

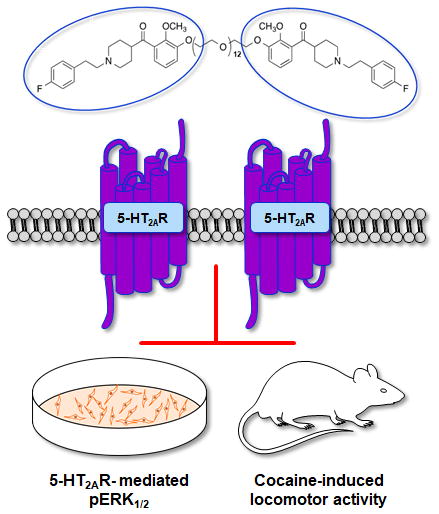
Abstract
The 5-HT2AR plays an important role in various neuropsychiatric disorders including cocaine use disorder and schizophrenia. Homodimerization of this receptor has been suggested but tools that allow direct assessment of 5-HT2AR:5-HT2AR homodimer relevance in these disorders are necessary. We chemically modified the selective 5-HT2AR antagonist M100907 to synthesize a series of homobivalent ligands connected by ethylene glycol linkers of varying lengths that may be useful tools to probe 5-HT2AR:5-HT2AR homodimer function. We tested these molecules for 5-HT2AR antagonist activity in a cell line stably expressing the functional 5-HT2AR, and quantified a post-receptor signaling target, activation (phosphorylation) of extracellular regulated kinases 1/2 (ERK1/2), in comparison to in vivo efficacy to alter spontaneous or cocaine-evoked locomotor activity in rats. All of the synthetic compounds inhibited 5-HT-mediated phosphorylation of ERK1/2 in the cellular signaling assay; the potency of the bivalent ligands varied as a function of linker length with the intermediate linker lengths being the most potent. The Ki values for the binding of bivalent ligands to 5-HT2AR were only slightly lower than the values for the parent (+)-M100907 compound, but retained significant selectivity for 5-HT2AR over 5-HT2BR or 5-HT2CR binding. In addition, the 11-atom-linked bivalent 5-HT2AR antagonist (2 mg/kg, i.p.) demonstrated efficacy on par with (+)-M100907 to inhibit cocaine-evoked hyperactivity. As we develop further strategies for ligand-evoked receptor assembly and analyses of diverse signaling and functional roles, these novel homobivalent 5-HT2AR antagonist ligands will serve as useful in vitro and in vivo probes of 5-HT2AR structure and function.
Keywords: Serotonin, Serotonin 5-HT2A receptor, bivalent ligand
Graphical Abstract
Introduction
Serotonin (5-HT) neurotransmission is critically involved in the regulation of normal behavior (e.g., cognition, mood, satiety, sexual behavior, sleep) and pathological disorders (e.g., anxiety, depression, schizophrenia, substance use disorder) and is therefore, an important medications target. Actions of 5-HT are mediated by at least 14 subtypes of 5-HT receptors, 13 of which are G protein coupled receptors (GPCRs) and are presently grouped into seven families (5-HT1R – 5-HT7R) according to their structural and functional characteristics.1 The metabotropic 5-HT2R family (5-HT2AR, 5-HT2BR, 5-HT2CR) plays an important role in the regulation of CNS function and dysfunction. The receptors in the 5-HT2 family couple predominantly to Gαq/11 proteins to activate phospholipase Cβ (PLCβ) resulting in downstream intracellular calcium (Cai++) release and phosphorylation of ERK1/2 (pERK1/2).2 Abnormalities of 5-HT2R function have been implicated in several neuropsychological and neurological disorders1, 3–6 and active initiatives are underway to develop novel 5-HT2R ligands as therapies for such disorders.7
The 5-HT2AR is of particular interest as a key target of atypical antipsychotics which are thought to improve symptoms and cognitive functioning in schizophrenia due to potent 5-HT2AR antagonist actions.8–10 Other selective 5-HT2AR antagonists show promise to improve symptomology in preclinical models of psychostimulant substance use disorder (i.e., cocaine, nicotine),11–14 anxiety,15 depression,16–18 and sleep disorders.19–21 Selective 5-HT2AR antagonists have been in clinical trials for neurological and/or psychiatric disorders including volinanserin [MDL100907, M100907; (R)-(+)-α-(2,3-dimethoxyphenyl)-1-[2-(4-fluorophenyl)ethyl]-4- piperidinemethanol; Aventis Pharmaceuticals; compound 1; Fig. 1)22, 23 and structurally similar 5-HT2AR antagonists/inverse agonists for schizophrenia24, 25 and sleep disorders.26, 27 Of these, the 5-HT2AR inverse agonist/antagonist pimavanserin (tradename NUPLAZID®; ACP-103; ACADIA Pharmaceuticals) has recently been approved by the FDA for Parkinson’s Disease psychosis4 and the 5-HT1AR agonist/5-HT2AR antagonist flibanserin (tradename Addyi®; BIMT 17; Sprout Pharmaceuticals) has been approved for the treatment of hypoactive sexual desire disorder in pre-menopausal women.28
Figure 1.
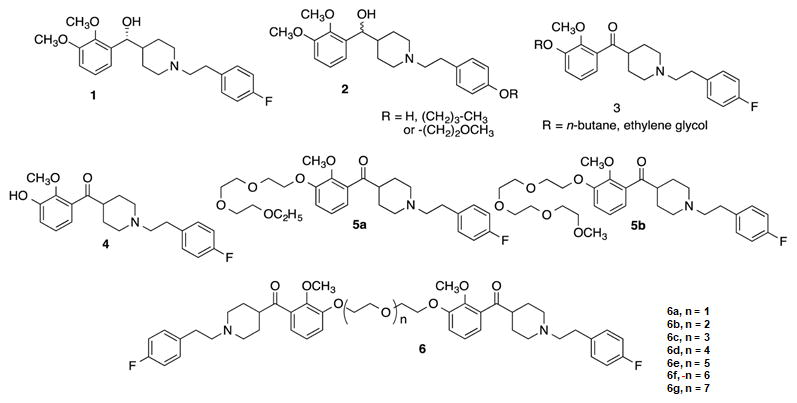
Chemical structures of (+)-M100907 (1) and synthesized analogs. Modifications at the fluorine site (2), modifications at the 3-methoxy site (3), monomer of des-3-methyl ketone M100907 (4), monovalent ligands with 10-atom (5a) and 13-atom (5b) ethylene glycol tethers, and structure of bivalent ligands with tethers of varying lengths (6) are displayed.
Traditional drug discovery efforts for GPCRs like the 5-HT2AR have been designed to target monomeric receptors and conceptualized as the pharmacophore interacting at one receptor binding site.29 However, in recent years, the importance of GPCR dimerization and oligomerization on receptor signaling, trafficking and localization has been demonstrated for a variety of GPCRs, highlighting the need to investigate receptor-receptor interactions in disease pathology (for review, see29). Existence of homodimeric 5-HT2AR:5-HT2AR interactions is supported by co-immunoprecipitation and fluorescence resonance energy transfer (FRET) experiments30 and 5-HT2AR homoreceptor complexes have been proposed as the minimum functioning unit of the PLCβ- and PLA2-mediated signaling pathways induced by 5-HT and synthetic 5-HT2AR agonists.31 Additionally, molecular dynamics modeling studies suggest that the putative 5-HT2AR ligand binding sites displace differently in simulations of monomers versus homodimers, which suggests that receptor:receptor interactions may prefer different 5-HT2AR ligands.32 Therefore, tools to explore 5-HT2AR:5-HT2AR relevance in signal transduction as well as behavioral outcomes are necessary.
One proposed approach to probe receptor:receptor signaling and function is through the use of bivalent ligands. These ligands are comprised of two pharmacophores covalently tethered via a suitable spacer which are hypothesized to interact with a binding site on each receptor in the dimer pair.33 Bivalent ligands have been synthesized to examine various neurotransmitter receptor systems,34 including 5-HT receptors35–37 such as the 5-HT4R38, 39 and 5-HT1BR.40–42 Here, we characterize homobivalent ligands with the pharmacophore of the piperidine M100907 which may serve as future tools to pharmacologically probe 5-HT2AR:5-HT2AR biology. The active (+)isomer of M100907 [(+)-M100907; compound 1, Fig. 1] binds the 5-HT2AR with high affinity and has >100 fold selectivity over the 5-HT2BR and 5-HT2CR.43, 44 The pharmacology of M100907 has been demonstrated in a wide diversity of in vitro45, 46 and in vivo11, 13, 23, 47, 48 studies.
We have previously reported the synthesis and initial characterization of several bivalent analogues based on a modified structure of M100907 (compound 1, Fig. 1).45 To select the best location for the attachment of the tether used to link the molecules, derivatives with substitutions of the p-fluorine (compound 2) or the methoxy group (compound 3) were tested for their ability to inhibit 5-HT-stimulated intracellular calcium (Cai++) release in a Chinese hamster ovary (CHO) cell line stably expressing the human 5-HT2AR (h5-HT2AR). The replacement of the fluorine with a hydroxyl or ether moiety (compound 2) resulted in significant loss of antagonist potency to inhibit 5-HT-induced Cai++ release.45 Attachment of an ethylene glycol at the OH of the catechol (compound 3) was found to retain significant activity.45 Thus, this site was selected as the tether attachment site for synthesizing bivalent molecules. An active first pass metabolite of M100907 (des-3-methyl-ketone-M100907; compound 4), which lacks a chiral center, also proved to be a potent (IC50 = 2.3 nM) 5-HT2AR antagonist in the Cai++ release bioassays.45 This molecule was selected as the starting material for the synthesis of tethered analogs to avoid generation of diasteromeric intermediates and eliminate the need for chiral resolution of the molecules. Two versions of compound 4 were synthesized with ethylene glycol groups (5a–b) and tested in the Cai++ bioassay to determine whether the polyether tether would have a deleterious effect on the activity of the ligand and found to retain nanomolar potency.45 Finally, the desired bivalent molecules (6a–g) were synthesized and tested. The bivalent ligands exhibited sub-micromolar potency to inhibit 5-HT-induced Cai++ release demonstrating the retention of antagonist properties.45 These studies suggested that intermediate tether lengths of 11–17 atoms in length are optimal for 5-HT2AR:5-HT2AR antagonism in the Cai++ assay.45
In the present report, we provide further functional characterization of these compounds. The compounds were additionally profiled through quantification of ERK1/2 phosphorylation which represents a distal downstream signaling outcome of 5-HT2AR activation. Phosphorylation of ERK1/2 serves as an integration point of multiple upstream signaling pathways, including G protein dependent- and independent signal transduction.46,1, 49, 50 In addition, affinity of these compounds for the 5-HT2AR and selectivity over the highly homologous 5-HT2BR or 5-HT2CR was determined. We found that the bivalent ligands (Compound 6 series) retained activity and selectivity similar to that of (+)-M100907 (Compound 1) and that the optimal tether length is between 8- and 17- atom linkers in the ERK1/2 activation cellular assay. Thus, a homobivalent ligand with an intermediate tether length (compound 6c) was selected for the first in vivo studies to evaluate the behavioral profile of a homobivalent 5-HT2AR antagonist molecule. These studies open the door to the development of new bivalent molecules with the potential to elucidate the neurobiological role of 5-HT2AR:5-HT2AR homodimers in the CNS.
Results and Discussion
The inhibitory potency of M100907 analogs (Fig. 1) was evaluated in an ERK1/2 activation assay in h5-HT2AR-CHO cells (Fig. 2; Table 1). Activation of ERK1/2 occurs by phosphorylation of the kinase which, in turn, phosphorylates other downstream targets to regulate gene expression and a variety of cellular processes (for review, see51). Serotonin induces a concentration-dependent increase in phosphorylated ERK1/2 (pERK1/2) with an EC50 of ~72 nM (Fig. 2A). Compound 1 and its analogs (4–6g; 10−10–10−4 M) were examined for their ability to antagonize ERK1/2 activation induced by a maximally effective concentration (1 μM) of 5-HT. The pIC50 and IC50 values are reported in Table 1. Compound 1, (+)-M100907,52 and compound 4, the des-3-methyl-ketone derivative starting material for synthesis of the bivalent ligands, displayed low nanomolar potency in inhibiting 5-HT-evoked ERK1/2 activation (Table 1, Fig. 2B).
Figure 2.
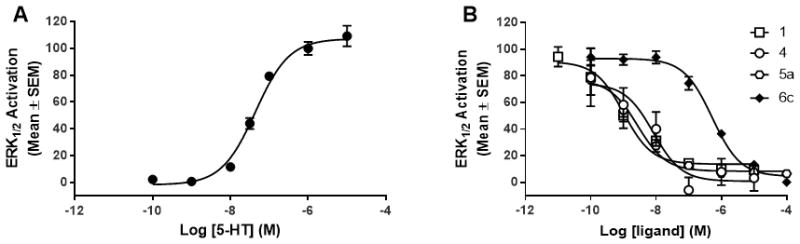
Representative ERK1/2 activation response in h5-HT2AR-CHO cells. [A] 5-HT evokes a concentration-dependent elevation of pERK1/2 expression (pEC50 = 7.14 ± 0.04; EC50 = 72.4 nM) and 1 μM 5-HT induces maximal ERK1/2 activation. [B] M100907 derivatives induce a concentration-dependent inhibition of 1 μM 5-HT. pIC50 and IC50 values are listed in Table 1.
Table 1.
Potency of M100907 derivatives on ERK1/2 activation in h5-HT2AR-CHO cells.a
| ID | Linker # | pIC50 ± SEM | IC50 (nM) |
|---|---|---|---|
| 1 | — | 8.29 ± 0.52 | 5.1 |
| 2 | — | N.T. | N.T. |
| 3 | — | N.T. | N.T. |
| 4 | — | 8.00 ± 0.05 | 10.1 |
| 5a | 10 | 8.11 ± 0.51 | 7.9 |
| 5b | 13 | 6.65 ± 0.04 | 225 |
| 6a | 5 | 6.53 ± 0.24 | 298 |
| 6b | 8 | 7.59 ± 0.53 | 25.6 |
| 6c | 11 | 6.75 ± 0.16 | 178 |
| 6d | 14 | 7.01 ± 0.13 | 98.2 |
| 6e | 17 | 6.78 ± 0.39 | 164 |
| 6f | 20 | 6.55 ± 0.12 | 284 |
| 6g | 23 | 6.72 ± 0.14 | 192 |
pIC50 is presented as mean ± SEM (n = 3–6). IC50 values were calculated from averaged pIC50 values.
“—” indicates that there is no atom linker on this compound. “NT” indicates compound has not been tested in this assay.
The evaluation of inhibitory potency of compounds 5a and 5b, derivatives of compound 4 with 10- or 13-atom ethylene glycol linkers, revealed that the addition of the 10-atom linker (5a) retained comparable potency compared to the parent compounds, while addition of a 13-atom linker (5b) reduced the potency ~22-fold compared to the ketone starting material (4). This reduction in potency is not surprising since others have also noted that mere addition of the polyether tether diminishes the functional activity of the ligand.33 Both 5a (10-atom) and 5b (13-atom) maintained sub-micromolar potency to inhibit 5-HT-mediated ERK1/2 activation, thus, these data suggest that the 10-atom chain may be of optimal length. The potency of the homobivalent ligands (6a–g) to inhibit 5-HT-mediated pERK1/2 was diminished compared to the parent compounds and varied modestly as a function of linker length (Table 1). Interestingly, the homobivalent with the shortest (5 atoms; 6a) and the longest (20–23 atoms; 6f and 6g) linkers had the lowest potency which suggests that there is an optimal linker length for maintenance of antagonist activity. Comparison of the inhibitory potency of the 6c, the 11-atom linked bivalent ligand, and 5a, its respective 10-atom monovalent counterpart, show that the potency of 6c is decreased by ~22-fold versus 5a, suggesting that addition of a second pharmacophore decreased the functional activity (Fig. 2B). To the contrary, comparison of the 14-atom linker bivalent ligand (6d) to its respective 13-atom monovalent counterpart (5b) resulted in a slight increase in potency (Table 1). These findings are consistent with previously published effects of these compounds in the Cai++ bioassay45 and, together, may suggest that linker length impacts the potency of these ligands and should be considered for future experiments.
The affinity and selectivity of the synthesized M100907 derivatives for the 5-HT2AR versus the highly homologous 5-HT2BR and 5-HT2CR, were assessed via radioligand binding assays at the Psychoactive Drug Screening Program (PSDP). The resultant Ki values for binding to 5-HT2AR, 5-HT2BR and 5-HT2CR, as assessed by displacement of [3H]-ketanserin (0.5 nM), [3H]-LSD (1 nM) and [3H]-mesulergine (0.5 nM), respectively, are shown in Table 2. All ligands displayed nanomolar affinity for the 5-HT2AR, but Ki values were ~4–8 fold lower than that previously reported for the high affinity (+)-M100907 isomer (Compound 1; Ki = 3 nM).53 According to the PDSP, variations of this magnitude are expected in Ki assessments, thus the differences in Ki values observed at 5-HT2AR for most of the M100907 analogs were modest versus compound 1 (Table 2). Only the bivalent ligand with the 20-atom linker (6f) exhibited a substantial decrease (~10 fold) in affinity for the (5-HT2AR, Ki = 32 nM) which coincides with the decrease in potency observed for this molecule in the ERK1/2 activation assay. In addition, the presence of the second pharmacophore in the 11- (6c) and 14-atom (6d) tethered bivalent ligands afforded no overt gain in affinity compared to their respective monovalent counterparts (5a, 5b). These lack of differences may just be due to the inherent variability in Ki measurements, and/or the lack of measurement of actual association/dissociation constants of the ligands; the binding analysis was done at equilibrium, as opposed to during the dynamic state related to the functional endpoints.54
Table 2.
Affinity profile of M100907 derivatives at the 5-HT2AR, 5-HT2BR, 5-HT2CR.
| ID | Linker # | 5-HT2AR | 5-HT2BR | 5-HT2CR | |||
|---|---|---|---|---|---|---|---|
| pKi1 | Ki (nM) | pKi2 | Ki (nM) | pKi3 | Ki (nM) | ||
| 1 | -- | 8.54 | 3.0 | 6.2 | 612 | 7.4 | 41 |
| 2 | -- | N.T. | N.T. | N.T. | |||
| 3 | -- | N.T. | N.T. | N.T. | |||
| 4 | -- | 8.8 | 1.6 | 6.6 | 261 | 7.8 | 15 |
| 5a | 10 | 8.3 | 5.4 | 6.2 | 701 | 6.5 | 312 |
| 5b | 13 | 8.2 | 6.9 | N.D.5 | 6.3 | 472 | |
| 6a | 5 | 8.54 | 3.1 | 6.7 | 202 | 7.6 | 27 |
| 6b | 8 | 8.14 | 7.2 | 6.3 | 476 | 7.2 | 64 |
| 6c | 11 | 8.24 | 5.8 | 6.9 | 126 | 7.1 | 79 |
| 6d | 14 | 8.54 | 3.4 | 6.8 | 157 | 7.1 | 84 |
| 6e | 17 | 8.54 | 3.0 | 6.6 | 267 | 7.3 | 50 |
| 6f | 20 | 7.54 | 32 | 5.9 | 1399 | 6.3 | 245 |
| 6g | 23 | 8.44 | 4.4 | 6.6 | 238 | 7.0 | 101 |
Ki determinations were generously provided by the NIMH Psychoactive Drug Screening Program.
Determined by displacement of [3H] ketanserin (0.5 nM) relative to displacement by 10 μM clozapine;
Determined by displacement of [3H] LSD (1.0 nM) relative to displacement by 10 μM methysergide;
Determined by displacement of [3H] mesulergine (0.5 nM) relative to displacement by 10 μM chlorpromazine.
Determined in two independent assays; N.D. = Not determined; Displacement by 10 μM of methylsergide was < 50%. N.T.= Not tested in this assay
The inclusion of the second pharmacophore in the bivalent molecules did not afford any improvement in selectivity of the compounds for the 5-HT2AR versus the 5-HT2BR or 5-HT2CR compared to the parent compounds. However, all the M100907 analogs maintained ~10-fold or greater affinity for the 5-HT2AR over the 5-HT2CR and >21-fold selectivity over the 5-HT2BR. Due to the high degree of homology between these receptors, the selectivity of 6c and 6d bivalent compounds for the 5-HT2AR was confirmed by assessment of these ligands in a functional assay of intracellular calcium (Cai++) release, in 5-HT2CR-expressing cells. Similar to compound 1, compounds 6c and 6d had no effect upon 5-HT2CR-mediated Cai++ release evoked by 1 μM 5-HT at concentrations <10 μM (data not shown).
The bivalent ligand with an intermediate (11-atom) linker (6c) was selected for initial in vivo efficacy studies based upon the in vitro results (above).45 Previous studies have reported that (+)-M100907 (Compound 1; 0.02–2 mg/kg) significantly suppresses cocaine-evoked, but not spontaneous, locomotor activity.48, 55 Therefore, the effects of compound 6c on spontaneous (saline) - versus cocaine-evoked locomotor activity was assessed in rats (Fig. 3). Locomotor activity was measured after (intraperitoneal) injection with vehicle (1% Tween 80 with 2% ethanol in 0.9% NaCl) or bivalent ligand 6c (2 mg/kg) 30 minutes prior to saline (1 ml/kg) or cocaine (15 mg/kg) injection. Data are presented as mean total horizontal ambulations summed for the entire 90-minute session (Fig. 3A) and mean total vertical activity summed for the entire 90 min session (Fig. 3B). A main effect of treatment was observed for both mean total horizontal ambulations [F(3,27) = 9.71, p<0.05; Fig. 3A] and mean total vertical activity [F(3,27) = 13.90, p<0.05; Fig. 3B] summed across the 90-min session. A priori comparisons indicated that the bivalent ligand 6c did not alter saline-evoked horizontal ambulation or vertical activity (n.s.; Fig. 3A and 3B). As expected, administration of vehicle plus cocaine (Veh + Coc) elevated total horizontal ambulation (Fig. 3A) and vertical activity (Fig. 3B) versus vehicle plus saline (Veh + Sal) administration for the entire 90-min session (p<0.05). A priori comparisons showed that the bivalent ligand 6c significantly suppressed cocaine-evoked horizontal ambulation (p<0.05; Fig. 3A) and vertical activity (p<0.05; Fig. 3C). A three-way mixed model ANOVA indicated no pretreatment x treatment x time interaction (F17,459) = 1.12, n.s.) for total horizontal ambulation (Fig. 3C). There was a pretreatment x treatment x time interaction (F17,459) = 1.74, p<0.05) for vertical activity (Fig. 3D). Taken together and similar to the previously published (+)-M100907,48 the bivalent ligand 6c modestly suppressed spontaneous locomotor activity and displayed efficacy to suppress cocaine-evoked locomotor activity in a time-dependent manner. Of note, the calculated Central Nervous System Multiparameter Optimization (CNS MPO) value for 6c was found to be 3. While this number is less than the optimal range of greater than 5, there are numerous CNS drugs with this value.56 These data provide the first positive confirmation that a newly synthesized homobivalent ligand displays efficacy to produce behavioral effects similar to the parent 5-HT2AR antagonist.
Figure 3.
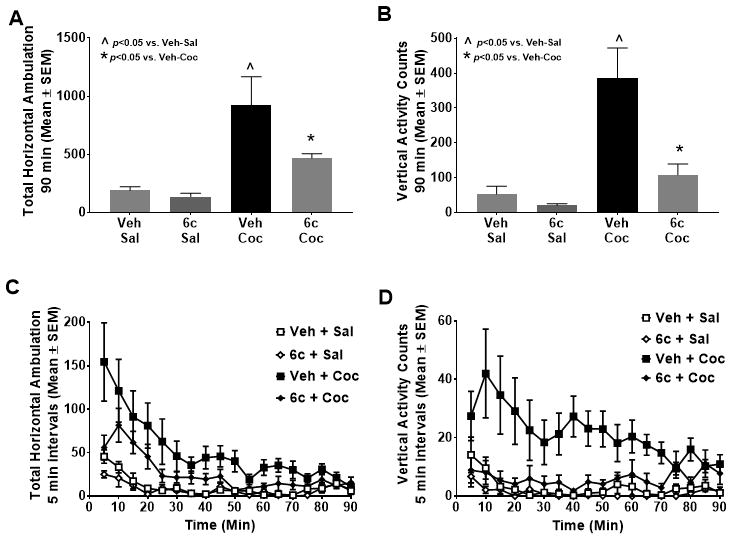
Effects of pretreatment with the bivalent derivative 6c. Rats (n=7–8/group) were treated (i.p.) with vehicle (Veh) or bivalent ligand 6c (2 mg/kg) 30 min prior to injection with saline (Sal; 1 ml/kg) or cocaine (Coc; 15 mg/kg) immediately before the test session commenced. Data are presented as mean (± SEM) total horizontal ambulation [A] or vertical activity counts [B] summed across the entire 90 min session. Total horizontal ambulation [C] and vertical activity [D] in 5-min time bins are presented.
The present data indicate that the series of covalently-linked homobivalent 5-HT2AR antagonist ligands based on the structure of the potent 5-HT2AR antagonist (+)-M100907 (Compound 1) retain potency and efficacy in vitro and in vivo. These ligands maintained nanomolar affinity for the 5-HT2AR, selectivity over the 5-HT2BR and 5-HT2CR, and nanomolar potency as 5-HT2AR antagonists in a cellular assay of ERK1/2 activation. Furthermore, these results suggest that tether lengths of 8–17 atoms in length are optimal to suppress ERK1/2 activation (present studies), which is consistent with our previous findings in the Cai++ assay.45 Encouragingly, we also demonstrated that the covalently-linked homobivalent ligand with optimal spacing suppresses cocaine-evoked behavior in vivo. Future studies are required to explore whether the addition of a second pharmacophore results in a second binding site which could represent the active site of a second interacting receptor, as might be expected given that the 5-HT2AR has been demonstrated to exist as a receptor homodimer.30–32, 57 Thus, these newly-designed bivalent ligands may provide useful tools to further explore 5-HT2AR function and homodimerization.
Materials and Methods
Cell lines and cell culture
A CHO-K1 cell line stably transfected with the 5-HT2AR (5-HT2AR-CHO cells; FA4 line) was a generous gift of K. Berg and W. Clarke (University of Texas Health Science Center at San Antonio). This line expresses transfected h5-HT2AR in the p198-DHFR-Hygro vector containing a hygromycin resistance gene.58 Reverse transcription of RNA followed by a quantitative real time PCR assay confirmed that 5-HT2AR-CHO cells expressed 5-HT2AR mRNA (estimated to be approximately 3–4% of the mRNA level of the housekeeping gene cyclophilin; data not shown), but did not express either 5-HT2BR or 5-HT2CR mRNA;59 the parental CHO-K1 cell line did not express detectable amounts of any 5-HT2R mRNAs.46 Cells were grown at 37°C, 5% CO2 and 85% relative humidity in GlutaMax α-MEM (Invitrogen, Carlsbad CA), 5% fetal bovine serum (Atlanta Biologicals, Atlanta GA), 100 μg/ml hygromycin (Mediatech, Manassas VA) and were passaged when they reached 80% confluence.
Ligands
Serotonin (5-HT; Acros Organics, ThermoFisher Scientific, Pittsburgh, PA) was dissolved in 1X Hank’s balanced salt solution (HBSS; Cellgro, Invitrogen) for in vitro studies. The (+)-M100907 (1) [R-(+)-(2,3-dimethoxyphenyl)-1-[2-(4-fluorophenylethyl)]-4-piperidine-methanol] was synthesized in the Drug Design and Synthesis Section, National Institute on Drug Abuse (National Institutes of Health) as described,52 and was dissolved in 1X HBSS for in vitro studies. The (+)-M100907 (1), des-3-methyl-ketone-M100907 (4), and the monovalent and bivalent M100907 derivative compounds (Series 5 and 6; see Fig. 1.) were synthesized as described previously45 and dissolved in 1X HBSS for in vitro studies. Compound 6c was dissolved in 1% Tween 80 + 2% ethanol in 0.9% NaCl for in vivo studies. (−)-Cocaine (National Institute on Drug Abuse, Research Triangle Park, NC, USA) was dissolved in 0.9% NaCl.
Plate immunoassay for ERK1/2 activation
We adapted a previously developed plate immunoassay,60 to measure levels of pERK1/2 expression following ligand administration, with optimized fixation and antibody incubation conditions for use in this cell line.46 Cells were plated in serum-replete medium at 16–20K cells in 150 μl in clear-sided, clear bottom 96-well tissue culture plates. Cells were grown for 24 hr in serum-replete medium, and then shifted overnight to serum-free medium. The day of the experiment, cells were fed with 80 μl of serum-free medium and returned to the incubator for 1–2 hrs, as adding medium alone caused a measurable pERK1/2 signal that subsided by 1 hr (data not shown), as seen by others.61
Test compounds were added as 20 μl of a 5x stock concentration and plates were incubated at 37°C for 15 min. Cells were then stimulated with 1 μM of 5-HT (25 μl of a 5 μM stock) and incubated at 37°C for 5 min. Full concentration-response curves (10−11-10−4 M) for 5-HT and each compound were performed in each experiment. Reactions were stopped by the addition of 2% paraformaldehyde (PFA) in phosphate buffered saline (PBS, pH 7.4). Cells were fixed for 45 min at room temperature (RT) then rinsed with PBS. Cells were then permeabilized with ice-cold methanol to ensure antibody access to intracellular antigens, washed with PBS and blocked for 45 min at RT with 0.1% fish gelatin (Sigma, St. Louis MO). Cells were then incubated with a 1:500 dilution of mouse monoclonal anti-pERK1/2 (p44/42; Cell Signaling, Danvers MA; #9106) overnight at 4°C with gentle shaking. Background was determined in wells incubated with no primary antibody. After washing with PBS, biotin-conjugated secondary antibody (Vector Labs, Burlingame CA; # BA-9200, 1:500 diluted in blocking solution) was added and incubated for 1 hr at RT. Following washing, alkaline phosphatase (AP) complexed with avidin (Vector Labs, #AK5000) was prepared according to the manufacturer’s directions, added to the wells and incubated for 1 hr at RT. After washing, 50 μl of the AP substrate para-nitrophenyl-phosphate (pNpp; Vector Labs, #SK-5900) with levamisole (an inhibitor of endogenous phosphatases; Vector Labs, #SP-5000; two drops/10 ml), freshly prepared in 100 mM sodium bicarbonate was added, and the plate was incubated at 37°C for 30 min. The absorbance of the yellow product para-nitrophenol (pNp) was measured at 405 nm (A405). Data were normalized to total cell mass in each well as measured by crystal violet staining (below) and expressed as A405/A590. The pIC50 values for pERK1/2 expression were determined using 3-parameter nonlinear regression analysis (GraphPad Prism 7.02) and calculated from at least three independent experiments, each conducted in technical replicates of 3–8, and are presented as the mean ± SEM.
Crystal violet staining
Total cell mass in each well was measured by crystal violet staining, a value proportional to cell number used to estimate cell number in each well. Upon completion of an experiment, wells were rinsed with water, air dried and 50 μl of crystal violet solution (0.1% in water) was added for 30 min at RT, followed by one additional rinse. Cell-absorbed dye was extracted by the addition of 10% acetic acid (30 min, RT) and absorbance read at 590 nm (A590).
Radioactive binding assays
Ki determinations were generously provided by the NIMH Psychoactive Drug Screening Program (PDSP; http://pdsp.med.unc.edu/). Briefly, binding assays were performed using crude membrane fractions from cell lines transiently or stably transfected with the appropriate receptor. The 5-HT2AR binding was determined by displacement of [3H] ketanserin (0.5 nM) relative to displacement by 10 μM clozapine. The 5-HT2BR binding was determined by displacement of [3H] LSD (1.0 nM) relative to displacement by 10 μM methysergide. The 5-HT2CR binding was determined by displacement of [3H] mesulergine (0.5 nM) relative to displacement by 10 μM chlorpromazine. An initial screen measured net displacement of bound ligand by 10 μM of each synthetic antagonist. Ki values were determined on all compounds that yielded > 50% displacement by performing competitive binding curves using 11 concentrations spanning six orders of magnitude, with triplicate determinations for each concentration. Binding determinations were repeated for the racemic M100907 and for the bivalent molecules and the resultant Ki values are represented as mean ± SEM.
Animals
A total of 32 male Sprague-Dawley rats (Harlan, Inc., Indianapolis, IN, USA) weighing 225–325 g at the start of the experiments were used. Rats were allowed to acclimate for 5–7 days in a colony room at a constant temperature (21–23°C) and humidity (45–50%) on a 12 hr light-dark cycle (lights on 0700–1900 hr). Rats were housed two rats per cage and food and water was available ad libitum. All experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (2011) and with the approval of the UTMB Institutional Animal Care and Use Committee. All efforts were made to minimize animal suffering, to reduce the number of animals used, and to utilize alternatives to in vivo techniques, when available.
In vivo assessment
Locomotor activity was monitored and quantified under low light conditions using a modified open field activity system (San Diego Instruments, San Diego, CA) according to previous publications with minor modifications.48, 62–64 Clear Plexiglass chambers (40 × 40 × 40 cm) were surrounded by a 4 × 4 photobeam matrix positioned 4 cm from the chamber floor. Consecutive photobeam breaks within the 16 × 16 cm of the activity monitor were recorded as central ambulations. Peripheral ambulations were counted as consecutive beam breaks in the surrounding perimeter. Central and peripheral ambulations were summed to provide a measure of total horizontal ambulation. Vertical activity was quantified as the sum of the upper photobeam breaks that occurred within the activity monitor every 5 min. Rats were acclimated to the colony room and following 1 week of handling, rats were habituated to the activity monitors for 3 h/day for 2 days before the test day. Using a between-subjects design, rats (n=7–8/group) received vehicle (1% Tween 80 + 2% ethanol in 0.9% NaCl, 1 ml/kg, i.p.), or compound 6c (2 mg/kg, i.p.), followed 30 minutes later by an injection of either saline (1 ml/kg, i.p.) or cocaine (15 mg/kg, i.p) and were immediately placed in activity monitors; locomotor activity was assessed for 90 min. Due to a misinjection, one rat was removed from vehicle plus cocaine group for a final n=7 in that group; all other groups were n=8.
Locomotor activity data are presented as mean total horizontal ambulation or vertical activity (± SEM) summed across the session the entire 90-min session or within 5 min time bins. The main effect of treatment on total horizontal ambulation and vertical activity were analyzed with a one-way ANOVA using the GLM procedure (SAS for Windows). Subsequent a priori comparisons between means for total horizontal ambulation and vertical activity were made using the Bonferroni correction. A three-way ANOVA was employed to analyze the between-subject factors of pretreatment (vehicle, 6c) and treatment (saline, cocaine) and the within-subject factor of time (5-min bins across 90 min session) using a mixed general linear model (SPSS). All statistical analyses were conducted with an experimentwise error rate of α=0.05
Acknowledgments
We thank Ms. Nicole C. Bremer and Drs. Patricia K. Seitz, Jennifer Jeng, and Rachel M. Hartley for providing assistance with the ERK1/2 activation experiments. Receptor binding profiles data was generously provided by the National Institute of Mental Health (NIMH) Psychoactive Drug Screening Program (PDSP), Contract # HHSN-271-2008-00025-C. The NIMH PDSP is directed by Dr. Bryan L. Roth at the University of North Carolina at Chapel Hill and Project Officer Dr. Jamie Driscoll at NIMH, Bethesda MD, USA.
Funding
This work was supported by the National Institute on Drug Abuse grants P50 DA0333935 (K.A.C., S.R.G., N.C.A.), T32 DA07287 (C.A.S.), and K05 DA020087 (K.A.C.). A portion of this work was supported by the NIH Intramural Research Programs of the National Institute on Drug Abuse and the National Institute of Alcohol Abuse and Alcoholism (K.C.R).
Footnotes
Author Contributions
C.A.S. performed the in vitro biological analyses, conducted pharmacological analyses and drafted the manuscript; M.J.S. and K.C.R. performed the chemical syntheses and analyses; R.G.F performed the in vivo biological analyses; C.S.W. and M.J.B. drafted the manuscript and provided biological interpretations; K.A.C., S.R.G., and N.C.A. conceptualized the project, oversaw experimental design/interpretation/analyses, and wrote/edited the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Hoyer D, Hannon JP, Martin GR. Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol Biochem Behav. 2002;71:533–554. doi: 10.1016/s0091-3057(01)00746-8. [DOI] [PubMed] [Google Scholar]
- 2.Felder CC, Kanterman RY, Ma AL, Axelrod J. Serotonin stimulates phospholipase A2 and the release of arachidonic acid in hippocampal neurons by a type 2 serotonin receptor that is independent of inositolphospholipid hydrolysis. Proc Natl Acad Sci U S A. 1990;87:2187–2191. doi: 10.1073/pnas.87.6.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roth BL, Hanizavareh SM, Blum AE. Serotonin receptors represent highly favorable molecular targets for cognitive enhancement in schizophrenia and other disorders. Psychopharmacology (Berl) 2004;174:17–24. doi: 10.1007/s00213-003-1683-8. [DOI] [PubMed] [Google Scholar]
- 4.Meltzer HY, Mills R, Revell S, Williams H, Johnson A, Bahr D, Friedman JH. Pimavanserin, a serotonin(2A) receptor inverse agonist, for the treatment of parkinson’s disease psychosis. Neuropsychopharmacology. 2010;35:881–892. doi: 10.1038/npp.2009.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bubar MJ, Cunningham KA. Prospects for serotonin 5-HT2R pharmacotherapy in psychostimulant abuse. Prog Brain Res. 2008;172:319–346. doi: 10.1016/S0079-6123(08)00916-3. [DOI] [PubMed] [Google Scholar]
- 6.Cunningham KA, Anastasio NC. Serotonin at the nexus of impulsivity and cue reactivity in cocaine addiction. Neuropharmacology. 2014;76(Pt B):460–478. doi: 10.1016/j.neuropharm.2013.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Howell LL, Cunningham KA. Serotonin 5-HT2 receptor interactions with dopamine function: implications for therapeutics in cocaine use disorder. Pharmacol Rev. 2015;67:176–197. doi: 10.1124/pr.114.009514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meltzer HY, Massey BW, Horiguchi M. Serotonin receptors as targets for drugs useful to treat psychosis and cognitive impairment in schizophrenia. Curr Pharm Biotechnol. 2012;13:1572–1586. doi: 10.2174/138920112800784880. [DOI] [PubMed] [Google Scholar]
- 9.Gray JA, Roth BL. Molecular targets for treating cognitive dysfunction in schizophrenia. Schizophr Bull. 2007;33:1100–1119. doi: 10.1093/schbul/sbm074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.TdP M-100907 (Aventis) Curr Opin Investig Drugs. 2001;2:123–132. [PubMed] [Google Scholar]
- 11.Anastasio NC, Stoffel EC, Fox RG, Bubar MJ, Rice KC, Moeller FG, Cunningham KA. Serotonin (5-hydroxytryptamine) 5-HT2A receptor: Association with inherent and cocaine-evoked behavioral disinhibition in rats. Behav Pharmacol. 2011;22:248–261. doi: 10.1097/FBP.0b013e328345f90d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burmeister JJ, Lungren EM, Kirschner KF, Neisewander JL. Differential roles of 5-HT receptor subtypes in cue and cocaine reinstatement of cocaine-seeking behavior in rats. Neuropsychopharmacology. 2004;29:660–668. doi: 10.1038/sj.npp.1300346. [DOI] [PubMed] [Google Scholar]
- 13.Nic Dhonnchadha BA, Fox RG, Stutz SJ, Rice KC, Cunningham KA. Blockade of the serotonin 5-HT2A receptor suppresses cue-evoked reinstatement of cocaine-seeking behavior in a rat self-administration model. Behav Neurosci. 2009;123:382–396. doi: 10.1037/a0014592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fletcher PJ, Rizos Z, Noble K, Soko AD, Silenieks LB, Le AD, Higgins GA. Effects of the 5-HT2C receptor agonist Ro60-0175 and the 5-HT2A receptor antagonist M100907 on nicotine self-administration and reinstatement. Neuropharmacology. 2012;62:2288–2298. doi: 10.1016/j.neuropharm.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 15.Weisstaub NV, Zhou M, Lira A, Lambe E, Gonzalez-Maeso J, Hornung JP, Sibille E, Underwood M, Itohara S, Dauer WT, Ansorge MS, Morelli E, Mann JJ, Toth M, Aghajanian G, Sealfon SC, Hen R, Gingrich JA. Cortical 5-HT2A receptor signaling modulates anxiety-like behaviors in mice. Science. 2006;313:536–540. doi: 10.1126/science.1123432. [DOI] [PubMed] [Google Scholar]
- 16.Berg KA, Harvey JA, Spampinato U, Clarke WP. Physiological and therapeutic relevance of constitutive activity of 5-HT 2A and 5-HT 2C receptors for the treatment of depression. Prog Brain Res. 2008;172:287–305. doi: 10.1016/S0079-6123(08)00914-X. [DOI] [PubMed] [Google Scholar]
- 17.Zaniewska M, McCreary AC, Wydra K, Filip M. Effects of serotonin (5-HT)2 receptor ligands on depression-like behavior during nicotine withdrawal. Neuropharmacology. 2010;58:1140–1146. doi: 10.1016/j.neuropharm.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 18.Celada P, Puig M, margos-Bosch M, Adell A, Artigas F. The therapeutic role of 5-HT1A and 5-HT2A receptors in depression. J Psychiatry Neurosci. 2004;29:252–265. [PMC free article] [PubMed] [Google Scholar]
- 19.Landolt HP, Wehrle R. Antagonism of serotonergic 5-HT2A/2C receptors: mutual improvement of sleep, cognition and mood? Eur J Neurosci. 2009;29:1795–1809. doi: 10.1111/j.1460-9568.2009.06718.x. [DOI] [PubMed] [Google Scholar]
- 20.Teegarden BR, Al SH, Xiong Y. 5-HT(2A) inverse-agonists for the treatment of insomnia. Curr Top Med Chem. 2008;8:969–976. doi: 10.2174/156802608784936700. [DOI] [PubMed] [Google Scholar]
- 21.Popa D, Lena C, Fabre V, Prenat C, Gingrich J, Escourrou P, Hamon M, Adrien J. Contribution of 5-HT2 receptor subtypes to sleep-wakefulness and respiratory control, and functional adaptations in knock-out mice lacking 5-HT2A receptors. J Neurosci. 2005;25:11231–11238. doi: 10.1523/JNEUROSCI.1724-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maurel-Remy S, Bervoets K, Millan MJ. Blockade of phencyclidine-induced hyperlocomotion by clozapine and MDL 100,907 in rats reflects antagonism of 5-HT2A receptors. Eur J Pharmacol. 1995;280:R9–11. doi: 10.1016/0014-2999(95)00333-g. [DOI] [PubMed] [Google Scholar]
- 23.de PT. M-100907 (Aventis) Curr Opin Investig Drugs. 2001;2:123–132. [PubMed] [Google Scholar]
- 24.Talvik-Lotfi M, Nyberg S, Nordstrom AL, Ito H, Halldin C, Brunner F, Farde L. High 5HT2A receptor occupancy in M100907-treated schizophrenic patients. Psychopharmacology (Berl ) 2000;148:400–403. doi: 10.1007/s002130050069. [DOI] [PubMed] [Google Scholar]
- 25.Meltzer HY, Arvanitis L, Bauer D, Rein W Meta-Trial Study Group. Placebo-Controlled Evaluation of Four Novel Compounds for the Treatment of Schizophrenia and Schizoaffective Disorder. Am J Psychiatry. 2004;161:975–984. doi: 10.1176/appi.ajp.161.6.975. [DOI] [PubMed] [Google Scholar]
- 26.Ancoli-Israel S, Vanover KE, Weiner DM, Davis RE, Van Kammen DP. Pimavanserin tartrate, a 5-HT(2A) receptor inverse agonist, increases slow wave sleep as measured by polysomnography in healthy adult volunteers. Sleep Med. 2011;12:134–141. doi: 10.1016/j.sleep.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Shamma HA, Anderson C, Chuang E, Luthringer R, Grottick AJ, Hauser E, Morgan M, Shanahan W, Teegarden BR, Thomsen WJ, Behan D. Nelotanserin, a novel selective human 5-hydroxytryptamine2A inverse agonist for the treatment of insomnia. J Pharmacol Exp Ther. 2010;332:281–290. doi: 10.1124/jpet.109.160994. [DOI] [PubMed] [Google Scholar]
- 28.Fisher WA, Pyke RE. Flibanserin efficacy and safety in premenopausal women with generalized acquired hypoactive sexual desire disorder. Sex Med Rev. 2017 doi: 10.1016/j.sxmr.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 29.George SR, O’Dowd BF, Lee SP. G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat Rev Drug Discov. 2002;1:808–820. doi: 10.1038/nrd913. [DOI] [PubMed] [Google Scholar]
- 30.Brea J, Castro M, Giraldo J, Lopez-Gimenez JF, Padin JF, Quintian F, Cadavid MI, Vilaro MT, Mengod G, Berg KA, Clarke WP, Vilardaga JP, Milligan G, Loza MI. Evidence for distinct antagonist-revealed functional states of 5-hydroxytryptamine(2A) receptor homodimers. Mol Pharmacol. 2009;75:1380–1391. doi: 10.1124/mol.108.054395. [DOI] [PubMed] [Google Scholar]
- 31.Iglesias A, Lage S, Cadavid MI, Loza MI, Brea J. Development of a Multiplex Assay for Studying Functional Selectivity of Human Serotonin 5-HT2A Receptors and Identification of Active Compounds by High-Throughput Screening. J Biomol Screen. 2016;21:816–823. doi: 10.1177/1087057116644162. [DOI] [PubMed] [Google Scholar]
- 32.Bruno A, Beato C, Costantino G. Molecular dynamics simulations and docking studies on 3D models of the heterodimeric and homodimeric 5-HT(2A) receptor subtype. Future Med Chem. 2011;3:665–681. doi: 10.4155/fmc.11.27. [DOI] [PubMed] [Google Scholar]
- 33.Shonberg J, Scammells PJ, Capuano B. Design strategies for bivalent ligands targeting GPCRs. ChemMedChem. 2011;6:963–974. doi: 10.1002/cmdc.201100101. [DOI] [PubMed] [Google Scholar]
- 34.Portoghese PS. From models to molecules: opioid receptor dimers, bivalent ligands, and selective opioid receptor probes. J Med Chem. 2001;44:2259–2269. doi: 10.1021/jm010158+. [DOI] [PubMed] [Google Scholar]
- 35.Bruno A, Guadix AE, Costantino G. Molecular dynamics simulation of the heterodimeric mGluR2/5HT(2A) complex. An atomistic resolution study of a potential new target in psychiatric conditions. J Chem Inf Model. 2009;49:1602–1616. doi: 10.1021/ci900067g. [DOI] [PubMed] [Google Scholar]
- 36.Choi SK, Green D, Ho A, Klein U, Marquess D, Taylor R, Turner SD. Designing selective, high affinity ligands of 5-HT1D receptor by covalent dimerization of 5-HT1F ligands derived from 4-fluoro-N-[3-(1-methyl-4-piperidinyl)-1H-indol-5-yl]benzamide. J Med Chem. 2008;51:3609–3616. doi: 10.1021/jm7011722. [DOI] [PubMed] [Google Scholar]
- 37.Heinrich T, Bottcher H, Schiemann K, Holzemann G, Schwarz M, Bartoszyk GD, van Amsterdam C, Greiner HE, Seyfried CA. Dual 5-HT1A agonists and 5-HT re-uptake inhibitors by combination of indole-butyl-amine and chromenonyl-piperazine structural elements in a single molecular entity. Bioorg Med Chem. 2004;12:4843–4852. doi: 10.1016/j.bmc.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 38.Lezoualc’h F, Jockers R, Berque-Bestel I. Multivalent-based drug design applied to serotonin 5-HT(4) receptor oligomers. Curr Pharm Des. 2009;15:719–729. doi: 10.2174/138161209787315602. [DOI] [PubMed] [Google Scholar]
- 39.Soulier JL, Russo O, Giner M, Rivail L, Berthouze M, Ongeri S, Maigret B, Fischmeister R, Lezoualc’h F, Sicsic S, Berque-Bestel I. Design and synthesis of specific probes for human 5-HT4 receptor dimerization studies. J Med Chem. 2005;48:6220–6228. doi: 10.1021/jm050234z. [DOI] [PubMed] [Google Scholar]
- 40.Perez M, Jorand-Lebrun C, Pauwels PJ, Pallard I, Halazy S. Dimers of 5HT1 ligands preferentially bind to 5HT1B/1D receptor subtypes. Bioorg Med Chem Lett. 1998;8:1407–1412. doi: 10.1016/s0960-894x(98)00222-4. [DOI] [PubMed] [Google Scholar]
- 41.Halazy S, Perez M, Fourrier C, Pallard I, Pauwels PJ, Palmier C, John GW, Valentin JP, Bonnafous R, Martinez J. Serotonin dimers: Application of the bivalent ligand approach to the design of new potent and selective 5-HT1B/1D agonists. J Med Chem. 1996;39:4920–4927. doi: 10.1021/jm960552l. [DOI] [PubMed] [Google Scholar]
- 42.Decker M, Lehmann J. Agonistic and antagonistic bivalent ligands for serotonin and dopamine receptors including their transporters. Curr Top Med Chem. 2007;7:347–353. doi: 10.2174/156802607779941297. [DOI] [PubMed] [Google Scholar]
- 43.Herth MM, Kramer V, Piel M, Palner M, Riss PJ, Knudsen GM, Rosch F. Synthesis and in vitro affinities of various MDL 100907 derivatives as potential 18F-radioligands for 5-HT2A receptor imaging with PET. Bioorg Med Chem. 2009;17:2989–3002. doi: 10.1016/j.bmc.2009.03.021. [DOI] [PubMed] [Google Scholar]
- 44.Knight AR, Misra A, Quirk K, Benwell KR, Revell DF, Kennett GA, Bickerdike MJ. Pharmacological characterisation of the agonist radioligand binding site of 5-HT2A, 5-HT2B and 5-HT2C receptors. Naunyn Schmiedebergs Arch Pharmacol. 2004;370:114–123. doi: 10.1007/s00210-004-0951-4. [DOI] [PubMed] [Google Scholar]
- 45.Shashack MJ, Cunningham KA, Seitz PK, McGinnis A, Smith TD, Watson CS, Gilbertson SR. Synthesis and evaluation of dimeric derivatives of 5-HT2A receptor (5-HT2AR) antagonist M100907. ACS Chem Neurosci. 2011;2:640–644. doi: 10.1021/cn200077q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seitz PK, Bremer NM, McGinnis AG, Cunningham KA, Watson CS. Quantitative changes in intracellular calcium and extracellular-regulated kinase activation measured in parallel in CHO cells stably expressing serotonin (5-HT) 5-HT2A or 5-HT2C receptors. BMC Neurosci. 2012;13:25. doi: 10.1186/1471-2202-13-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pockros LA, Pentkowski NS, Swinford SE, Neisewander JL. Blockade of 5-HT2A receptors in the medial prefrontal cortex attenuates reinstatement of cue-elicited cocaine-seeking behavior in rats. Psychopharmacology (Berl ) 2011;213:307–320. doi: 10.1007/s00213-010-2071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McMahon LR, Cunningham KA. Antagonism of 5-hydroxytryptamine(2a) receptors attenuates the behavioral effects of cocaine in rats. J Pharmacol Exp Ther. 2001;297:357–363. [PubMed] [Google Scholar]
- 49.Raymond JR, Mukhin YV, Gelasco A, Turner J, Collinsworth G, Gettys TW, Grewal JS, Garnovskaya MN. Multiplicity of mechanisms of serotonin receptor signal transduction. Pharmacol Ther. 2001;92:179–212. doi: 10.1016/s0163-7258(01)00169-3. [DOI] [PubMed] [Google Scholar]
- 50.Kurrasch-Orbaugh DM, Parrish JC, Watts VJ, Nichols DE. A complex signaling cascade links the serotonin2A receptor to phospholipase A2 activation: the involvement of MAP kinases. J Neurochem. 2003;86:980–991. doi: 10.1046/j.1471-4159.2003.01921.x. [DOI] [PubMed] [Google Scholar]
- 51.Roskoski R., Jr ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012;66:105–143. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 52.Ullrich T, Rice KC. A practical synthesis of the serotonin 5-HT2A receptor antagonist MDL 100907, its enantiomer and their 3-phenolic derivatives as precursors for [11C]labeled PET ligands. Bioorg Med Chem. 2000;8:2427–2432. doi: 10.1016/s0968-0896(00)00175-9. [DOI] [PubMed] [Google Scholar]
- 53.Kehne JH, Baron BM, Carr AA, Chaney SF, Elands J, Feldman DJ, Frank RA, van Giersbergen PL, McCloskey TC, Johnson MP, Mccarty DR, Poirot M, Senyah Y, Siegel BW, Widmaier C. Preclinical characterization of the potential of the putative atypical antipsychotic MDL 100,907 as a potent 5-HT2A antagonist with a favorable CNS safety profile. J Pharmacol Exp Ther. 1996;277:968–981. [PubMed] [Google Scholar]
- 54.Christopoulos A, Parsons AM, Lew MJ, El-Fakahany EE. The assessment of antagonist potency under conditions of transient response kinetics. Eur J Pharmacol. 1999;382:217–227. doi: 10.1016/s0014-2999(99)00550-6. [DOI] [PubMed] [Google Scholar]
- 55.Szucs RP, Frankel PS, McMahon LR, Cunningham KA. Relationship of cocaine-induced c-Fos expression to behaviors and the role of serotonin 5-HT2A receptors in cocaine-induced c-Fos expression. Behav Neurosci. 2005;119:1173–1183. doi: 10.1037/0735-7044.119.5.1173. [DOI] [PubMed] [Google Scholar]
- 56.Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem Neurosci. 2010;1:435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Teitler M, Klein MT. A new approach for studying GPCR dimers: drug-induced inactivation and reactivation to reveal GPCR dimer function in vitro, in primary culture, and in vivo. Pharmacol Ther. 2012;133:205–217. doi: 10.1016/j.pharmthera.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berg KA, Clarke WP, Chen Y, Ebersole BJ, McKay RDG, Maayani S. 5-hydroxytryptamine type 2A receptors regulate cyclic AMP accumulation in a neuronal cell line by protein kinase C- dependent and calcium/calmodulin-dependent mechanisms. Mol Pharmacol. 1994;45:826–836. [PubMed] [Google Scholar]
- 59.Berg KA, Stout BD, Maayani S, Clarke WP. Differences in rapid desensitization of 5-hydroxytryptamine2A and 5-hydroxytryptamine2C receptor-mediated phospholipase C activation. J Pharmacol Exp Ther. 2001;299:593–602. [PubMed] [Google Scholar]
- 60.Bulayeva NN, Gametchu B, Watson CS. Quantitative measurement of estrogen-induced ERK 1 and 2 activation via multiple membrane-initiated signaling pathways. Steroids. 2004;69:181–192. doi: 10.1016/j.steroids.2003.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-Containing NMDA Receptors in Ras-ERK Signaling and AMPA Receptor Trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 62.Anastasio NC, Gilbertson SR, Bubar MJ, Agarkov A, Stutz SJ, Jeng YJ, Bremer NM, Smith TD, Fox RG, Swinford SE, Seitz PK, Charendoff MN, Craft JW, Laezza F, Watson CS, Briggs JM, Cunningham KA. Peptide inhibitors disrupt the serotonin 5-HT2C receptor interaction with phosphatase and tensin homolog to allosterically modulate cellular signaling and behavior. J Neurosci. 2013;33:1615–1630. doi: 10.1523/JNEUROSCI.2656-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cunningham KA, Anastasio NC, Fox RG, Stutz SJ, Bubar MJ, Swinford SE, Watson CS, Gilbertson SR, Rice KC, Rosenzweig-Lipson S, Moeller FG. Synergism between a serotonin 5-HT2A receptor (5-HT2AR) antagonist and 5-HT2CR agonist suggests new pharmacotherapeutics for cocaine addiction. ACS Chem Neurosci. 2013;4:110–121. doi: 10.1021/cn300072u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cunningham KA, Fox RG, Anastasio NC, Bubar MJ, Stutz SJ, Moeller FG, Gilbertson SR, Rosenzweig-Lipson S. Selective serotonin 5-HT2C receptor activation suppresses the reinforcing efficacy of cocaine and sucrose but differentially affects the incentive-salience value of cocaine- vs. sucrose-associated cues. Neuropharmacology. 2011;61:513–523. doi: 10.1016/j.neuropharm.2011.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]