doi: 10.1016/j.immuni.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu J, Yamane H, Cote-Sierra J, Guo L, Paul WE. GATA-3 promotes Th2 responses through three different mechanisms: induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res. 2006;16:3–10. doi: 10.1038/sj.cr.7310002. [DOI] [PubMed] [Google Scholar]
- 39.Yeh WI, McWilliams IL, Harrington LE. IFNgamma inhibits Th17 differentiation and function via Tbet-dependent and Tbet-independent mechanisms. J Neuroimmunol. 2014;267:20–7. doi: 10.1016/j.jneuroim.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325:1001–5. doi: 10.1126/science.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kusam S, Toney LM, Sato H, Dent AL. Inhibition of Th2 differentiation and GATA-3 expression by BCL-6. J Immunol. 2003;170:2435–41. doi: 10.4049/jimmunol.170.5.2435. [DOI] [PubMed] [Google Scholar]
- 42.Ye BH, Cattoretti G, Shen Q, Zhang J, Hawe N, de Waard R, et al. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation. Nat Genet. 1997;16:161–70. doi: 10.1038/ng0697-161. [DOI] [PubMed] [Google Scholar]
- 43.Liu X, Lu H, Chen T, Nallaparaju KC, Yan X, Tanaka S, et al. Genome-wide analysis identifies Bcl6-controlled regulatory networks during T follicular helper cell differentiation. Cell Rep. 2016;14:1735–47. doi: 10.1016/j.celrep.2016.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, et al. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205:1063–75. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jin W, Dong C. IL-17 cytokines in immunity and inflammation. Emerg Microbes Infect. 2013;2:e60. doi: 10.1038/emi.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C, et al. ROR-gamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology. 2009;136:257–67. doi: 10.1053/j.gastro.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 47.von Vietinghoff S, Ley K. IL-17A controls IL-17F production and maintains blood neutrophil counts in mice. J Immunol. 2009;183:865–73. doi: 10.4049/jimmunol.0804080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Onishi RM, Gaffen SL. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology. 2010;129:311–21. doi: 10.1111/j.1365-2567.2009.03240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–10. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med. 2011;17:983–8. doi: 10.1038/nm.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. 2011;17:975–82. doi: 10.1038/nm.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Silvestri M, Rossi GA, Cozzani S, Pulvirenti G, Fasce L. Age-dependent tendency to become sensitized to other classes of aeroallergens in atopic asthmatic children. Ann Allergy Asthma Immunol. 1999;83:335–40. doi: 10.1016/S1081-1206(10)62674-9. [DOI] [PubMed] [Google Scholar]
- 53.Migueres M, Davila I, Frati F, Azpeitia A, Jeanpetit Y, Lheritier-Barrand M, et al. Types of sensitization to aeroallergens: definitions, prevalences and impact on the diagnosis and treatment of allergic respiratory disease. Clin Transl Allergy. 2014;4:16. doi: 10.1186/2045-7022-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.