

Abstract
Background
The mediodorsal (MD) thalamus plays a critical role in cognition through its extensive innervation of the medial prefrontal cortex (mPFC), but how the two structures cooperate at the single-cell level to generate associated cognitive functions and other mPFC-dependent behaviors remains elusive. A principal importance for organizing cortical activity is maintaining the proper balance between excitation and inhibition (E/I balance). Further, the PFC E/I balance has been implicated in successful execution of multiple PFC-dependent behaviors in both animal research and the context of human psychiatric disorders.
Methods
Here, we utilized a pharmacogenetic strategy to decrease MD activity in adult male rats, and evaluated the consequences for E/I balance in PFC pyramidal neurons, as well as cognition, social interaction and anxiety.
Results
We found that dampening MD activity caused significant reductions in GABAergic signaling, increased E/I balance in the mPFC, and was concomitant with abnormalities in these behaviors. Further, by selectively activating parvalbumin (PV) interneurons in the mPFC with a novel pharmacogenetic approach, we restored GABAergic signaling and E/I balance, as well as ameliorated all behavioral impairments.
Conclusions
These findings underscore the importance of thalamocortical activation of mPFC GABAergic interneurons in a broad range of mPFC-dependent behaviors. Further, it highlights this circuitry as a platform for therapeutic investigation in psychiatric diseases that involve impairments in PFC-dependent behaviors.
Keywords: synaptic function, GABAergic inhibition, PVIs, thalamus, prefrontal cortex, pharmacogenetics, cognition
Background
Research on thalamocortical relays has been pivotal in framing our understanding of how sensory information flows from the periphery, to the thalamus, and to its final cortical destination (1). In the medial prefrontal cortex (mPFC), this characterization has been challenging, due to the complexity of information it receives and must represent in a flexible and dynamic manner. A prime opportunity to advance our understanding of mPFC function is by focusing on the mechanisms of communication with one of its primary gateways, the mediodorsal thalamus (MD). However, despite recent progress (2–4), our grasp on the nature of the interaction between these two structures remains limited.
Cortical neurons are organized into functional microcolumns, where certain columns represent different types of information in a supramodal manner (5, 6). Layer III pyramidal neurons (PNs) in the mPFC receive long-range inputs from a wealth of structures, including limbic regions like the hippocampus and amygdala, neuromodulatory regions like the Ventral Tegmental Area, Locus Coeruleus, and Dorsal Raphe as well as from the MD (7, 8). Layer III PNs can send information horizontally within their lamina, as well as synapse onto downstream PNs in Layer V (9). Layer V PNs, also receive these same long-range inputs from limbic, neuromodulatory regions, and the thalamus (10, 11), and integrate that information with the inputs from Layer III PNs. Using this convergent information, they are tasked with “deciding” whether or not to fire and transmit a response to downstream motor centers. A state that may aid Layer V PNs in performing such calculations is maintaining a high level of GABAergic inhibition from nearby interneurons relative to excitation (E/I balance) (12).
Two cognitive abilities that are among the most consistently associated with damage to either structure are working memory (WM) (13, 14) and cognitive flexibility (15, 16). Both represent building blocks of higher-level cognitive processes, and are often severely impacted in the context of psychiatric disease. Additionally, pharmacological studies (17–19) and electrophysiological recordings (20) during behavior suggest these cognitive domains might be intimately linked to GABAergic signaling in PFC. Layer III parvalbumin interneurons (PVIs) receive the largest density of innervation from MD fibers (21), and synapse directly on Layer V PNs (22). Additionally, mPFC PVIs may be more effectively recruited by incoming excitatory stimuli (23), including MD afferents (24). Here, we sought to explore the interplay of these phenomena and demonstrate a causal link between MD hypofunction, mPFC Layer V PN E/I balance, GABAergic signaling alterations, and behavioral impairments.
We hypothesized that MD projections provide stronger glutamatergic activation to mPFC PVIs, modulating GABAergic output onto Layer V PNs, thereby regulating E/I balance and optimizing mPFC-dependent behavior. Conversely, when thalamic function is compromised, this causes mPFC E/I balance disruption and cognitive dysfunction. To explore this, we utilized a pharmacogenetic approach to transiently decrease MD activity, induce cognitive impairments, and characterize their mechanistic underpinnings with whole-cell recordings. Following MD silencing, we observed reductions in GABAergic neurotransmission, and a shift in Layer V PN E/I balance, concomitant with deficits in WM and set-shifting performance. We utilized pharmacogenetic and pharmacological approaches, and both restored GABAergic signaling and normalized E/I balance, as well as ameliorated select impairments in cognition. Our findings revealed that this pathway is more broadly implicated in other mPFC-dependent behaviors including social interaction and anxiety, highlighting the importance of thalamocortical activation of mPFC PVIs in a myriad of behaviors.
Methods
Detailed methods are described in the Supplemental Materials. Briefly, young adult male Sprague-Dawley rats were injected with adeno-associated vectors into the MD (AP: - 3.3, ML: 0.7, DV: -5.3) or MD and mPFC (AP: 3.2, ML: 0.5, and DV: 2.5). With exception of those for electrophysiological recordings, all animals were sacrificed for histological confirmation of viral expression. For quantification of the PV-hM3Dq virus, mPFC slices from animals injected with the PV-hM3Dq-GFP vector were collected, and incubated with primary antibody against PV (rabbit anti-PV, 1:2,000, Abcam) and a red fluorescent secondary (DyLight 594-conjugated goat Anti-Rabbit, 1:500, Jackson Laboratories).
The WM task was modified from a previous report (25). Briefly, animals were trained to alternate in a T-maze following a 10 second (s) delay to obtain a food reward (frosted cheerios). Animals were injected I.P. 45 minutes prior to each testing day with CNO (3 mg/kg), Indiplon (1 mg/kg, Tocris Bioscience), or an equivalent amount of physiological saline. Groups were compared on days to acquisition, and average percentage of correct trials at each delay interval. The set-shifting task was carried out as described in (26). Animals were trained to dig in bowls that varied in either scent (garlic or coriander) or digging medium (sand or bedding). To measure sociability and social novelty, animals were tested in a three-chamber paradigm (27). Anxiety was measured using an elevated plus maze (EPM), and time spent in the open versus closed arms was quantified for five minutes. Locomotor activity was assessed using an open-field chamber for 15 minutes.
Prior to mPFC whole-cell recordings, viral expression and location were confirmed in MD-containing sections. Electrophysiological parameters were recorded in both current clamp and voltage clamp configurations. For all experiments, to measure the contribution of MD afferent activity to the parameter of interest, we compared the activity of cells with or without Clozapine-N-Oxide (CNO, 1 or 5 μM). All electrophysiological and behavioral data were analyzed using SPSS Statistics. Most measures are presented as mean ± SEM. All procedures involving animals were approved by the Drexel University IACUC and conducted in accordance to the National Institute of Health guidelines.
Results
Expression of inhibitory Gi-coupled DREADD decreases MD neuronal activity
To decrease the activity of MD neurons, rats received a bilateral MD injection (Fig 1A), resulting in expression of the inhibitory hM4Di DREADD receptor fluorescently tagged with mCherry (AAV8-CAMKIIα-hM4D(Gi)-mCherry, MD-hM4Di; Fig 1B) or a control virus (AAV8-CaMKIIα-eYFP, MD-YFP). To confirm the functionality of the hM4Di receptors, we used whole-cell patch-clamp recordings. The membrane potential of YFP-labeled control cells was not significantly altered by CNO administration (both p > 0.05; Fig 1C). However, when hM4Di receptors are bound by the CNO, this should activate Gi-mediated signaling and induce hyperpolarization of transfected neurons (28, 29). Accordingly, bath application of CNO hyperpolarized hM4Di neurons (p = 0.037; Fig 1D), which recovered to baseline following washout (p > 0.05).
Figure 1. DREADD expression and function in the MD Thalamus.
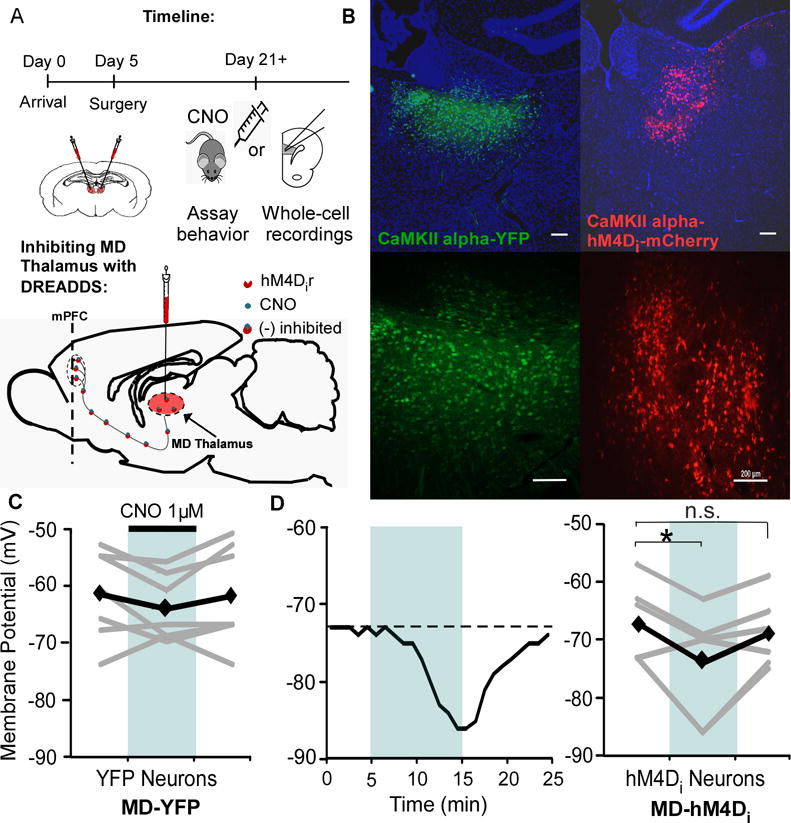
(A) Experimental timeline and schematic of viral injection and DREADD approach.
(B) Expression of the control virus (AAV8-CaMKIIα-eYFP, left) and inhibitory DREADD virus (AAV8-CAMKIIα-hM4D(Gi)-mCherry, right) with DAPI staining in blue. Scale bar = 200 μm.
(C) The membrane potential of YFP control cells in the MD did not change in response to 1 μM CNO bath application (1 μM, Rest = -61.7 ± 3.0 mV; CNO = -64.3 ± 2.2 mV; Washout = -62.0 ± 3.3 mV; paired t test, both p > 0.05; n = 7). Black represents average, while grey is individual cells.
(D) Left, representative response of an hM4Di neuron to bath application of CNO. Right, CNO decreased the membrane potential of hM4Di neurons (Rest = -67.2 ± 2.8 mV; CNO = -74.0 ± 3.9 mV; paired t test, * p < 0.05; n = 7) that recovered following washout (Washout = -68.8 ± 2.5 mV; paired t test, p > 0.05).
CNO application in the mPFC reduces terminal release from MD axons
Here, we wanted to assess whether we could utilize pharmacogenetics as a strategy to evaluate the consequences of downregulating MD activity on individual mPFC neurons. Rats received a bilateral MD transfection with the hM4Di virus (MD-hM4Di). Following three weeks, animals were sacrificed, and mPFC slices were collected (Fig 2A). Given that MD neurons are glutamatergic, we measured the change in spontaneous excitatory post-synaptic currents (sEPSCs) in response to CNO with the goal of decreasing glutamatergic release from severed pre-synaptic MD terminals in the mPFC. Consistent with previous work demonstrating decreased axonal release (30), CNO significantly reduced the sEPSC frequency in both PNs (p = 0.012) and fast-spiking interneurons (p = 0.038; Fig 2B). In accordance with a pre-synaptic alteration, the sEPSC amplitudes were not altered (p > 0.05 for both; Supplemental Figures (S)1B and C). To evaluate whether CNO has non-specific post-synaptic effects on mPFC neurons we measured action potentials, and found the spiking of both neuronal types was not altered by CNO (both p > 0.05; Fig 2C and D). Additionally, there were no differences in sEPSC frequency in PNs following CNO bath application in slices from control animals (p > 0.05, Fig 2E and G). Hyperpolarization of hM4Di-expressing neurons involves activation of GIRK channels at the soma (28, 29), but the mechanisms of axonal inhibition remain uncharacterized. To assess the contribution of pre-synaptic GIRK channels to the CNO’s effect on sEPSC frequency, again we utilized MD-hM4Di animals. When we included tertiapin-Q (tert-Q, 100 nM), a non-specific GIRK channel inhibitor, in the bath, we found that the decrease in sEPSC was occluded in PNs. Interestingly, in the presence of tert-Q, CNO increased sEPSC frequency (p > 0.05, Fig 2F and H), suggesting when GIRK channels are inhibited, activation of hM4Di receptors may lead to a different downstream signaling cascade. We did not observe any differences in sEPSC amplitude in response to tertiapin-Q, indicating that the increase in sEPSC frequency is likely mediated pre-synaptically (p > 0.05; Figure S1D).
Figure 2. Bath application of CNO in the mPFC reduces glutamate release from MD pre-synaptic terminals.
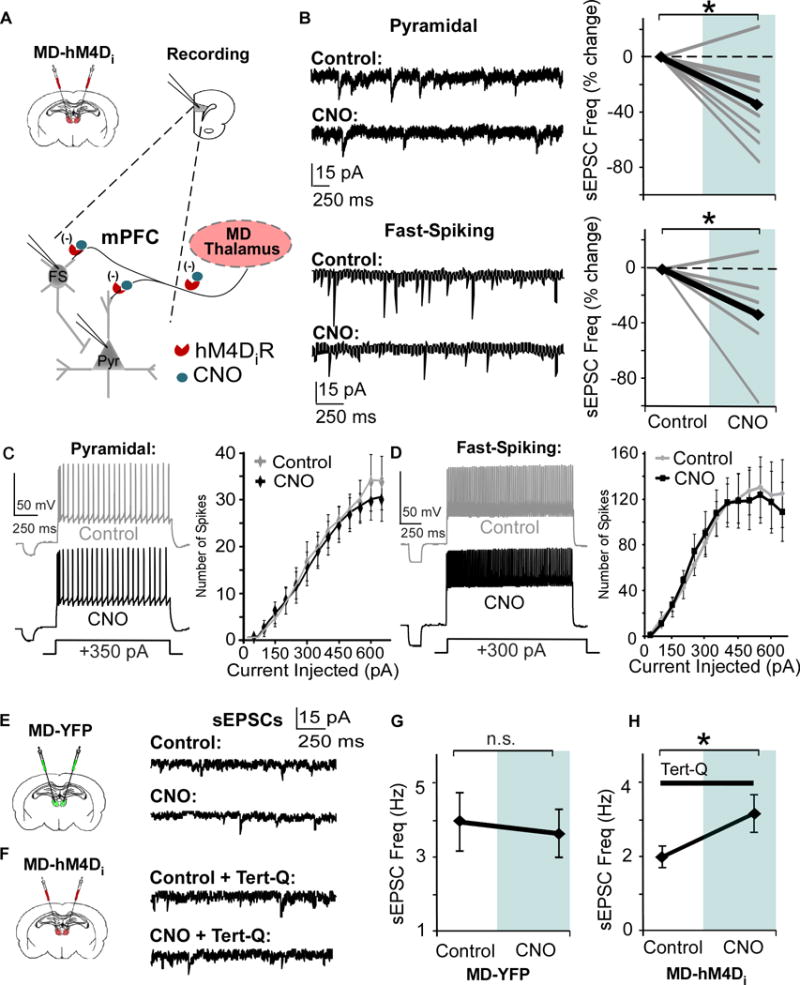
(A) Strategy for decreasing thalamic activity by inhibiting MD terminals in the mPFC. Graphs show a bilateral injection of the CaMKIIα-hM4D-mCherry or CaMKIIα-eYFP virus and patch-clamp recording in mPFC slices.
(B) Bath application of 1 μM CNO signficantly decreases sEPSC frequency in PNs and Fast-Spiking (FS) interneurons (Pyramidal, CNO = -34.8% ± 9.9; FS, CNO = -33.3% ± 12.7; paired t test, both * p < 0.05; pyramidal n = 9, FS n = 7, data shown as percent change from baseline).
(C) and (D) Spike number in response to depolarizing current injections is not altered by CNO in both PNs and FS interneurons (Repeated Measures ANOVA for final three current injections, Pyramidal, Control = 8.3 ± 0.76 pA; CNO = 8.3 ± 0.91 pA; paired t test, p > 0.05; FS, Control = 18.3 ± 1.64 pA; CNO = 16.6 ± 1.64 pA; paired t test, p > 0.05; pyramidal cells n = 9, FS interneurons n = 9).
(E) Left, animals received an injection of the control virus in the MD. Right, representative sEPSCs in mPFC pyramidal cells before and after bath application of CNO.
(F) Left, animals received an injection of the hM4D virus in the MD. Right, representative sEPSCs in mPFC pyramidal cells before and after CNO, with Tert-Q (100 nM) included in the bath.
(G) There was no difference in sEPSCs frequency before and after CNO in mPFC PNs in animals injected with the control virus in the MD (Control = 3.96 ± 0.79 Hz; CNO = 3.64 ± 0.66 Hz; paired t-test, p > 0.05).
(H) Including tert-Q in the bath solution occludes the decrease in sEPSC frequency (B), and sEPSC frequency increases with CNO in mPFC PNs in animals injected with the hM4D virus in the MD (Control = 2.00 ± 0.29 Hz; CNO = 3.17 ± 0.51 Hz; paired t-test, p > 0.05).
Decreasing MD activity disrupts the E/I ratio in mPFC PNs
To investigate how decreasing MD afferent activity impacts intra-mPFC E/I balance, we recorded evoked EPSC and IPSCs, and determined the ratio of the two amplitudes from individual neurons (eEPSCs, eIPSCs, Fig 3B). Paired-pulse stimuli were delivered in layer II/III and Layer V PNs were clamped at -60 mV and 0 mV (the reversal potentials for GABAA and AMPA-mediated currents respectively) to evoke EPSCs and IPSCs. Following inhibition of MD terminals with CNO, there was no difference in the eEPSC amplitude (p > 0.05; Fig 3C), however the eIPSC amplitude was significantly reduced (p = 0.022), resulting in a shift in the E/I ratio (p = 0.002; Fig 3D). However, there was no difference in the paired-pulse ratio (PPR) in either eEPSCs or eIPSCs (both p > 0.05; Fig 3E), suggesting no changes in intra-mPFC pre-synaptic release probability.
Figure 3. MD inhibition alters mPFC E/I ratio by decreasing GABAergic signaling, which is rescued by indiplon.
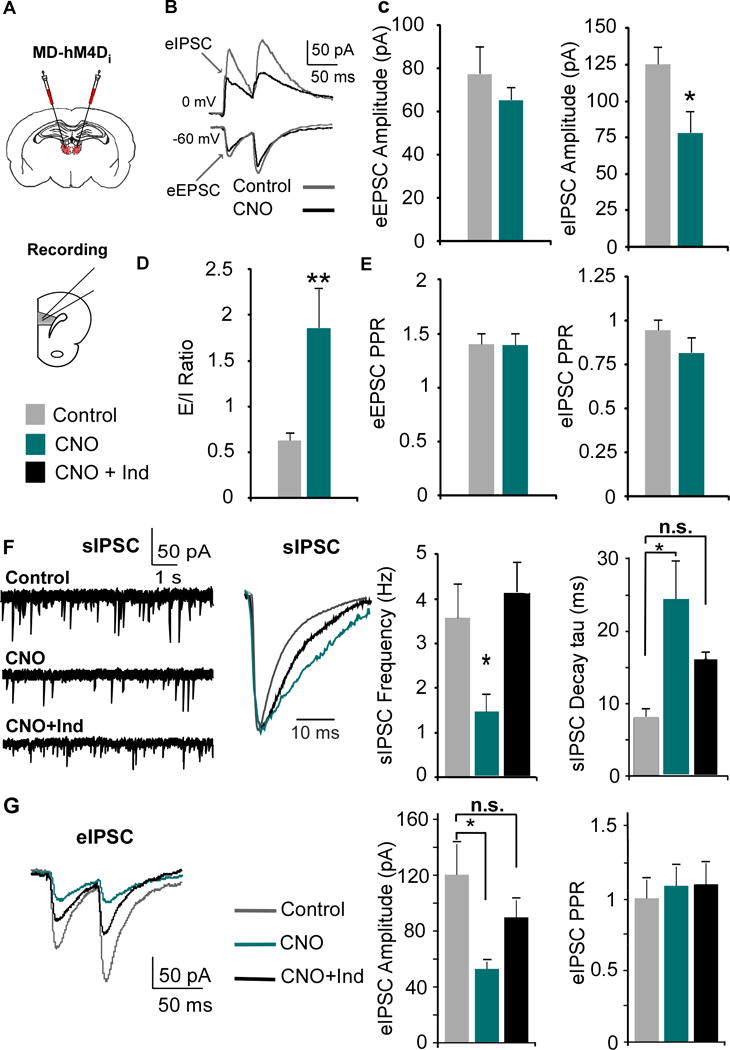
(A) The MD was transfected with the inhibitory CaMKIIα-hM4D-mCherry virus, and mPFC slices were collected for recordings three weeks post-injection.
(B) EPSCS and IPSCS were evoked by stimulating layer II/III with paired pulses.
(C) The eIPSC, but not eEPSC, amplitude was significantly decreased by CNO (EPSC: Control = 77.58 ± 12.82 pA; CNO = 65.18 ± 5.87 pA; Student’s t test, * p < 0.05; Control n = 11, CNO n = 10; IPSC: Control = 125.3 ± 12.23 pA; CNO = 77.9 ± 15.05 pA; Student’s t test, p < 0.05).
(D) Decreasing MD activity increases the E/I ratio (eEPSC/eIPSC amplitude, Control = 0.63 ± 0.08; CNO = 1.86 ± 0.43; Mann-Whitney test, ** p < 0.01).
(E) There were no changes in the PPR (amplitude of the second over the first pulse) in either the eEPSC or eIPSC (Student’s t-test, both p > 0.05).
(F) The sIPSC frequency was decreased by CNO and rescued by co-application with Indiplon (1 μM, Control = 3.61 ± 0.73 Hz; CNO = 1.49 ± 0.36 Hz; CNO + Ind = 4.13 ± 0.63 Hz; ANOVA followed by Tukey’s post hoc, F(2,25) = 6.004, * p < 0.05; n = 10 and 8 for Control and CNO resprectively). CNO also increased the average decay of the sIPSCs which was partially rescued by Indiplon (tau; sIPSCs: Control = 7.912 ± 1.26 ms; CNO = 24.50 ± 5.04 ms; CNO + Ind = 15.97 ± 1.90 ms; ANOVA followed by Tukey’s post hoc, F(2,24) = 8.91, ** p < 0.01).
(G) Indiplon rescued the decreased evoked IPSC amplitude following CNO administration (Control = 120.3 ± 22.0 pA; CNO = 53.0 ± 5.08 pA; CNO + Ind = 89.8 ± 12.1 pA; ANOVA followed by Tukey’s post hoc, F(2,27) = 5.18, group effect p = 0.012, * p < 0.05; Control n = 10, CNO n = 10, CNO + Ind = 10).
Given the selective reduction in eIPSC amplitude, we hypothesized that MD afferents provide stronger stimulation to GABAergic interneurons, which would be reflected in spontaneous IPSCs (sIPSCs) in mPFC PNs. Following bath application of CNO in mPFC slices, we observed significant reductions in sIPSC frequency (p = 0.034; Fig 3F), but no differences in sIPSC amplitude (Fig S1). Next, we measured the average tau in the remaining sIPSCs, and observed that the time constant was significantly longer in comparison to controls (p = 0.001; Fig 3F). The predominant GABAA receptor (GABAAR) subtype present in the adult rat brain is the α1-GABAAR (31), which is characterized by a fast decay. Subunit composition of GABAARs confers the decay kinetics of IPSCs (32, 33), thus given the larger tau, we hypothesized there was reduced activation of α1-GABAARs. To examine this, we co-applied CNO and indiplon (Ind), an α1-GABAAR PAM, and observed a rescue of sIPSC frequency as well as a partial rescue of the sIPSC decay compared to control (p > 0.05 for both). To measure whether indiplon could rescue the eIPSC amplitude, we recorded eIPSCs in PNs (Fig 3G). We found that CNO decreased the eIPSC amplitude, which was rescued by indiplon (p = 0.012), although there were no differences between groups in PPR (p > 0.05). Similar reductions in GABAergic signaling were observed following systemic CNO administration (Fig S2).
Decreasing MD activity leads to deficits in WM that are rescued by indiplon
Given the consistency of WM impairments following MD lesions (34), this provided an ideal paradigm in which to probe the contribution of mPFC disinhibition to WM deficits. We utilized a delayed non-match to sample paradigm, where rats were trained to alternate for a food reward in a T-maze (Fig 4A). Animals in the control groups (MD-hM4Di animals treated with saline: MD-hM4Di-SAL and MD-YFP animals treated with saline or CNO: MD-YFP-SAL and MD-YFP-CNO respectively) exhibited no differences in training or testing, so these groups were pooled (see Supplemental). These animals were initially compared to MD-hM4Di animals treated with CNO (MD-hM4Di-CNO, 3 mg/kg). When we measured the learning ability of these groups, there were no differences in task acquisition (p = 0.352; Fig 4A), prior to any drug treatment. However, when we administered CNO to reduce MD activity and quantified performance, we found a significant group effect at the 60s delay interval (ANOVA p = 0.033). Post-hoc analyses revealed MD-hM4Di-CNO animals performed worse in comparison with controls at the longest delay (p = 0.034; Fig 4B). Given the ability of indiplon to ameliorate GABAergic deficits observed during whole-cell recordings, we treated rats with both CNO and indiplon (3 mg/kg and 1 mg/kg respectively, MD-hM4Di-CNO+Ind), and animals were indistinguishable from controls at the 60s delay (p = 0.883). Together, these data suggest that MD disruption may alter WM through GABAergic hypofunction in the mPFC. Additionally, naïve rats treated with indiplon (naïve-Ind) exhibited no differences in performance in comparison to controls.
Figure 4. WM impairment induced by MD inhibition is rescued by the GABAergic PAM, indiplon.
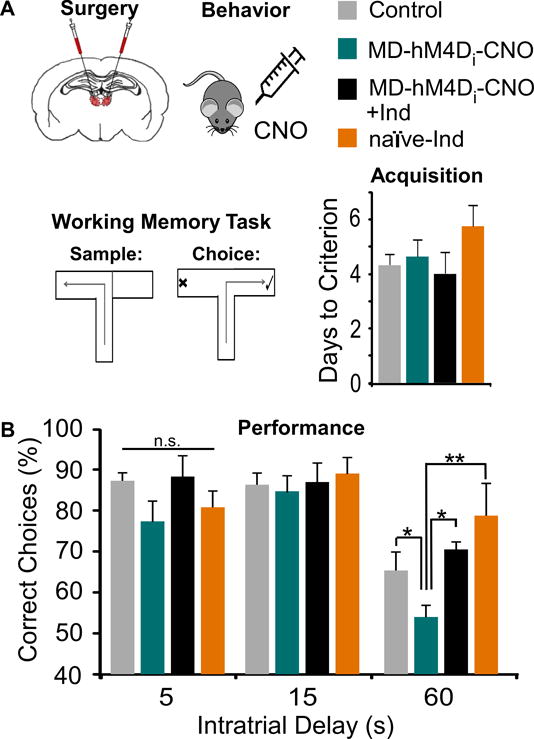
(A) Animals received a bilateral transfection of the MD were treated systemically with Saline, CNO, indiplon, or CNO+indiplon, and were tested on a T-maze task. There were no differences in days required to acquire the task (ANOVA, F(3,29) = 1.134, p > 0.05; Control n = 11, MD-hM4Di-CNO n = 5, MD-hM4Di-CNO+Ind n = 6, naïve-Ind n = 4).
(B) MD inhibition (MD-hM4Di-CNO) impaired WM at the 60, but not 5 and 15, second delay interval. This deficit was ameliorated by treating rats with indiplon (MD-hM4Di-CNO+Ind), whereas indiplon alone (naïve-Ind) did not disrupt performance (Repeated Measures ANOVA followed by Tukey’s post hoc, F(3,20) = 3.55, * p < 0.05, ** p < 0.01).
Expression of a novel DREADD construct
The concurrent findings of decreased GABAergic signaling and WM deficits, along with the ability to rescue these deficits by treatment with a GABAergic PAM, following MD silencing suggest causal links among these phenomena. To explore this with regional and cellular specificity, we designed and utilized a novel DREADD construct, where an hM3Dq DREADD was inserted into a PV-GFP viral vector (35). Following injection of the PV-hM3Dq-GFP into the mPFC, we observed robust GFP staining in the prelimbic mPFC (Fig 5A). To determine whether the construct was expressed in PVIs, we quantified the amount of colocalization with a PV antibody. We found that the hM3Dq DREADD (green) was expressed in approximately 82% of PV labeled cells (red, colocalization: orange, Fig 5B), while approximately 67 % of hM3Dq tagged neurons were PV-labeled. Using whole-cell recordings, about 70 % of hM3Dq-labeled neurons recorded from exhibited a fast-spiking firing pattern (Fig S3). Correspondingly, activating transfected neurons by bath application of CNO caused a significant increase in sIPSC frequency in layer V PNs (p = 0.041; Fig 5C).
Figure 5. Pharmacogenetic activation of PVIs rescues E/I ratio.
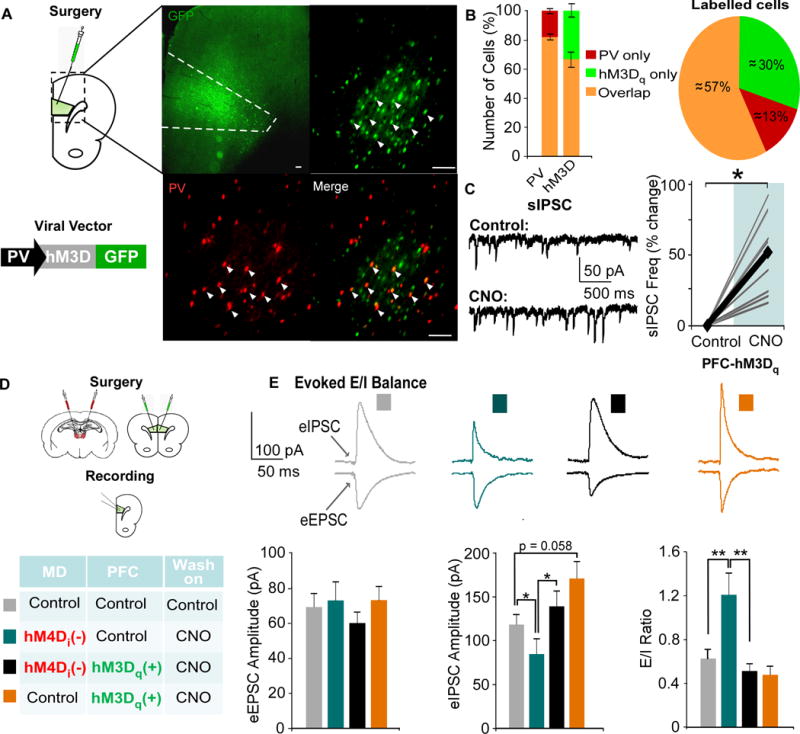
(A) Animals received an injection of the excitatory PV-hM3Dq-GFP vector in the mPFC. Co-staining with a PV-antibody (red) revealed colocalization (orange, arrowhead) with the PV-hM3Dq -GFP vector (primary: rabbit anti-PV, 1:2,000, Abcam; secondary: DyLight 594-conjugated goat Anti-Rabbit, 1:500, JacksonImmuno Laboratories).
(B) Cells were counted, and the amount of PV, hM3Dq, and overlapping cells were quantified.
(C) PNs exhibited a signficant increase in sIPSC frequency following bath application of CNO (Control = 4.99 ± 1.70Hz, CNO = 6.73 ± 1.99 Hz; an average of over 50% increase, paired t-test, * p < 0.05; n = 10; data shown as percent change from baseline).
(D) Animals received a bilateral transfection of the MD and mPFC with the combinations designated in the table.
(E) Single pulses were delivered to layer II/III to evoke EPSCs and IPSCs in each group. The eIPSC amplitude was significantly decreased by inhibiting MD activity (teal), and this was restored by activating PVIs (black, Kruskal Wallis Test followed by post hoc analyses, x2(3) = 11.332, * p < 0.05, ** p < 0.01; MD-Con-PFC-Con n = 15, MD-hM4D-PFC-Con n = 15, MD-hM4D-PFC-hM3D n = 19, MD-Con-PFC-hM3D n = 13). MD inhibition alone (orange, MD-Con-PFC-hM3D also increased the E/I ratio, which was rescued by activating mPFC PVIs (MD-hM4D-PFC-hM3D, Kruskal Wallis Test, x2(3) = 10.438, *p < 0.05, ** p < 0.01, *** p < 0.001).
Pharmacogenetic activation of PVIs rescues E/I ratio in mPFC PNs
To explore whether activation of PVIs could restore E/I ratio deficits observed previously, we combined our previous hM4Di injection into the MD with a bilateral injection the PV-hM3Dq-GFP virus or a control vector into the mPFC (Fig 5D). Identical to previous observations, there were no differences across groups in the eEPSC amplitude (p = 0.715; Fig 5E), but significant differences in the eIPSC amplitude and E/I ratio (eIPSC, p = 0.010; E/I ratio, p = 0.015). Inhibiting MD activity selectively decreased the eIPSC amplitude and (MD-hM4d-PFC-Con, p = 0.029) and increased the E/I ratio (p = 0.008). However, activation of PVIs restored the eIPSC amplitude and normalized the E/I ratio (MD-hM4D-PFC-hM3D, comparison to controls, p = 0.811, p = 1.000 respectively). In animals where we activated PVIs alone (MD-Con-PFC-hM3D), they exhibited a non-significant trend toward an increased eIPSC amplitude (p = 0.058), but not an altered E/I ratio (p = 0.981) ex vivo.
Elevating mPFC PVI activity restores certain behavioral deficits induced by MD inhibition
Rats received the same pattern of injections as described above, and we assessed WM (Fig 6). Similarly, while the shorter delays of 5s and 15s were spared (Fig 6B), differences in performance emerged at the 60s interval. MD inhibition reduced the percentage of correct trials compared to controls (p = 0.029), however, activating PVIs was sufficient to fully recover this deficit at 60s (comparison to controls p = 0.524). Interestingly, activating PVIs alone suppressed normal WM capacity selectively at 60s (p = 0.006).
Figure 6. The MD regulates WM and select components of cognitive flexibility through modulation of PVIs in the mPFC.
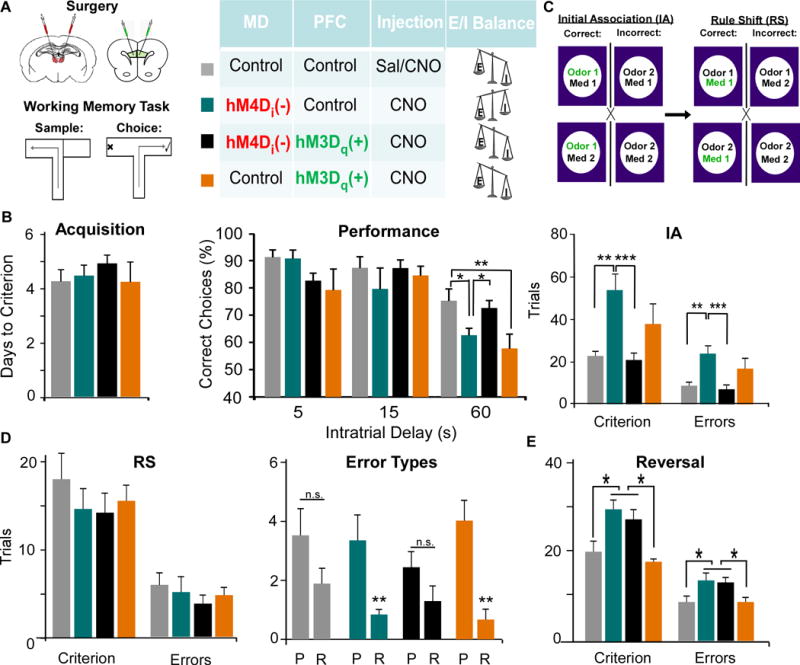
(A) Rats received a bilateral injection of viruses in the MD and mPFC with the viral combinations designated in the diagram.
(B) MD inhibition (teal) impaired WM at the 60 second delay interval, and this was rescued by activation of PVIs (black), while activation of PVIs alone (orange) impaired performance (ANOVA followed by Dunnet’s post hoc test, F(3,24) = 4.493, * p < 0.05, ** p < 0.01, MD-Con-PFC-Con n = 9, MD-hM4D-PFC-Con n = 7, MD-hM4D-PFC-hM3D n = 8, MD-Con-PFC-hM3D n = 5).
(C) Animals were trained to discriminate between scents and digging mediums to obtain a food reward. MD-hM4D-PFC-Con rats were impaired at learning the initial association (IA), and made more errors, but this was rescued by mPFC PVI activation MD-hM4D-PFC-hM3D. Activating PVIs alone MD-Con-PFC-hM3D increased Trials to Criterion and Errors. (Trials to Criterion: ANOVA followed by Tukey’s post hoc, F(3,24) = 6.861; Errors: ANOVA followed by Tukey’s post hoc, F(3,24) = 6.542, ** p < 0.01, *** p < 0.001, MD-Con-PFC-Con n = 8, MD-hM4D-PFC-Con, n = 7, MD-hM4D-PFC-hM3D, n = 7, MD-Con-PFC-hM3D, n = 6).
(D) There was no difference in trials to criterion, when all errors were pooled (ANOVA, Criterion F(3,24) = 0.632, Errors F(3,24) = 0.462). Only MD-hM4D-PFC-Con and MD-Con-PFC-hM3D groups exhibited an increase in perseverative compared to random errors (Mann Whitney Test, ** p < 0.01).
(E) There was an increase in trials to criterion and errors during the reversal following MD inhibition, that was not rescued by mPFC PVI activation (Trials to Criterion: ANOVA, F(3,17) = 4.720; Errors: ANOVA, F(3,17) = 4.947 *p < 0.05, MD-Con-PFC-Con n = 4, MD-hM4D-PFC-Con n = 7, MD-hM4D-PFC-hM3D n = 10, MD-Con-PFC-hM3D n = 5).
We assessed animals on a set-shifting task where animals were trained to dig in bowls that varied in either scent (garlic, coriander) or medium (sand, bedding). Animals first learned an initial association (IA) where a certain scent was consistently rewarded (Fig 6C). Interestingly, dampening MD activity (MD-hM4D-PFC-Con) and activating PVIs alone (MD-Con-PFC-hM3D) disrupted learning the IA, increasing the trials to criterion (comparison to controls p = 0.004 and p = 0.003 respectively) as well as the total errors (comparison to controls p = 0.006 and p = 0.003 respectively). Similar to WM, combined MD silencing and mPFC PVI activation (MD-hM4D-PFC-hM3D) fully recovered learning of the IA (comparison to controls p = 0.974). Then, animals had to learn a rule shift where the digging medium now predicted the location of the food reward. Although there were no differences among groups in the trials to criterion or total errors (p = 0.602 and p = 0.711 respectively; Fig 6D), only silencing of MD activity (MD-hM4D-PFC-Con) and elevating PVI activity (MD-Con-PFC-hM3D) significantly increased perseverative compared to random errors (p = 0.005 and p = 0.001 respectively). Following the rule shift, animals underwent a reversal, where the previously unrewarded digging medium became the rewarded bowl. Unlike the rule shift, reversal learning is underlain by activity in the MD to orbital frontal cortex (OFC) pathway (36–38). There were significant group differences in trials to criterion (p = 0.011; Fig 6E), and in total errors (p = 0.009). We observed an increase in trials to criterion and errors in MD inhibited animals (MD-hM4D-PFC-Con) compared to controls (MD-Con-PFC-Con, p = 0.030 and p = 0.025 respectively), while there were no differences between this group and the rescue group (MD-hM4D-PFC-hM3D, p > 0.05). Additionally, as predicted, activation of mPFC PVIs alone did not alter reversals (p > 0.05).
Next, we explored whether our findings from cognition extended to other behaviors sensitive to mPFC disruption. First, we measured social interaction using a three-chamber sociability task (Fig 7A). We observed that MD inhibited animals (MD-hM4D-PFC-Con) showed a reduction in social preference (p = 0.040; Fig 7B), due to decreased time in the social chamber compared to controls (MD-Con-PFC-Con, p = 0.022). Elevating PVI activity (MD-Con-PFC-hM3D) also reduced social preference (p = 0.034; Fig 7B). Similar to the normalizing effects of activating PVIs in MD inhibited animals on cognition, MD-hM4D-PFC-hM3D rats were not significantly different from controls in social preference or zone time (p = 0.713 and 0.535 respectively; Fig 7B). Although there were no differences among groups in percentage of time spent in the novel social chamber (Fig 7C), we found differences in total zone time between the familiar rat and novel rat chamber only in control animals (MD-Con-PFC-Con, p = 0.031) and the MD inhibited rats (MD-hM4D-PFC-Con, p = 0.002), suggesting MD inhibition does not disrupt social novelty. However, in MD-hM4D-PFC-hM3D animals, this preference was lost, in contrast to the beneficial effects of this manipulation in all other behavioral measures (p = 0.798), and the social novelty preference was also absent in MD-Con-PFC-hM3D rats (p = 0.715; Fig 7C).
Figure 7. The MD thalamus regulates social interaction and anxiety behavior through modulation of PVIs in the mPFC.
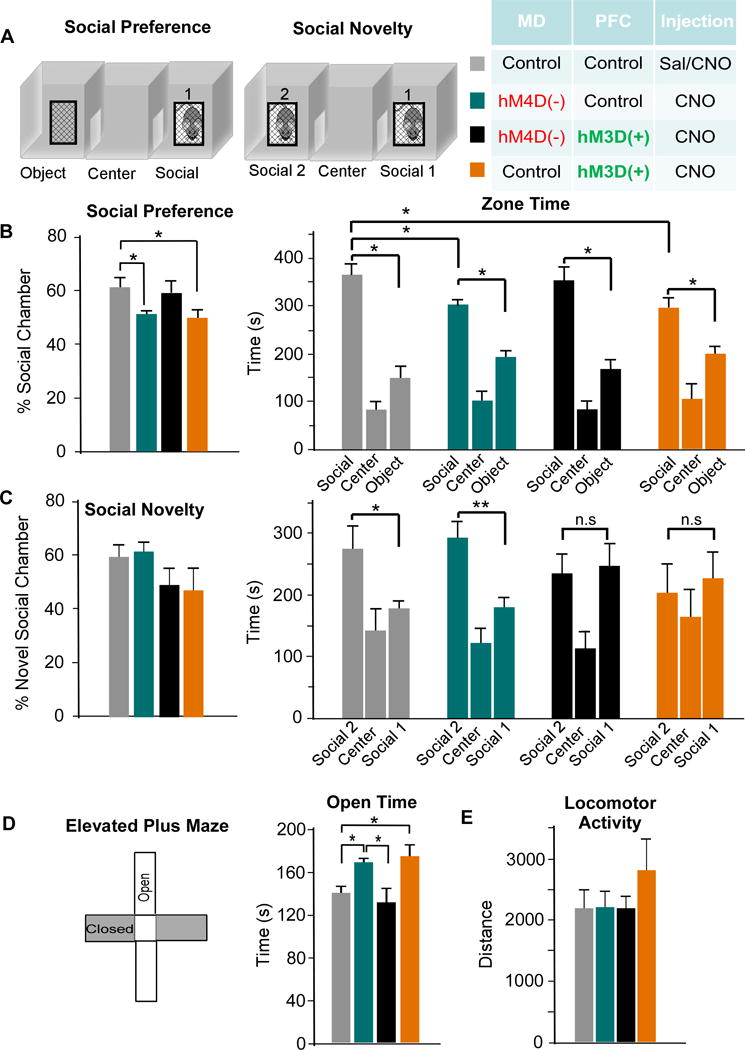
(A) Rats were tested on the three-chamber sociability task. During the social preference portion, zone time was measured in the object, center, and social chamber. For social novelty. Rats were exposed to a novel rat, and time in each zone was measured.
(B) MD inhibition (teal) reduced the social preference due to decreased time spent in the social zone. This was rescued by activation of PVIs (black), while activation of PVIs alone (orange) also disrupted social preference (ANOVA, F(3,24) = 2.132 * p < 0.05, MD-Con-PFC-Con n = 6, MD-hM4D-PFC-Con n = 7, MD-hM4D-PFC-hM3D n = 9, MD-Con-PFC-hM3D n = 5).
(C) There was no difference in the percent time spent in the novel social chamber (ANOVA, F(3,24) = 1.502, p > 0.05). In comparison to the control group, MD-Con-PFC-Con, MD inhibition group, MD-hM4D-PFC-Con, showed a similar preference in zone time for the novel rat chamber (* p < 0.05). However, this zone time preference was absent in MD-hM4D-PFC-hM3D and MD-Con-PFC-hM3D- rats.
(D) Left, rats were tested on elevated plus maze and time spent in open arms was quantified. Right, MD inhibition, MD-hM4D-PFC-Con, and activation of PVIs, MD-Con-PFC-hM3D, showed a similar increase in open time compared to controls, MD-Con-PFC-Con (ANOVA, F(3,25) = 3.079 * p < 0.05), while combined MD inhibition and PVI activation, MD-hM4D-PFC-hM3D, normalized this phenotype.
(E) There were no differences in locomotor activity across all groups (ANOVA, F(3,26) = 0.892 p > 0.05).
To examine the contribution of changes in anxiety levels to social interaction and novelty deficits, we tested animals on the EPM. MD-hM4D-PFC-Con and MD-Con-PFC-hM3D treated rats showed an increase in open-arm time compared to controls (MD-Con-PFC-Con) suggesting an anxiolytic effect in these two groups (MD-hM4D-PFC-Con, p = 0.010; MD-Con-PFC-hM3D, p = 0.025; Fig 7D). Activating PVIs normalized the anxiolytic effect of MD inhibition (MD-hM4D-PFC-hM3D, p = 0.652, comparison to controls). Finally, we found no baseline changes in the ambulatory distance across all groups during open-field testing (p = 0.458; Fig 7E).
Conclusions
Using pharmacogenetic inhibition of MD afferents, we decreased MD axonal release in the mPFC, and observed significant reductions in inhibitory currents, decreased eIPSC amplitude, and an increased E/I ratio. This corresponded with significant behavioral impairments in WM, set-shifting, and social interaction. Further, pharmacological and pharmacogenetic approaches selected to increase GABAergic signaling were sufficient to alleviate physiological and behavioral deficits, highlighting a causal link between MD dysfunction, GABAergic signaling in the mPFC, and behavioral dysfunction.
With CNO bath application, we observed reduced pre-synaptic glutamate release from MD axon terminals. This was likely facilitated by GIRK channels, as determined by the occlusion of this effect with application of tert-Q prior to CNO. These findings are consistent with data indicating the presence of pre-synaptic GIRK channels (39), and their efficacy in reducing glutamate release (40). To our knowledge, this is the first demonstration of a mechanism of action for hM4Di receptors at pre-synaptic terminals. The reduction in sEPSCs may not represent relative strength of MD inputs in vivo to each neuronal subtype, and rather was used as a model to evaluate how mPFC excitatory and inhibitory neuronal activity may change in response to MD inhibition. However, the magnitude of the reduction does correspond with recent work suggesting the density of thalamic afferents provides almost 30% of all total inputs, and outnumbers that of intra-mPFC connections (41).
Disentangling primary deficits in inhibition versus excitation due to homeostatic mechanisms that alter both in disease states remains challenging (42, 43). However, E/I balance deficits have been proposed as an etiology of cognitive symptoms in psychiatric diseases, such as SZ and autism (44, 45) both diseases that harbor prominent GABAergic deficits (46, 47). We found that MD inhibition recapitulated certain core phenotypes by reducing inhibition in Layer V PNs with associated decreases in α1-GABAAR-mediated sIPSCs. The selective GABAergic hypofunction observed is likely attributable to distinct properties of thalamocortical synapses to PNs versus PVIs, such as AMPAR composition or density of pre-synaptic release sites (48, 49). If AMPARs on PVIs have larger conductances compared to those on PNs, we would expect that decreasing the activity of thalamocortical inputs may have larger consequences for PVI activity.
GABAergic inhibition is proposed to tighten PN spatial selectivity in the PFC (50) and correspondingly, mPFC disinhibition is associated with impaired information processing (52), and spatial tuning and WM deficits (20). Selective WM impairments at longer delays have been observed following pharmacogenetic thalamic inhibition in mice (34), as well as with computational modeling of the consequences of mPFC disinhibition on WM maintenance (53), consistent with our results. To assess the contribution of GABAergic hypofunction to the observed deficits, we treated rats with both CNO and indiplon, a potent α1-GABAAR PAM (54). Similar to the physiological recovery in GABAergic signaling, we observed a full amelioration of WM performance, illustrating the importance of α1-GABAARs on mPFC PNs in mnemonic maintenance at longer delays.
We also employed a novel approach for increasing GABAergic signaling, utilizing an excitatory DREADD targeted to mPFC PVIs. Combining this with MD inhibition, we were able to selectively increase the eIPSC amplitude and fully restore the normal E/I balance, with corresponding behavioral augmentation. Importantly, elevating PVI activity normalized all observed mPFC-dependent WM and cognitive flexibility impairments in MD inhibited animals. We predict that high levels of inhibition suppress the activity of functional units of neurons or microcolumns representing distracting information. In concert with the appropriate patterns of excitation, dynamic regulation of these two opposing forces likely support the maintenance of delay-dependent neuronal activity and sustain rule representations in the mPFC as was demonstrated recently (2, 4). Enabling excitatory reverberation to be preferentially sustained within microcolumns representing the correct information could allow the mPFC to optimally process incoming task-related information that can be fed forward to drive the appropriate behavior (Fig 8).
Figure 8. MD thalamus regulates mPFC E/I balance to impact cognition.
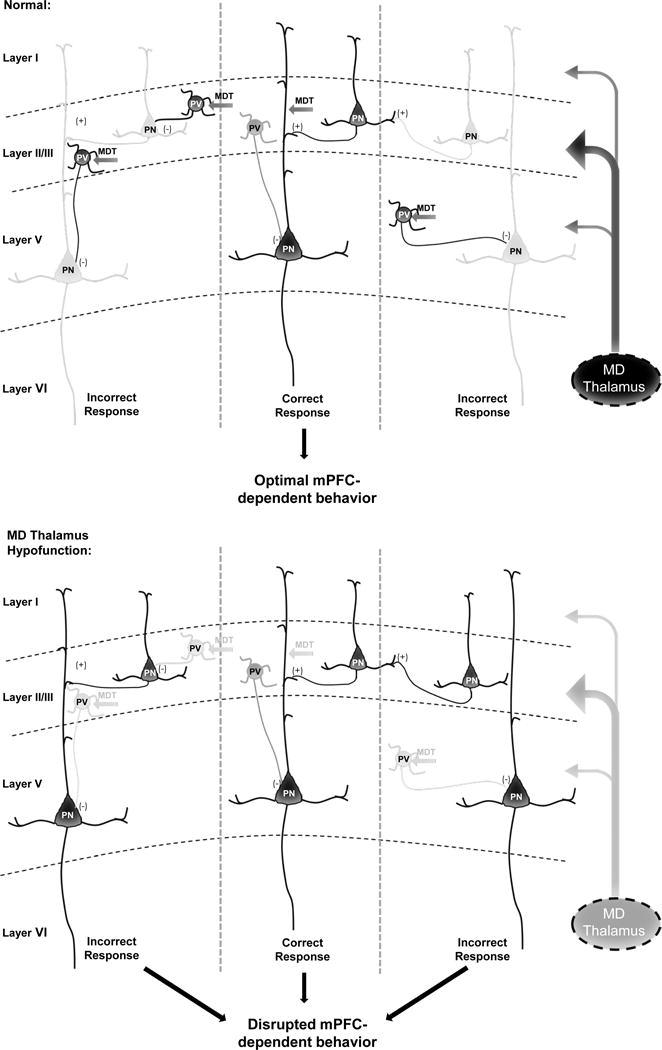
Under normal conditions, the MD thalamus provides a stronger drive to Layer II/III PVIs vs. PNs in the mPFC to maintain a high level of inhibition relative to excitation in Layer V PNs. Potentially, this allows for lateral inhibition of functional microcolumnar units representing distracting information, such that only relevant information is fed forward to optimize behaviors such as WM, cognitive flexibility, and social interaction. However, based on our findings, when MD activity is decreased, activation of PVIs by MD afferents is removed, leading to disinhibition of neurons in microcolumns representing distracting or irrelevant information, and disrupting behavior. Dark grey and black represent high levels of activity, versus the light grey that indicates reduced or low activity. This working model is based on our data and known projections of the MD and connectivity in the mPFC (10, 11, 21, 22).
Previous work has also linked mPFC E/I balance disruption to impairments in sociability (52, 55), and consistent with this, we found that social preference, which was disrupted by MD inhibition, was also ameliorated by increasing PVI activity. We determined that these disruptions in sociability were not due to increased anxiety in the affected groups, given that both groups with social interaction deficits exhibited an anxiolytic phenotype on the EPM. This could suggest increases in risk-taking or impulsive behavior in these groups, but this requires further study.
When we silenced the MD or activated mPFC PVIs prior to a digging-based set-shifting task (26), we found a deficit in learning the IA of a digging-based set-shifting task. This contrasts with previous reports demonstrating no differences in learning simple discriminations following MD or PFC lesions (15, 56), but corresponds with primate research demonstrating the involvement of the MD in encoding and acquisition of strategies (57). In agreement with previous studies, these two groups also showed an increase in perseverative compared to random errors (15, 56). Our findings also confirm a segregation of function between the MD-mPFC versus the MD-OFC pathway in mediating extradimensional and intradimensional shifts respectively (36–38). Future studies exploring the reliance of reversal learning on OFC E/I balance are warranted.
Although increasing mPFC PVI activity was not sufficient to alter the E/I ratio in the slice, it was sufficient to disrupt almost all behaviors examined. We predict that the effects of hM3Dq receptor activation may be more potent in vivo, mimicking behavioral studies using GABA agonists in naïve animals to induce pharmacological mPFC lesions (58). If so, this highlights a cohesive thread among the physiological and the behavioral results: that the successful performance in a broad range of mPFC-dependent behaviors depends critically upon the maintenance of proper mPFC E/I balance. Further, disturbances to that precise balance in either direction, such as increasing it through inhibiting thalamic activity, or reducing it by overactivating mPFC PVIs, will disrupt mPFC circuit function and behavior.
Supplementary Material
Acknowledgments
This work was supported by the Dean’s Fellowship for Themed or Collaborative Research of the Graduate School of Biomedical Sciences and Professional Studies of Drexel University College of Medicine to Brielle Ferguson; NIH/NIMH, F31MH111361 to Brielle Ferguson; NIH /NIMH, R01MH085666 and NIH/NIMH R21MH111609 to Wen-Jun Gao; Helen S. Vernik Schizophrenia Pilot Research Project from the Department of Psychiatry, Drexel University College of Medicine to Wen-Jun Gao and Brielle Ferguson; and the NARSAD Independent Investigator Award, 2015 to Wen-Jun Gao.
We thank Ms. Sarah Monaco and Yelena Gulchina for comments on the manuscript.
Footnotes
Conflict of Interests:
The authors declare no competing financial interests.
Author contributions:
B.R.F. and W.J.G. designed research; B.R.F. performed research and analyzed data; B.R.F. and W.J.G. wrote the paper.
References
- 1.Guillery RW, Sherman SM. Thalamic relay functions and their role in corticocortical communication: generalizations from the visual system. Neuron. 2002;33:163–175. doi: 10.1016/s0896-6273(01)00582-7. [DOI] [PubMed] [Google Scholar]
- 2.Bolkan SS, Stujenske JM, Parnaudeau S, Spellman TJ, Rauffenbart C, Abbas AI, et al. Thalamic projections sustain prefrontal activity during working memory maintenance. Nat Neurosci. 2017 doi: 10.1038/nn.4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wimmer RD, Schmitt LI, Davidson TJ, Nakajima M, Deisseroth K, Halassa MM. Thalamic control of sensory selection in divided attention. Nature. 2015;526:705–709. doi: 10.1038/nature15398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmitt LI, Wimmer RD, Nakajima M, Happ M, Mofakham S, Halassa MM. Thalamic amplification of cortical connectivity sustains attentional control. Nature. 2017;545:219–223. doi: 10.1038/nature22073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Opris I, Santos L, Gerhardt GA, Song D, Berger TW, Hampson RE, et al. Prefrontal cortical microcircuits bind perception to executive control. Scientific reports. 2013;3:2285. doi: 10.1038/srep02285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swadlow HA, Gusev AG, Bezdudnaya T. Activation of a cortical column by a thalamocortical impulse. J Neurosci. 2002;22:7766–7773. doi: 10.1523/JNEUROSCI.22-17-07766.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoover WB, Vertes RP. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain structure & function. 2007;212:149–179. doi: 10.1007/s00429-007-0150-4. [DOI] [PubMed] [Google Scholar]
- 8.Kuroda M, Ojima H, Igarashi H, Murakami K, Okada A, Shinkai M. Synaptic relationships between axon terminals from the mediodorsal thalamic nucleus and layer III pyramidal cells in the prelimbic cortex of the rat. Brain Res. 1996;708:185–190. doi: 10.1016/0006-8993(95)01438-1. [DOI] [PubMed] [Google Scholar]
- 9.Thomson AM, Bannister AP. Interlaminar connections in the neocortex. Cereb Cortex. 2003;13:5–14. doi: 10.1093/cercor/13.1.5. [DOI] [PubMed] [Google Scholar]
- 10.Kuroda M, Murakami K, Oda S, Shinkai M, Kishi K. Direct synaptic connections between thalamocortical axon terminals from the mediodorsal thalamic nucleus (MD) and corticothalamic neurons to MD in the prefrontal cortex. Brain Res. 1993;612:339–344. doi: 10.1016/0006-8993(93)91683-j. [DOI] [PubMed] [Google Scholar]
- 11.Kuroda M, Murakami K, Kishi K, Price JL. Thalamocortical synapses between axons from the mediodorsal thalamic nucleus and pyramidal cells in the prelimbic cortex of the rat. The Journal of comparative neurology. 1995;356:143–151. doi: 10.1002/cne.903560110. [DOI] [PubMed] [Google Scholar]
- 12.Isaacson JS, Scanziani M. How inhibition shapes cortical activity. Neuron. 2011;72:231–243. doi: 10.1016/j.neuron.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kesner RP, Hunt ME, Williams JM, Long JM. Prefrontal cortex and working memory for spatial response, spatial location, and visual object information in the rat. Cereb Cortex. 1996;6:311–318. doi: 10.1093/cercor/6.2.311. [DOI] [PubMed] [Google Scholar]
- 14.Stokes KA, Best PJ. Mediodorsal thalamic lesions impair “reference” and “working” memory in rats. Physiol Behav. 1990;47:471–476. doi: 10.1016/0031-9384(90)90111-g. [DOI] [PubMed] [Google Scholar]
- 15.Block AE, Dhanji H, Thompson-Tardif SF, Floresco SB. Thalamic-prefrontal cortical-ventral striatal circuitry mediates dissociable components of strategy set shifting. Cereb Cortex. 2007;17:1625–1636. doi: 10.1093/cercor/bhl073. [DOI] [PubMed] [Google Scholar]
- 16.Ragozzino ME, Detrick S, Kesner RP. Involvement of the prelimbic-infralimbic areas of the rodent prefrontal cortex in behavioral flexibility for place and response learning. J Neurosci. 1999;19:4585–4594. doi: 10.1523/JNEUROSCI.19-11-04585.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sawaguchi T, Matsumura M, Kubota K. Delayed response deficits produced by local injection of bicuculline into the dorsolateral prefrontal cortex in Japanese macaque monkeys. Exp Brain Res. 1989;75:457–469. doi: 10.1007/BF00249897. [DOI] [PubMed] [Google Scholar]
- 18.Auger ML, Floresco SB. Prefrontal cortical GABA modulation of spatial reference and working memory. Int J Neuropsychopharmacol. 2015;18 doi: 10.1093/ijnp/pyu013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paine TA, Slipp LE, Carlezon WA., Jr Schizophrenia-like attentional deficits following blockade of prefrontal cortex GABAA receptors. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2011;36:1703–1713. doi: 10.1038/npp.2011.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao SG, Williams GV, Goldman-Rakic PS. Destruction and creation of spatial tuning by disinhibition: GABA(A) blockade of prefrontal cortical neurons engaged by working memory. J Neurosci. 2000;20:485–494. doi: 10.1523/JNEUROSCI.20-01-00485.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rotaru DC, Barrionuevo G, Sesack SR. Mediodorsal thalamic afferents to layer III of the rat prefrontal cortex: synaptic relationships to subclasses of interneurons. The Journal of comparative neurology. 2005;490:220–238. doi: 10.1002/cne.20661. [DOI] [PubMed] [Google Scholar]
- 22.Kuroda M, Yokofujita J, Oda S, Price JL. Synaptic relationships between axon terminals from the mediodorsal thalamic nucleus and gamma-aminobutyric acidergic cortical cells in the prelimbic cortex of the rat. The Journal of comparative neurology. 2004;477:220–234. doi: 10.1002/cne.20249. [DOI] [PubMed] [Google Scholar]
- 23.Povysheva NV, Gonzalez-Burgos G, Zaitsev AV, Kroner S, Barrionuevo G, Lewis DA, et al. Properties of excitatory synaptic responses in fast-spiking interneurons and pyramidal cells from monkey and rat prefrontal cortex. Cereb Cortex. 2006;16:541–552. doi: 10.1093/cercor/bhj002. [DOI] [PubMed] [Google Scholar]
- 24.Floresco SB, Grace AA. Gating of hippocampal-evoked activity in prefrontal cortical neurons by inputs from the mediodorsal thalamus and ventral tegmental area. J Neurosci. 2003;23:3930–3943. doi: 10.1523/JNEUROSCI.23-09-03930.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clapcote SJ, Lipina TV, Millar JK, Mackie S, Christie S, Ogawa F, et al. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron. 2007;54:387–402. doi: 10.1016/j.neuron.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 26.Cho KK, Hoch R, Lee AT, Patel T, Rubenstein JL, Sohal VS. Gamma rhythms link prefrontal interneuron dysfunction with cognitive inflexibility in Dlx5/6(+/−) mice. Neuron. 2015;85:1332–1343. doi: 10.1016/j.neuron.2015.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, et al. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes, brain, and behavior. 2004;3:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- 28.Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roth BL. DREADDs for Neuroscientists. Neuron. 2016;89:683–694. doi: 10.1016/j.neuron.2016.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahler SV, Vazey EM, Beckley JT, Keistler CR, McGlinchey EM, Kaufling J, et al. Designer receptors show role for ventral pallidum input to ventral tegmental area in cocaine seeking. Nat Neurosci. 2014;17:577–585. doi: 10.1038/nn.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fritschy JM, Mohler H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. The Journal of comparative neurology. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- 32.McClellan AM, Twyman RE. Receptor system response kinetics reveal functional subtypes of native murine and recombinant human GABAA receptors. J Physiol. 1999;515(Pt 3):711–727. doi: 10.1111/j.1469-7793.1999.711ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lavoie AM, Tingey JJ, Harrison NL, Pritchett DB, Twyman RE. Activation and deactivation rates of recombinant GABA(A) receptor channels are dependent on alpha-subunit isoform. Biophysical journal. 1997;73:2518–2526. doi: 10.1016/S0006-3495(97)78280-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parnaudeau S, O’Neill PK, Bolkan SS, Ward RD, Abbas AI, Roth BL, et al. Inhibition of mediodorsal thalamus disrupts thalamofrontal connectivity and cognition. Neuron. 2013;77:1151–1162. doi: 10.1016/j.neuron.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nathanson JL, Jappelli R, Scheeff ED, Manning G, Obata K, Brenner S, et al. Short Promoters in Viral Vectors Drive Selective Expression in Mammalian Inhibitory Neurons, but do not Restrict Activity to Specific Inhibitory Cell-Types. Frontiers in neural circuits. 2009;3:19. doi: 10.3389/neuro.04.019.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chudasama Y, Robbins TW. Dissociable contributions of the orbitofrontal and infralimbic cortex to pavlovian autoshaping and discrimination reversal learning: further evidence for the functional heterogeneity of the rodent frontal cortex. J Neurosci. 2003;23:8771–8780. doi: 10.1523/JNEUROSCI.23-25-08771.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dias R, Robbins TW, Roberts AC. Dissociation in prefrontal cortex of affective and attentional shifts. Nature. 1996;380:69–72. doi: 10.1038/380069a0. [DOI] [PubMed] [Google Scholar]
- 38.Parnaudeau S, Taylor K, Bolkan SS, Ward RD, Balsam PD, Kellendonk C. Mediodorsal thalamus hypofunction impairs flexible goal-directed behavior. Biol Psychiatry. 2015;77:445–453. doi: 10.1016/j.biopsych.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ponce A, Bueno E, Kentros C, Vega-Saenz de Miera E, Chow A, Hillman D, et al. G-protein-gated inward rectifier K+ channel proteins (GIRK1) are present in the soma and dendrites as well as in nerve terminals of specific neurons in the brain. J Neurosci. 1996;16:1990–2001. doi: 10.1523/JNEUROSCI.16-06-01990.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ladera C, del Carmen Godino M, Jose Cabanero M, Torres M, Watanabe M, Lujan R, et al. Pre-synaptic GABA receptors inhibit glutamate release through GIRK channels in rat cerebral cortex. Journal of neurochemistry. 2008;107:1506–1517. doi: 10.1111/j.1471-4159.2008.05712.x. [DOI] [PubMed] [Google Scholar]
- 41.DeNardo LA, Berns DS, DeLoach K, Luo L. Connectivity of mouse somatosensory and prefrontal cortex examined with trans-synaptic tracing. Nat Neurosci. 2015;18:1687–1697. doi: 10.1038/nn.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoftman GD, Datta D, Lewis DA. Layer 3 Excitatory and Inhibitory Circuitry in the Prefrontal Cortex: Developmental Trajectories and Alterations in Schizophrenia. Biol Psychiatry. 2017;81:862–873. doi: 10.1016/j.biopsych.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nelson SB, Valakh V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron. 2015;87:684–698. doi: 10.1016/j.neuron.2015.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao R, Penzes P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Current molecular medicine. 2015;15:146–167. doi: 10.2174/1566524015666150303003028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes, brain, and behavior. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coghlan S, Horder J, Inkster B, Mendez MA, Murphy DG, Nutt DJ. GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neuroscience and biobehavioral reviews. 2012;36:2044–2055. doi: 10.1016/j.neubiorev.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35:57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hestrin S. Different glutamate receptor channels mediate fast excitatory synaptic currents in inhibitory and excitatory cortical neurons. Neuron. 1993;11:1083–1091. doi: 10.1016/0896-6273(93)90221-c. [DOI] [PubMed] [Google Scholar]
- 49.Kloc M, Maffei A. Target-specific properties of thalamocortical synapses onto layer 4 of mouse primary visual cortex. J Neurosci. 2014;34:15455–15465. doi: 10.1523/JNEUROSCI.2595-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang XJ, Tegner J, Constantinidis C, Goldman-Rakic PS. Division of labor among distinct subtypes of inhibitory neurons in a cortical microcircuit of working memory. Proc Natl Acad Sci U S A. 2004;101:1368–1373. doi: 10.1073/pnas.0305337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Le Roux N, Amar M, Moreau A, Baux G, Fossier P. Impaired GABAergic transmission disrupts normal homeostatic plasticity in rat cortical networks. Eur J Neurosci. 2008;27:3244–3256. doi: 10.1111/j.1460-9568.2008.06288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murray JD, Anticevic A, Gancsos M, Ichinose M, Corlett PR, Krystal JH, et al. Linking microcircuit dysfunction to cognitive impairment: effects of disinhibition associated with schizophrenia in a cortical working memory model. Cereb Cortex. 2014;24:859–872. doi: 10.1093/cercor/bhs370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petroski RE, Pomeroy JE, Das R, Bowman H, Yang W, Chen AP, et al. Indiplon is a high-affinity positive allosteric modulator with selectivity for alpha1 subunit-containing GABAA receptors. J Pharmacol Exp Ther. 2006;317:369–377. doi: 10.1124/jpet.105.096701. [DOI] [PubMed] [Google Scholar]
- 55.Selimbeyoglu A, Kim CK, Inoue M, Lee SY, Hong ASO, Kauvar I, et al. Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2-deficient mice. Science translational medicine. 2017;9 doi: 10.1126/scitranslmed.aah6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Birrell JM, Brown VJ. Medial frontal cortex mediates perceptual attentional set shifting in the rat. J Neurosci. 2000;20:4320–4324. doi: 10.1523/JNEUROSCI.20-11-04320.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mitchell AS, Gaffan D. The magnocellular mediodorsal thalamus is necessary for memory acquisition, but not retrieval. J Neurosci. 2008;28:258–263. doi: 10.1523/JNEUROSCI.4922-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoon T, Okada J, Jung MW, Kim JJ. Prefrontal cortex and hippocampus subserve different components of working memory in rats. Learning & memory. 2008;15:97–105. doi: 10.1101/lm.850808. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.