

Abstract
Combination immunotherapies utilizing complementary modalities that target distinct tumor attributes or immunosuppressive mechanisms, or engage different arms of the antitumor immune response, can elicit greater therapeutic efficacy than the component monotherapies. Increasing the number of agents included in a therapeutic cocktail can further increase efficacy, however, this approach poses numerous challenges for clinical translation. Here, a novel platform to simplify combination immunotherapy by covalently linking immunotherapeutic agonists to the costimulatory receptors CD134 and CD137 into a single heterodimeric drug, “OrthomAb”, is shown. This reagent not only retains costimulatory T cell activity, but also elicits unique T cell functions that are not programmed by either individual agonist, and preferentially expands effector T cells over Tregs. Finally, in an aggressive melanoma model OrthomAb elicits better therapeutic efficacy compared to the unlinked agonists. This demonstration that two drugs can be combined into one provides a framework for distilling complex combination drug cocktails into simpler delivery platforms.
Electronic supplementary material
The online version of this article (10.1007/s00262-018-2116-1) contains supplementary material, which is available to authorized users.
Keywords: Cancer immunotherapy, Costimulation, CD134, OX40, CD137, 4-1BB
Introduction
Biologics that target tumor cell surface proteins are effective in treating certain cancers. For instance, the anti-CD20 mAb (rituximab) elicits ~ 50% response rates in non-Hodgkin’s B cell lymphoma patients who previously failed chemotherapy [1, 2]. Nevertheless, relapse can occur due to outgrowth of resistant tumors [3], and many cancers are not amenable to treatment with these types of mAbs that target antigens expressed on both tumors and the healthy tissues from which they arise. Alternatively, immunomodulatory mAbs can target tumors indirectly by activating antitumor immune responses. Thus, although tumors express mutated proteins that give rise to neoepitopes that can be recognized by CTL [4, 5], they employ a variety of immunosuppressive mechanisms to evade T cell-mediated elimination [6–8]. For example, T cell function can be dampened through engagement of checkpoint receptors by ligands expressed on APC [9], tumor cells or tumor-infiltrating myeloid cells [10]. So far, mAb antagonists to the checkpoint molecules CTLA-4 and PD-1/PD-L1 have been approved to treat patients with melanoma and other advanced cancers [11–14], and antagonists to other checkpoint molecules such as LAG-3, VISTA, Tim-3 and TIGIT are under development [15, 16]. Another mechanism that contributes to ineffectual antitumor T cell responses is presentation of tumor epitopes by APC expressing low levels of costimulatory ligands [17, 18]. To counter this, agonist mAbs to costimulatory TNFR superfamily members such as CD134 (OX40), CD137 (4-1BB), GITR and CD27 [19–22] have been developed that are therapeutic in pre-clinical mouse models and currently undergoing testing in human cancer clinical trials.
Combining immunomodulatory mAbs within the same class can augment therapeutic outcomes. For example, since anti-CTLA-4 and -PD-1/PD-L1 are generally thought to act during distinct early and late stages of the antitumor response, respectively [10, 23, 24], it is perhaps not surprising that combining the two checkpoint inhibitors boosts therapeutic response compared to either alone [25]. We found over a decade ago that combining anti-CD134 plus -CD137 elicits robust CD8 CTL responses and tumor immunity [26, 27], a result confirmed by others and in multiple systems [28, 29]. Importantly, this CD134 CD137 “dual costimulation” also programs CD4 T cells to develop cytotoxic tumor cell killing potential [30], as well as deliver potent therapeutic help [31]. Thus, dual costimulation induces key antitumor responses.
While cancer immunotherapy is clearly moving towards the implementation of combination therapies, there are regulatory complexities involved in approval for multi-agent clinical testing [32], as well as increased potential for adverse events [25, 32]. Although 2-drug combination therapies are being tested clinically (e.g., [25]), and CD134 CD137 dual costimulation is in Phase 1 (NCT02315066), combinations involving three or more agents may have even greater efficacy [28, 33, 34]. Thus, a major limitation is the lack of novel approaches to combination therapy that avoid complex mixtures of reagents. Here, we have addressed this scientific gap by covalently linking CD134 and CD137 agonists into a single drug platform, termed “OrthomAb”, which not only retains the costimulatory activity of the unlinked agonists, but additionally programs unique T cell functions and preferentially expands effector T cells over Tregs. Importantly, OrthomAb also elicits therapeutic tumor immunity in an aggressive melanoma model. This approach provides a blueprint for distilling complex combination immunotherapies into more clinically-feasible delivery approaches.
Materials and methods
Animals
Six- to 8-week-old male or female C57BL/6 (B6) mice (Jackson Laboratory) and OVA (257SIINFEKL264)-specific OT-I TCR transgenic Rag1−/− B6 mice (bred in-house) were used as splenocyte sources for in vitro cultures. 6- to 8-week-old female B6 mice were used for B16–F10 tumor therapy experiments, that were performed as previously described [31]. All mice were maintained in the UConn Health Animal Facility in accordance with National Institutes of Health and UConn Health Institutional Animal Care and Use Committee (IACUC) guidelines.
OrthomAb synthesis
The heterobifunctional chemical linkers trans-cyclooctene-PEG4-NHS ester (TCO) and methyltetrazine-PEG5-NHS ester (MTz) (both from KeraFast) were coupled to the mAbs anti-CD137 (clone 3H3, rat IgG2a) and -CD134 (clone OX86, rat IgG1) (10 mg each, both from BioXCell) in 4 ml PBS at a linker:mAb molar ratio of 7:1 at room temperature for 1 h. Coupled mAbs were desalted using Amicon Ultra-4 Centrifugal Filter Units with Ultracel-10 membranes, concentrated to a volume of 1 ml each, and then clicked together by mixing followed by 1 h incubation at room temperature. The resulting heteroconjugate (OrthomAb) was then isolated from the heterogeneous mixture of reaction products (that also included unlinked monomers and higher-order multimers) using a BioLogic DuoFlow QuadTec 10 medium-pressure liquid chromatography system (BioRad) to perform 3 successive rounds of size-exclusion chromatography with a HiPrep 16/60 Sephacryl S-300 HR column (GE Healthcare Life Sciences). Chromatographic tracings of A280 versus time and SDS–PAGE were used to visualize species present in each fraction, and appropriate fractions were pooled for subsequent purifications. Purity of the final isolated OrthomAb heterodimer was determined using ImageJ densitometry software (NIH), and concentration was determined by Pierce BCA Protein Assay (Thermo Fisher Scientific). An isotype control heteroconjugate was generated using similar methodology and anti-HRP (clone HRPN, IgG1) and anti-TNP (clone 2A3, rat IgG2a) (both from BioXCell).
In vitro costimulation assays
B6 or OT-I splenocytes (1 × 105 cells in 200 µl RPMI plus 10% FBS per well in a 96-well plate) were stimulated for the indicated times with the indicated amounts of anti-CD3 mAb (clone 145-2C11, BD Biosciences) or SIINFEKL peptide (NE BioLabs), respectively, plus OrthomAb, unlinked costimulators or control polyclonal rat IgG. Secreted cytokines in culture supernatants were measured using ELISA kits from BD Biosciences (for IFN-γ, IL-2 and IL-6) and R&D Systems (for IL-17) as per the manufacturers’ instructions. Cytokine concentrations were calculated using MARS Data Analysis Software from absorbance values measured using a CLARIOstar microplate reader (BMG LABTECH). Flow cytometry was used to measure cell proliferation (dilution of CellTrace Violet, Thermo Fisher Scientific), and induction of CD134 (OX86), CD137 (1AH2) and CD25 (PC61.5) surface expression on conventional (Foxp3neg) CD4+ and CD8+ T cells and Foxp3+CD4+ T cells. Intracellular staining for Foxp3 (FJK-16 s), GzmB (NGBZ) and Eomes (Dan11mag) was performed following fixation and permeabilization using Foxp3 staining buffer (Tonbo Biosciences). Antibodies were purchased from BD Biosciences, eBioscience, or Tonbo Biosciences, and data were acquired using an LSR II (BD Biosciences) or MACSQuant Analyzer 10 (Miltenyi Biotec), and analyzed using FlowJo software (FlowJo, LLC).
Tumor immunotherapy
B16-F10 melanoma cells (1 × 105, American Type Culture Collection) that were passaged less than 1 month were intradermally injected into the back of B6 mice. Costimulation therapy was administered as indicated when tumors became visible (day 2 or 3, when tumors were at least 1 mm x 1 mm surface area), and tumor growth monitored every 1–2 days at the indicated times. Surface area (mm2) was calculated by multiplying the longest diameter and the diameter perpendicular to it. Area under the curve (AUC) analysis [40] was performed as previously described [31].
Statistics
Graphs were generated and statistical analyses performed using GraphPad Prism (GraphPad Software, Inc.). Comparisons between two groups were performed using unpaired, two-tailed, t tests plus Welch’s correction. Comparisons between three or more groups were performed using one-way ANOVA plus Tukey’s multiple comparison test. Comparisons between titration curves or time courses of two groups were performed using two-way ANOVA plus Sidak’s multiple comparison test. Comparisons between titration curves or time courses of three of more groups were performed using two-way ANOVA plus Tukey’s multiple comparison test. Quantitative data are expressed as mean value ± SEM or SD for datasets with n ≥ 3 or n = 2, respectively. Tumor survival curve comparisons were performed using the log-rank (Mantel-Cox) test.
Results
OrthomAb was synthesized by conjugating anti-CD134 with anti-CD137 using MTz and TCO couplers that were attached to the mAbs at an optimized 7:1 ratio (depicted in Fig. 1a) via click chemistry [35]. SDS–PAGE indicated that the conjugation reaction contained heterodimers (~ 300 kDa), higher-order multimers and residual monomers (Fig. 1b). The heterodimers were enriched by three successive rounds of FPLC size-exclusion chromatography using Sephacryl 300 (Fig. 1c–e) to yield a final OrthomAb product that was > 90% dimer (Fig. 1f).
Fig. 1.
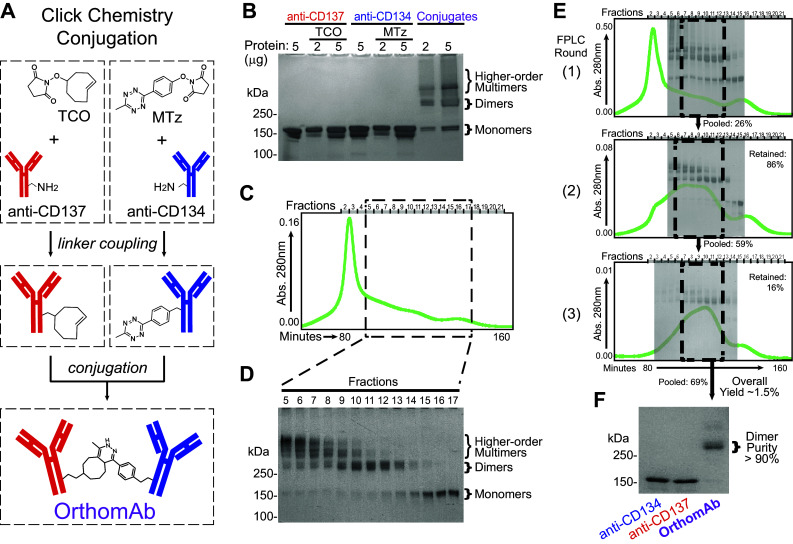
OrthomAb synthesis and purification. a Schematic depicting click conjugation of anti-CD134 and -CD137. b Non-reducing SDS–PAGE showing species at different stages of synthesis, with the two right-most lanes showing a representative unpurified reaction product mixture. Chromatographic tracing of A280 versus time (c) and SDS–PAGE (d) indicate the species present in each fraction. e Chromatographic tracings with SDS–PAGE image overlays depicting species present at each purification round for a representative triple-purified OrthomAb batch, with calculated yields shown at each step. f Representative SDS–PAGE showing purified OrthomAb dimer
To test if OrthomAb costimulates T cells, C57BL/6 (B6) splenocytes were stimulated in vitro with a very low concentration of soluble anti-CD3 mAb (50 ng/ml) that only partially induced CD25 (Fig. 2a). Importantly, addition of OrthomAb to the cultures (1.25 µg/ml) substantially increased CD25 expression on both CD4 and CD8 T cells (Fig. 2a), and also increased proliferation (CellTrace Violet dilution) of conventional CD4 and CD8 T cells in a dose-dependent manner (Fig. 2b, upper and middle panels). Strikingly, however, Treg proliferation appeared to be inhibited by OrthomAb (Fig. 2b, lower panels), which prompted further analysis of the effect of OrthomAb and unlinked costimulators on the different T cell subsets. In contrast to conventional T cells that express CD134 and CD137 following TCR stimulation, Foxp3+ Tregs constitutively express CD134 and CD137, and agonists to both can impact Treg expansion and function [36–39]. Unlinked CD134 and CD137 agonists individually, as well as in combination, augmented the expansion of anti-CD3-stimulated Foxp3+ Tregs, whereas at the doses used only anti-CD137 and the unlinked combination boosted Foxp3neg (conventional) CD4 and CD8 T cell expansion (Fig. 2c, gray versus black bars). Notably, although OrthomAb boosted the expansion of conventional CD4 T cells beyond that elicited by unlinked CD137 agonist and the unlinked combination, it actually elicted weaker Treg expansion in comparison to the unlinked combination (Fig. 2c, bottom panel). CellTrace proliferation analysis revealed that OrthomAb, but not unlinked single and dual costimulators, actually inhibited anti-CD3-induced Treg cell division (Fig. 2d, e). The net outcome of this effect of OrthomAb on the different T cell subsets was an increased ratio of Foxp3neg effector T cells to Foxp3+ Tregs with OrthomAb compared to unlinked single and dual agonists (Fig. 2f).
Fig. 2.
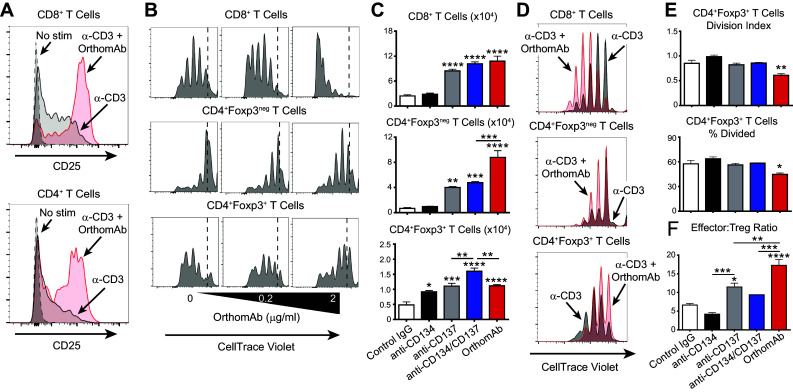
OrthomAb augments effector T cell activation and expansion. B6 splenocytes (1 × 105) were incubated with 0 or 50 ng/ml (a) or 20 ng/ml (b) soluble anti-CD3 mAb ± OrthomAb. a Surface CD25 expression on CD8+ and CD4+ T cells at 24 h post anti-CD3 stimuation ± OrthomAb (1.25 µg/ml). b Anti-CD3-stimulated splenocytes labeled with CellTrace Violet were costimulated with 0, 0.2 or 2 µg/ml OrthomAb, and cell division (CellTrace dilution) of CD8+, CD4+Foxp3neg, and CD4+Foxp3+ T cells measured at 72 h. Data shown are representative of multiple similar experiments. In a separate experiment (c–f), B6 splenocytes (1 × 105) were labeled with CellTrace Violet and cultured 72 h with 15 ng/ml soluble anti-CD3 and control IgG (2 µg/ml), anti-CD134 (2 µg/ml), anti-CD137 (2 µg/ml), anti-CD134 plus anti-CD137 (1 µg/ml each) or OrthomAb (2 µg/ml), n = 4 per group. c Total numbers of CD8+, CD4+Foxp3neg, and CD4+Foxp3+ T cells. d CellTrace dilution histogram overlays for cultures treated with soluble anti-CD3 (black) or soluble anti-CD3 plus OrthomAb (red). e Division Index and % Divided CD4+Foxp3+ Tregs were calculated using FlowJo Proliferation Tool. f Ratio of effector T cells (CD3+CD8+ + CD3+CD4+Foxp3neg) to regulatory T cells (CD3+CD4+Foxp3+). In this and subsequent figures *, **, *** and **** indicate p < 0.05, 0.01, 0.001 and 0.0001, respectively, compared to the control or delineated group (One-way ANOVA)
A key clinical consideration is the capacity of OrthomAb to impact the amount and type of cytokine secreted by stimulated T cells. This was first tested using Rag−/− OT-I TCR transgenic CD8 T cells stimulated in vitro with cognate SIINFEKL peptide (Fig. 3). OrthomAb addition to the SIINFEKL-stimulated cell cultures elicited significantly greater secretion of IL-2 and IFN-γ compared to isotype control IgG, a control conjugate of irrelevant specificity, or media alone (Fig. 3a). Second, in a titratable manner, OrthomAb augmented secretion of IL-2, IFN-γ and, unexpectedly, IL-6 (Fig. 3b). Further, we tested the individual costimulators against OrthomAb for cytokine production (Fig. 4a, b) and CD25 expression (Fig. 4c), and the data show that OrthomAb induced responses far beyond anti-CD134 or -CD137. Similarly, OrthomAb elicited much higher levels of IFN-γ, IL-6 and IL-17 secretion from soluble anti-CD3-stimulated B6 splenocytes compared to unlinked anti-CD134 and -CD137 (Fig. 5a), and curiously, only OrthomAb elicited IL-6 secretion in the absence of anti-CD3 (Fig. 5b, bottom panel). Finally, OrthomAb elicited much greater secretion of IFN-γ, IL-6 and IL-17 from soluble anti-CD3-stimulated B6 splenocytes compared to the unlinked anti-CD134 plus anti-CD137 combination (Fig. 5c). Thus, these data suggest that OrthomAb has a unique capacity to influence not only proliferation but also the amount and type of cytokine produced, which may prove efficacious during a biological response.
Fig. 3.
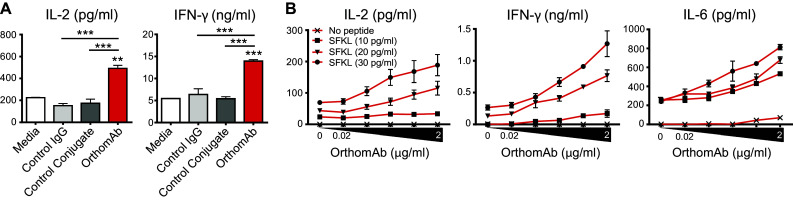
OrthomAb boosts cytokine secretion by peptide-stimulated CD8 T cells. OT-I Rag KO splenocytes (1 × 105) were cultured 24 h with SIINFEKL (SFKL) peptide at 50 pg/ml (a) or the indicated concentrations (b) plus 2 µg/ml isotype control IgG, control conjugate or OrthomAb. a Supernatant IL-2 and IFN-γ measured by ELISA, mean ± SD, n = 2–4, representative of at least 3 independent, similar experiments. b Supernatant IL-2, IFN-γ, and IL-6 with the indicated SFKL peptide and OrthomAb concentrations
Fig. 4.
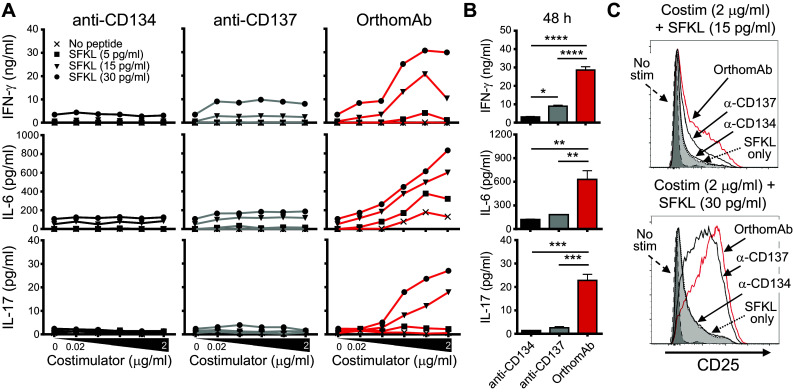
OrthomAb boosts cytokine secretion by peptide-stimulated CD8 T cells more effectively than anti-CD134 and anti-CD137. OT-I Rag KO splenocytes (1 × 105) were cultured 48 h with SFKL peptide and costimulators at the indicated concentrations. a Supernatant IFN-γ, IL-6, and IL-17 with titrated concentrations of SFKL peptide and costimulators. b Supernatant IFN-γ, IL-6, and IL-17 pooled from the 30 pg/ml SFKL peptide plus 0.2, 0.6, and 2.0 µg/ml costimulator wells in (a). c CD25 expression on OT-I CD8+ T cells. Data are representative of multiple similar trials
Fig. 5.
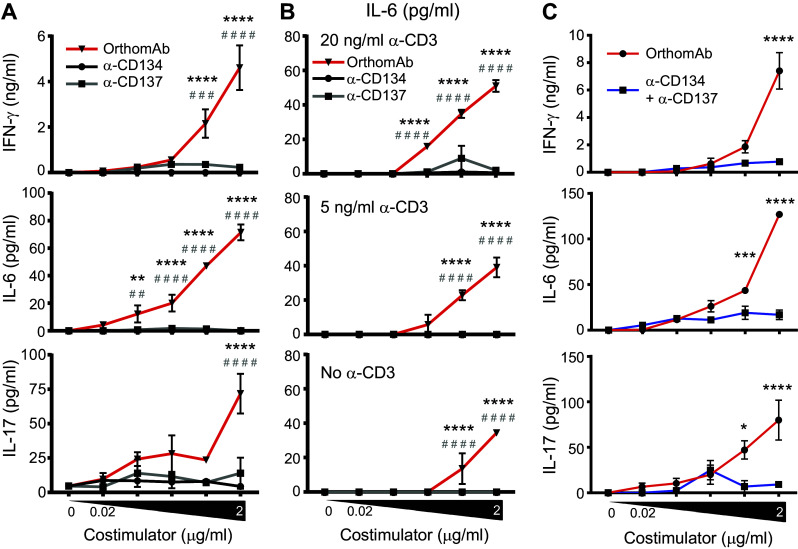
OrthomAb boosts cytokine secretion by anti-CD3-stimulated T cells. B6 splenocytes (1 × 105) were cultured with soluble anti-CD3 at 15 ng/ml (a), 0, 5 or 20 ng/ml (b) or 20 ng/ml (c) plus the indicated concentrations of costimulators for 48 (a, c) or 24 h (b). Significance between OrthomAb and anti-CD134 or -CD137 are indicated by * and #, respectively (a, b), and between OrthomAb and unlinked anti-CD134 plus -CD137 indicated by * (c). N = 3 in each panel, which are representative of multiple similar trials
Next, the aggressive B16-F10 (B16) melanoma model was used to examine if OrthomAb could costimulate T cells in vivo and elicit antitumor immunity. In the first study, mice harboring established B16 tumors were treated with OrthomAb (150 µg) or control conjugate on day 3 (after tumors became visible) and then again on day 6. OrthomAb reduced tumor size relative to the control, with statistically significant differences on days 4–6, 9 and 10, and reduced overall tumor burden (area under the curve [40]), through day 13 (supplementary Fig. 1a). Analysis of TIL on day 18 revealed that the percentages of CD8+ and CD4+ TIL that were double-positive for the cytolytic granule protein granzyme B (GzmB) and the T-box transcription factor Eomesodermin (Eomes) that programs CTL function [41] were ~ twofold higher (p < 0.001) with OrthomAb compared to control-treated tumors (supplementary Fig. 1b). In a second experiment, mice with established B16 tumors were treated with OrthomAb, anti-CD134, anti-CD137 or control IgG on days 2 (after the tumors became visible), 5 and 8. At the doses used, monotherapy with the individual costimulators elicited, at best, only modest reductions in tumor growth (Fig. 6a) as quantified by either tumor size at day 14 (Fig. 6b) or area under the curve through day 14 (Fig. 6c). In contrast, OrthomAb significantly reduced tumor burden ~ twofold (Fig. 6a–c) and extended survival compared to control IgG (Fig. 6d).
Fig. 6.
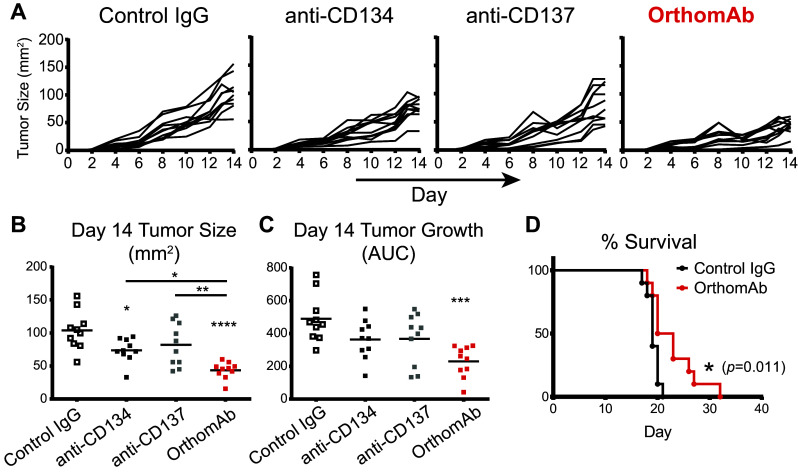
OrthomAb elicits tumor immunity. B6 mice were injected intra-dermally with 1 × 105 B16 tumor cells on day 0, and on day 2 treated with 150 µg control IgG, anti-CD134, anti-CD137 or OrthomAb, and given identical boosters on days 5 and 8. N = 10 per group. a Individual tumor growth curves. Scatter plots of day 14 tumor sizes (b) and area under the curve (AUC) values (c). d Survival curves for control IgG versus OrthomAb
Discussion
Combination immunotherapies can more effectively treat cancer compared to monotherapies [25], and increasing the number of agents included in these therapeutic cocktails can further boost efficacy [28, 33, 34]. Nevertheless, this approach, while feasible in mouse models, poses various challenges for human clinical translation, including regulatory complexities involved in approval for multi-agent clinical testing [32]. The OrthomAb approach provides a means to streamline combination immunotherapy by covalently joining two separate agents, CD134 and CD137 agonists, into a single drug platform. Ultimately, this approach could be replicated by conjugating other agents that, when administered with OrthomAb, would in essence distill a 4-drug combination therapy into a simpler 2-drug combination. This would be advantageous in several ways, including reducing the number of clinical trial arms.
In contrast to other well-established dual-specific immunotherapeutics, such as bispecific antibodies that bind a tumor surface antigen (CD20, EpCAM, HER2, or others) as well as CD3 to enable tumor cell killing by tethering CTL to tumor cells [42], OrthomAb is a tetravalent heteroconjugate that comprised mAb agonists to two separate costimulatory receptors that are each expressed on T cells. This might co-cluster CD134 and CD137 within the same synapse, initiating a unique or “hybrid” downstream signal that programs unexpected, but potentially useful, T cell functions. This possibility is consistent with our current findings that OrthomAb elicits greater secretion of the T cell effector cytokines IFN-γ and IL-17 compared to unlinked CD134 and CD137 single and dual mAb agonists. Alternatively, simply multimerizing individual agonists may be sufficient to augment costimulatory activity. Nevertheless, OrthomAb was unique in its ability to elicit IL-6 secretion, even in the absence of TCR stimulation. Given that IL-6 is known to inhibit Treg stability and function [43–46], this result might also help to explain another unexpected property of OrthomAb, which is its ability to favor the expansion of effector T cells over Tregs (in comparison to unlinked dual agonists). This effect appears fortuitous given that a major issue in developing T cell-based immunotherapies is the potential of modalities to promote the expansion of effector T cells while minimizing that of Tregs, particularly given that both use IL-2 as a growth factor, and thus likely contributes to the ability of OrthomAb to elicit better antitumor efficacy compared to the individual unlinked agonists. Ultimately, the mechanisms underlying these responses elicited by OrthomAb may be highly complex given that multiple T cell and non-T cell populations express CD134 and CD137, and hence the responses of certain cell types may be the consequence of either direct effects by OrthomAb or indirect effects mediated by other responding cells.
In sum, fusing two anti-cancer biologics into a single drug platform provides a novel blueprint for distilling complex combination therapies into a smaller number of therapeutic agents, thus simplifying clinical translation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Abbreviations
- Eomes
Eomesodermin
- GzmB
Granzyme B
- MTz
Methyltetrazine-PEG5-NHS ester
- TCO
Trans-cyclooctene-PEG4-NHS ester
Author contributions
ATV, AJA and JMR were involved in the study conception and design. JMR, AJA, PM, AM and JS were involved in data acquisition and analysis. AJA, ATV and JMR drafted the manuscript. All authors contributed intellectually during the course of the research as well as in critical revision of the manuscript.
Funding
This work was supported by National Institutes of Health Grants R01CA109339 and R01AI094640 as well as a SPARK Award from UConn Health (to Adam J. Adler and Anthony T. Vella).
Compliance with ethical standards
Conflict of interest
Adam J. Adler and Anthony T. Vella have filed a patent application on OrthomAb. All other authors declare that they have no conflict of interest.
Ethical approval
All experiments involving mice were conducted in accordance with the ethical standards established by the National Institutes of Health and UConn Health, and were approved by the UConn Health Institutional Animal Care and Use Committee (IACUC).
Contributor Information
Adam J. Adler, Phone: 860-679-7992, Email: aadler@uchc.edu
Anthony T. Vella, Phone: 860-679-4364, Email: vella@uchc.edu
References
- 1.Maloney DG, Grillo-Lopez AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, Janakiraman N, Foon KA, Liles TM, Dallaire BK, Wey K, Royston I, Davis T, Levy R. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood. 1997;90(6):2188–2195. [PubMed] [Google Scholar]
- 2.McLaughlin P, Grillo-Lopez AJ, Link BK, Levy R, Czuczman MS, Williams ME, Heyman MR, Bence-Bruckler I, White CA, Cabanillas F, Jain V, Ho AD, Lister J, Wey K, Shen D, Dallaire BK. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol. 1998;16(8):2825–2833. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]
- 3.Stolz C, Schuler M. Molecular mechanisms of resistance to Rituximab and pharmacologic strategies for its circumvention. Leuk Lymphoma. 2009;50(6):873–885. doi: 10.1080/10428190902878471. [DOI] [PubMed] [Google Scholar]
- 4.Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. 2015;125(9):3413–3421. doi: 10.1172/JCI80008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tran E, Robbins PF, Rosenberg SA. ‘Final common pathway’ of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol. 2017;18(3):255–262. doi: 10.1038/ni.3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 7.Adler AJ. Mechanisms of T Cell tolerance and suppression in cancer mediated by tumor-associated antigens and hormones. Curr Cancer Drug Targets. 2007;7:3–14. doi: 10.2174/156800907780006931. [DOI] [PubMed] [Google Scholar]
- 8.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182(2):459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, Freeman GJ, Sharpe AH. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. 2017;214(4):895–904. doi: 10.1084/jem.20160801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 15.Lines JL, Sempere LF, Broughton T, Wang L, Noelle R. VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol Res. 2014;2(6):510–517. doi: 10.1158/2326-6066.CIR-14-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44(5):989–1004. doi: 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sotomayor EM, Borrello I, Tubb E, Rattis FM, Bien H, Lu Z, Fein S, Schoenberger S, Levitsky HI. Conversion of tumor-specific CD4 + T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat Med. 1999;5(7):780–787. doi: 10.1038/10503. [DOI] [PubMed] [Google Scholar]
- 18.Diehl L, den Boer AT, Schoenberger SP, van der Voort EI, Schumacher TN, Melief CJ, Offringa R, Toes RE. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat Med. 1999;5(7):774–779. doi: 10.1038/10495. [DOI] [PubMed] [Google Scholar]
- 19.Weinberg AD, Morris NP, Kovacsovics-Bankowski M, Urba WJ, Curti BD. Science gone translational: the OX40 agonist story. Immunol Rev. 2011;244(1):218–231. doi: 10.1111/j.1600-065X.2011.01069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ascierto PA, Simeone E, Sznol M, Fu YX, Melero I. Clinical experiences with anti-CD137 and anti-PD1 therapeutic antibodies. Semin Oncol. 2010;37(5):508–516. doi: 10.1053/j.seminoncol.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Cohen AD, Diab A, Perales MA, Wolchok JD, Rizzuto G, Merghoub T, Huggins D, Liu C, Turk MJ, Restifo NP, Sakaguchi S, Houghton AN. Agonist anti-GITR antibody enhances vaccine-induced CD8(+) T-cell responses and tumor immunity. Cancer Res. 2006;66(9):4904–4912. doi: 10.1158/0008-5472.CAN-05-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts DJ, Franklin NA, Kingeter LM, Yagita H, Tutt AL, Glennie MJ, Bullock TN. Control of established melanoma by CD27 stimulation is associated with enhanced effector function and persistence, and reduced PD-1 expression of tumor infiltrating CD8(+) T cells. J Immunother. 2010;33(8):769–779. doi: 10.1097/CJI.0b013e3181ee238f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity. 2002;16(1):23–35. doi: 10.1016/S1074-7613(01)00259-X. [DOI] [PubMed] [Google Scholar]
- 24.Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity. 2016;44(5):955–972. doi: 10.1016/j.immuni.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee SJ, Myers L, Muralimohan G, Dai J, Qiao Y, Li Z, Mittler RS, Vella AT. 4–1BB and OX40 dual costimulation synergistically stimulate primary specific CD8 T cells for robust effector function. J Immunol. 2004;173(5):3002–3012. doi: 10.4049/jimmunol.173.5.3002. [DOI] [PubMed] [Google Scholar]
- 27.Lee SJ, Rossi RJ, Lee SK, Croft M, Kwon BS, Mittler RS, Vella AT. CD134 costimulation couples the CD137 pathway to induce production of supereffector CD8 T cells that become IL-7 dependent. J Immunol. 2007;179(4):2203–2214. doi: 10.4049/jimmunol.179.4.2203. [DOI] [PubMed] [Google Scholar]
- 28.Cuadros C, Dominguez AL, Lollini PL, Croft M, Mittler RS, Borgstrom P, Lustgarten J. Vaccination with dendritic cells pulsed with apoptotic tumors in combination with anti-OX40 and anti-4–1BB monoclonal antibodies induces T cell-mediated protective immunity in Her-2/neu transgenic mice. Int J Cancer. 2005;116(6):934–943. doi: 10.1002/ijc.21098. [DOI] [PubMed] [Google Scholar]
- 29.Gray JC, French RR, James S, Al-Shamkhani A, Johnson PW, Glennie MJ. Optimising anti-tumour CD8 T-cell responses using combinations of immunomodulatory antibodies. Eur J Immunol. 2008;38(9):2499–2511. doi: 10.1002/eji.200838208. [DOI] [PubMed] [Google Scholar]
- 30.Qui HZ, Hagymasi AT, Bandyopadhyay S, St Rose MC, Ramanarasimhaiah R, Menoret A, Mittler RS, Gordon SM, Reiner SL, Vella AT, Adler AJ. CD134 plus CD137 dual costimulation induces Eomesodermin in CD4 T cells to program cytotoxic Th1 differentiation. J Immunol. 2011;187(7):3555–3564. doi: 10.4049/jimmunol.1101244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mittal P, St Rose MC, Wang X, Ryan JM, Wasser JS, Vella AT, Adler AJ. Tumor-Unrelated CD4 T cell help augments CD134 plus CD137 dual costimulation tumor therapy. J Immunol. 2015;195(12):5816–5826. doi: 10.4049/jimmunol.1502032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verweij J, de Jonge M, Eskens F, Sleijfer S. Moving molecular targeted drug therapy towards personalized medicine: issues related to clinical trial design. Mol Oncol. 2012;6(2):196–203. doi: 10.1016/j.molonc.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uno T, Takeda K, Kojima Y, Yoshizawa H, Akiba H, Mittler RS, Gejyo F, Okumura K, Yagita H, Smyth MJ. Eradication of established tumors in mice by a combination antibody-based therapy. Nat Med. 2006;12(6):693–698. doi: 10.1038/nm1405. [DOI] [PubMed] [Google Scholar]
- 34.Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, Williams RT, Rakhra K, Zhang MH, Rothschilds AM, Kumari S, Kelly RL, Kwan BH, Abraham W, Hu K, Mehta NK, Kauke MJ, Suh H, Cochran JR, Lauffenburger DA, Wittrup KD, Irvine DJ. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med. 2016;22(12):1402–1410. doi: 10.1038/nm.4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed Engl. 2001;40(11):2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 36.Takeda I, Ine S, Killeen N, Ndhlovu LC, Murata K, Satomi S, Sugamura K, Ishii N. Distinct roles for the OX40-OX40 ligand interaction in regulatory and nonregulatory T cells. J Immunol. 2004;172(6):3580–3589. doi: 10.4049/jimmunol.172.6.3580. [DOI] [PubMed] [Google Scholar]
- 37.Zhang P, Gao F, Wang Q, Wang X, Zhu F, Ma C, Sun W, Zhang L. Agonistic anti-4–1BB antibody promotes the expansion of natural regulatory T cells while maintaining Foxp3 expression. Scand J Immunol. 2007;66(4):435–440. doi: 10.1111/j.1365-3083.2007.01994.x. [DOI] [PubMed] [Google Scholar]
- 38.Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, Killeen N, Ishii N, Chang Li X. OX40 costimulation turns off Foxp3 + Tregs. Blood. 2007;110(7):2501–2510. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akhmetzyanova I, Zelinskyy G, Littwitz-Salomon E, Malyshkina A, Dietze KK, Streeck H, Brandau S, Dittmer U. CD137 agonist therapy can reprogram regulatory T cells into cytotoxic CD4 + T cells with antitumor activity. J Immunol. 2016;196(1):484–492. doi: 10.4049/jimmunol.1403039. [DOI] [PubMed] [Google Scholar]
- 40.Duan F, Simeone S, Wu R, Grady J, Mandoiu I, Srivastava PK. Area under the curve as a tool to measure kinetics of tumor growth in experimental animals. J Immunol Methods. 2012;382(1–2):224–228. doi: 10.1016/j.jim.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 41.Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, Banica M, DiCioccio CB, Gross DA, Mao CA, Shen H, Cereb N, Yang SY, Lindsten T, Rossant J, Hunter CA, Reiner SL. Control of effector CD8 + T cell function by the transcription factor Eomesodermin. Science. 2003;302(5647):1041–1043. doi: 10.1126/science.1090148. [DOI] [PubMed] [Google Scholar]
- 42.Chames P, Baty D (2009) Bispecific antibodies for cancer therapy: the light at the end of the tunnel? MAbs. 1 (6):539–547 [DOI] [PMC free article] [PubMed]
- 43.Lin G, Wang J, Lao X, Wang J, Li L, Li S, Zhang J, Dong Y, Chang AE, Li Q, Li S. Interleukin-6 inhibits regulatory T cells and improves the proliferation and cytotoxic activity of cytokine-induced killer cells. J Immunother. 2012;35(4):337–343. doi: 10.1097/CJI.0b013e318255ada3. [DOI] [PubMed] [Google Scholar]
- 44.Sharma MD, Huang L, Choi JH, Lee EJ, Wilson JM, Lemos H, Pan F, Blazar BR, Pardoll DM, Mellor AL, Shi H, Munn DH. An inherently bifunctional subset of Foxp3 + T helper cells is controlled by the transcription factor eos. Immunity. 2013;38(5):998–1012. doi: 10.1016/j.immuni.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nish SA, Schenten D, Wunderlich FT, Pope SD, Gao Y, Hoshi N, Yu S, Yan X, Lee HK, Pasman L, Brodsky I, Yordy B, Zhao H, Bruning J, Medzhitov R. T cell-intrinsic role of IL-6 signaling in primary and memory responses. Elife. 2014;3:e01949. doi: 10.7554/eLife.01949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhanumathy KK, Zhang B, Ahmed KA, Qureshi M, Xie Y, Tao M, Tan X, Xiang J. Transgene IL-6 enhances DC-stimulated CTL responses by counteracting CD4 + 25 + Foxp3 + regulatory T cell suppression via IL-6-induced Foxp3 downregulation. Int J Mol Sci. 2014;15(4):5508–5521. doi: 10.3390/ijms15045508. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.