

Abstract
It has been known for over a century that the hippocampus, the center for learning and memory in the brain, is selectively vulnerable to ischemic damage, with the CA1 being more vulnerable than the CA3. It is also known that leucine enkephalin, or YGGFL, is neuroprotective. We hypothesized that the extracellular hydrolysis of YGGFL may be greater in the CA1 than the CA3, which would lead to the observed difference in susceptibility to ischemia. In rat organotypic hippocampal slice cultures, we estimated the Michaelis constant and the maximum velocity for membrane-bound aminopeptidase activity in the CA1 and CA3 regions. Using electroosmotic push-pull perfusion and offline capillary liquid chromatography we inferred enzyme activity based on the production rate of GGFL, a natural and inactive product of the enzymatic hydrolysis of YGGFL. We found nearly three-fold higher aminopeptidase activity in the CA1 than the CA3. The aminopeptidase inhibitor bestatin significantly reduced hydrolysis of YGGFL in both regions by increasing apparent Km. Based on propidium iodide cell death measurements 24 hours after oxygen-glucose deprivation, we demonstrate that inhibition of aminopeptidase activity using bestatin selectively protected CA1 against delayed cell death due to oxygen-glucose deprivation and that this neuroprotection occurs through enkephalin-dependent pathways.
Keywords: organotypic hippocampal slice culture, oxygen-glucose deprivation, enkephalin, Michaelis-Menten, push-pull perfusion, extrasynaptic transmission, ischemia
Graphical abstract
INTRODUCTION
According to the American Stroke Association, ischemic stroke makes up approximately 87% of all stroke cases. It occurs due to obstruction of blood flow to the brain, resulting in oxygen and glucose deprivation. For over 130 years, it has been known that ischemia and other pathological insults result in damage to the hippocampus, the center of learning and memory1. The hippocampus responds selectively to these insults, with the CA1 region being the most susceptible to ischemic injury. This was first documented in a human patient in 19622 and demonstrated in transient ischemia models of rats and gerbils in the 1980s3, 4, showing a time course of “delayed neuronal death.” This delay is marked by an initial onset of damage within the first 24 hours post-insult and results in maximum damage around 48–72 hours5. This allows for a window during which treatment can reduce or reverse the damage. There have been numerous molecular studies focused on excitotoxicity and abnormal calcium influx, oxidative stress and reactive oxygen species (ROS)6, as well as apoptotic processes and structural changes (reviewed in Dirnagl et al.7 and Schmidt-Kastner et al.8). Changes at the protein level have also been observed, including suppressed protein synthesis in the CA1 at 6 hours and 3 days post-ischemia9–11 as well as post-translational modification, e.g. ubiquitination12, ROS-induced carbonyl modification13, and sumoylation14.
Despite the characterization of these events between initial onset of ischemia and subsequent acute and chronic effects, the exact mechanism behind the selective vulnerability of CA1 is not well understood. There is, however, some consensus that this vulnerability involves calcium-mediated processes8, 15–18. The CA1, CA3, and dentate gyrus all exhibit increases in intracellular calcium 10 min after the initial onset of oxygen-glucose deprivation (OGD) in acute slices19. The CA1, however, displays a significantly higher level of intracellular Ca2+ than the other two regions. Mitani et al. found no differences in hypoxia-induced glutamate levels released to the extracellular space in the three regions20, suggesting that the differences in intracellular Ca2+ levels are not due to glutamate. However, the difference was eliminated in the presence of either nifedipine or verapamil, two L-type voltage gated Ca2+ channel (L-VGCC) antagonists19. Interestingly, opioid peptides, such as enkephalins and dynorphins, are thought to decrease Ca2+ levels by direct inhibition of L-VGCCs and indirect activation of K+ channels through activation of δ-opioid receptors (DORs)21, 22. DOR expression has been found in both pyramidal and non-pyramidal cell types in the hippocampus23, 24 (reviewed in Gendron et al.25) and it has been suggested that DOR action is coupled to different second messengers at each site23. Zhang et al. demonstrated that DOR activation protects cortical neurons against glutamate-induced injury26. Severe hypoxia has been shown to decrease endogenous YGGFL while hypoxic preconditioning increases both DOR mRNA and protein as well as reverses the reduction of YGGFL that was caused by severe hypoxia27, 28. Preconditioning the neurons with opioid peptides prior to ischemia results in reduced brain infarct volume and improved neurological functions 24-hours post-occlusion in male rats29. Elevated endogenous opioid peptides had a similar effect on reducing infarct volume30. The range of of DOR actions include 1) maintaining ionic homeostasis, 2) inhibiting excitatory transmitter release, 3) increasing antioxidants, and 4) regulating anti-apoptotic and pro-survival pathways, to name a few31. All of this evidence suggests that opioid peptides and DOR activation play crucial roles in protecting against ischemic damage.
It is known that enkephalins are stored in large dense-core vesicles and are released extrasynaptically, resulting in volume transmission32–34. While there are numerous studies on enkephalins and their activation of receptors such as DOR, the fate of opioid peptides once they are released into the ECS is less well known. The effects of enkephalins, unlike those of many classical neurotransmitters (e.g. glutamate), are not limited by reuptake but solely by diffusion and extracellular degradation. Ectopeptidases are membrane-bound enzymes whose catalytic domain faces the extracellular space (ECS)35. These enzymes degrade peptides in the ECS. Recently, it was revealed that they play a greater role in regulating peptide activities crucial to physiological function (reviewed in Konkoy et al. and Ou et al.36, 37). Not only that, but the activity of these enzymes can be altered as a result of either trophic or pathological triggers. Acute immobilization stress caused changes in enkephalinase and oxytocinase, two ectopeptidases that regulate anxiolytic peptides38. Ischemic preconditioning, a neuroprotective treatment, restores the activity of two ectopeptidases that hydrolyze amyloid beta, a toxic peptide implicated in Alzheimer’s disease39. The modulation of ectopeptidase activity is a largely unexplored mechanism of neuropeptide control.
Our group showed previously40 that exogenously-applied YGGFL is primarily hydrolyzed at the Tyr-Gly bond into GGFL in the rat hippocampus. Cleavage at this bond inactivates enkephalins41. There are two prominent ectopeptidases that cleave at the Tyr-Gly bond with high affinity for enkephalins in the rat hippocampus: aminopeptidase N (APN, EC 3.4.11.2) and puromycin-sensitive aminopeptidase (PSA, EC 3.4.11.14)42. We questioned whether or not the ectopeptidase catalyzed degradation of enkephalins is different between the CA1 and CA3 regions of the rat hippocampus. However, in order to best answer this question, we need tools that are capable of making enzyme measurements in their native state in the tissue.
Traditional methods require homogenization of tissue and result in loss of any matrix information as well as all spatial and temporal resolution (reviewed in Ou et al.37). In situ zymography was the first method to study localized enzyme activity in intact tissue43–47. The limitation of this method, however, is the need for frozen brain sections rather than live tissue. Thus, there is a need for biochemical and biophysical tools to probe membrane-bound enzymes in their native, unperturbed states with adequate spatial resolution to study regional differences in enzyme activity. Our lab reported the first quantitative measurements of extracellular enzyme kinetics in live tissue based on the development of electroosmotic (EO) sampling40, 48, 49 from organotypic hippocampal slice cultures (OHSCs). OHSCs live on a membrane under which is growth medium. The growth medium can be replaced by a synthetic physiological fluid like artificial cerebrospinal fluid, aCSF. The aCSF can be augmented by peptide substrates. Fluid was drawn through OHSCs by the application of an external electric field, creating fluid movement via electroosmosis. Coupling EO sampling to an online microfluidic system, Wu et al. measured the apparent Michaelis constant (Km) and maximum reaction rate (Vmax) for sequential degradation of exogenous coenzyme A to cysteamine in OHSCs49. Xu et al. coupled EO sampling to capillary liquid chromatography with electrochemical detection and established Michaelis-Menten parameters for aminopeptidase activity in whole-tissue OHSCs40. Interestingly, they found significant hydrolysis of exogenous YGGFL by a bestatin-sensitive aminopeptidase.
Despite its successes, EO sampling lacked the spatial resolution required to determine the differences among the different regions of the hippocampus. Thus, a second capillary probe was added to permit perfusion and sampling from specific regions of the hippocampal culture instead of sampling from the entire culture. This technique is called electroosmotic push-pull perfusion (EOPPP). Rupert et al. perfused the OHSCs with neuropeptide galanin and reported qualitative differences in galanin products in CA1 and CA3 using EOPPP followed by MALDI-TOF/TOF50. No quantitative information on enzyme activity was reported.
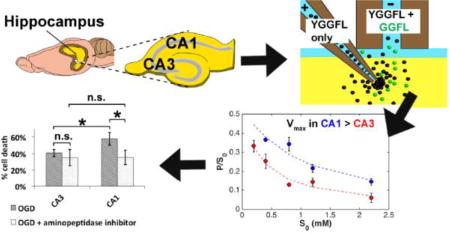
In this paper, we report quantitative measurements of enzyme activity for hydrolysis of YGGFL to GGFL in the CA1 and CA3 regions of the rat hippocampus using EOPPP with offline capillary liquid chromatography (cLC), complemented with finite element method (FEM) calculations. We derived the values of Vmax and Km by fitting an integrated form of the classical Michaelis-Menten equation to both experimentally- and computationally-determined parameters. We found nearly three-fold higher activity of this enzyme in the CA1 region. This observation led to the hypothesis that higher aminopeptidase activity may contribute to selective CA1 vulnerability to ischemic damage. We tested this hypothesis using 20-, 30-, and 40-min oxygen glucose deprivation (an ex vivo model for stroke) combined with cell-death analyses by propidium iodide staining. We found that inhibiting aminopeptidase activity with bestatin selectively reduced cell death in CA1 as a result of OGD. This neuroprotection was reversed by the δ-opioid receptor antagonist naltrindole. This is the first report of spatially resolved, quantitative enzyme activity using natural substrates in live tissue.
RESULTS AND DISCUSSION
Sensitivity and selectivity
In all EOPPP experiments, we perfuse cultures with a solution containing varying concentrations of the substrate YGGFL (S), a matching concentration of a peptidase-resistant internal standard,DYDAGDFDL (IS), and a fluorescent dye, Texas Red-modified 3kDa dextran (TR3) to visualize the process under a microscope. Xu et al. showed that no hydrolysis occurs in the perfusate after peptides are extracted from the tissue using EO sampling, implying that the effect of soluble cytosolic peptidases in the extracted fluid is negligible40. Additionally, Rupert et al. showed that EOPPP causes minimal cell death in the surrounding tissue under certain conditions51. We employed these conditions here.
Using EOPPP, we perfused the CA1 and CA3 regions of OHSCs (see Figure 1) with varying concentrations of S and IS. Perfusion at each concentration of substrate was performed at least eight times. We found the major hydrolysis product of YGGFL to be GGFL in the rat hippocampal slice culture, in agreement with previous findings by us40 and others52..
Figure 1.

(A) Bright-field image of organotypic hippocampal slice culture. Sampling regions from the CA1 and CA3 are circled. (B) Schematic showing the EOPPP process. The substrate YGGFL (LE in this figure), ISDYDAGDFDL, and fluorescent dye TR3 were passed through the culture via electroosmosis. (C) Chromatograms of calibration standard (black), CA1 sample (red), and CA3 sample (blue). Peaks were measured using UV detection at 214 nm. The major hydrolysis product was determined to be GGFL (indicated by arrows). GGFM (an external standard), YGGFL, and IS peaks were also quantified.
We wanted to assess how selective the determination of aminopeptidase activity on YGGFL in tissue cultures is by determining the ratio of the sum of YGGFL and GGFL concentrations in collected perfusate to the initial concentration of substrate YGGFL. The IS has a similar diffusion coefficient to YGGFL but it is not hydrolyzed enzymatically. Thus the collected IS concentration represents the concentration of YGGFL that would have been collected in the absence of enzyme activity. It accounts for the dilution experienced by the electroosmotically introduced substrate solution as well as substrate loss by diffusion away from the sampling capillary. It is used as a surrogate for the initial substrate concentration exposed to the enzymes in the tissue, S0. We perfused CA1 and CA3 regions in cultures at various initial YGGFL and IS concentrations in the presence and absence of the inhibitor bestatin. The hypothesis is that if the sum of the concentration of the product, P, plus the remaining substrate concentration, S, was a significant fraction of the concentration of IS collected ((S+P)/S0) and that fraction was independent of the presence of the inhibitor bestatin, then we could use the measurement to assess differences in aminopeptidase activity in CA1 and CA3.
EOPPP was carried out in CA1 and CA3 using a range of initial concentrations in the source capillary, which we denote S*0 from 0.2 to 2.2 mM. The asterisk differentiates this term, the initial substrate concentration in the source capillary from the initial concentration in the tissue ECS, S0. (A more thorough discussion of how we account for the natural dilution of substrate when it is delivered electroosmotically is in the Methods section.) We found that the average (S+P)/S0 is 69.6 ± 0.5% for this S*0 range with no inhibitor treatment. This is in agreement with the prior work cited above. In order to assess the dependence of ((S+P)/S0) on inhibitor, we pooled all of the data together (two regions, five S*0, and with/without inhibitor) and carried out a linear regression of the observed measurements of (S+P)/S0 on the categorical variables “Region” (CA1 = 0, CA3 = 1), “Inhibitor” (absence = 0, presence = 1), and the continuous variable S*0 (0.2, 0.4, 0.8, 1.0, and 2.2 mM). Because the data also seemed to show that the dependence of ((S+P)/S0) on S*0 was dependent on region, we used the product “region × S*0” as another independent variable. Table 1 summarizes the results of the regression.
Table 1.
Summary of regression statistics.
| Coefficient | Std. Error | t value | p value | |
|---|---|---|---|---|
| Intercept | 0.36 | 0.05 | 7.14 | 2.97 × 10−9 |
| Region | 0.31 | 0.07 | 4.45 | 4.55 × 10−5 |
| S*0 | 0.32 | 0.04 | 7.19 | 2.44 × 10−9 |
| Inhibitor | −0.02 | 0.04 | −0.51 | 0.610 |
| Region × S*0 | −0.26 | 0.07 | −3.89 | 2.83 × 10−4 |
|
|
Regression results for CA3 (eq. 1a) and CA1 (eq. 1b) can be summarized by two equations:
| [1a] |
| [1b] |
There are several observations to be made from this analysis. Foremost, the presence of the inhibitor does not significantly affect ((S+P)/S0). In addition, for S*0 values near 1 mM the sensitivity to aminopeptidase activity is high, as in both CA1 and CA3 the fraction of YGGFL hydrolyzed to the product GGFL is on the order of 70%. On the other hand, at the lowest value of S*0, 0.2 mM, while the CA3 region still maintains a reasonable ratio of ((S+P)/S0) of 66%, in the CA1 is only 42%. As a result, in the determination of Michaelis-Menten parameters, we did not use the data for S*0 = 0.2 mM in the CA1.
The fact that the presence of the inhibitor does not significantly affect the ratio ((S+P)/S0) means that the remaining 30% of the total activity is due to a bestatin-insensitive enzyme or enzymes. This remaining activity could be due to an enzyme that cleaves the substrate YGGFL at a different site (such that GGFL is not produced) and/or an enzyme that cleaves GGFL into smaller fragments. Because the ratio P/S0 is more selective for the activity of the bestatin-sensitive aminopeptidase than the ratio of S/S0, in our analysis below we primarily focus on Michaelis-Menten parameters derived from the former ratio. It is true, however, that despite quantitative differences, the qualitative conclusions drawn from examining P/S0 and S/S0 measurements are the same (to be discussed in the next section).
Differential aminopeptidase activity in the hippocampus
Using the Lambert W code in Matlab R2015b we derived apparent Vmax and Km, V’max and K’m, for each region from fitting the integrated Michaelis-Menten equation (eq. 3 in the Experimental section) to P/S0 vs S*0 (Figure 2). The results from the fit are summarized in Table 2. We distinguish these maximal rates and Michaelis-Menten constants with a prime because they were derived using S*0, the initial laboratory concentrations of the substrate (in the fill solution of the source capillary), as opposed to actual substrate concentrations in the tissue. We showed using in silico experiments53 that both the substrate concentration and local velocities vary within the interrogated region. Fortunately, despite this complexity, the actual Vmax and Km in the tissue ECS are related to the inferred V’max and K’m from the fitting by simple calibration factors. In fact, Vmax and Km in the tissue ECS can be obtained by dividing the V’max and K’m by 10.6 ± 0.3 and 2.8 ± 0.2 (mean ± SE), respectively53. The values of V’max and K’m as well as Vmax and Km are summarized in Table 2.
Figure 2.

Plots of P/S0 as a function of S*0 for both CA1 (blue) and CA3 (red) regions. Experimental values are shown as scatter plots with error bars (SEM) and predicted values from the integrated Michaelis Menten function are shown as dashed lines.
Table 2.
Michaelis-Menten parameters in CA3 and CA1 regions of the rat hippocampus (mean ± SEM), derived from fitting the integrated Michaelis Menten to the P/S0perfusion data.
| Region | V’max (µM/s) | Vmax (µM/s) | K’m (mM) | Km (µM) |
|---|---|---|---|---|
| CA3 | 33 ± 6 | 3.1 ± 0.6 | 0.2 ± 0.1 | 70 ± 40 |
| CA1 | 90 ± 10 | 9 ± 1 | 0.4 ± 0.2 | 100 ± 70 |
As seen in Table 2, the Vmax in CA1 is nearly three-fold higher than that in CA3. Since Vmax is the maximum rate of reaction, this indicates that a higher level of aminopeptidase activity is present in the CA1, which could indicate a higher concentration of enzymes and/or a more active isoform of the enzyme. This regional difference was significant with a p-value < 0.0001 (one-tailed t-test). There was no significant difference between the values for Km, however (p = 0.14, two-tailed t-test). Interestingly, Km values in both regions are comparable to that of APN reported in the literature (45–60 µM)41.
In order to characterize the aminopeptidase, we incubated the OHSCs with 100 µM of the aminopeptidase inhibitor bestatin prior to sampling. Xu et al. demonstrated previously that it was the only inhibitor in a set of inhibitors that significantly decreased the production of GGFL from YGGFL in OHSCs40. As described above, experimental P/S0 and S*0 were used with the integrated Michaelis-Menten equation (eq. 3) to determine V’max. Because bestatin is known to be a competitive inhibitor, the integrated Michaelis-Menten is similar to eq. 3 with the exception that K’m is replaced by K’mapp. By fitting the integrated Michaelis-Menten equation to the inhibitor data for both regions, we determined the values for K’mapp. Given that the calibration factor of K’mapp/Kmapp is 2.8 ± 0.253, we can obtain Kmapp accordingly. Furthermore, because we know Km from our data without the inhibitor, we can determine Ki for the two regions by using the simple relationship . These results are summarized in Table 3. The amount of GGFL was significantly reduced compared to amount of IS collected. Bestatin inhibited aminopeptidase activity in both CA1 and CA3 regions of the rat hippocampus during the sampling experiments by dramatically increasing the Michaelis-Menten constant from 100 ± 70 µM to 1.0 ± 0.2 mM in the CA1 and 70 ± 40 µM to 0.6 ± 0.2 mM in the CA3. The Kmapp and Ki values for the two regions are not significantly different (p = 0.31 and 0.43, respectively, two-tailed t-test). Interestingly, the Ki values, although accompanied by significant uncertainty, are comparable to the Ki of bestatin for APN reported in the literature (4 µM)41.
Table 3.
Kmapp in both CA1 and CA3 after addition of the inhibitor bestatin (mean ± SEM).
| Region | K’mapp (mM) | Kmapp(mM) | Ki (µM) |
|---|---|---|---|
| CA3 + bestatin | 1.7 ± 0.5 | 0.6 ± 0.2 | 11 ± 7 |
| CA1 + bestatin | 2.7 ± 0.5 | 1.0 ± 0.2 | 12 ± 9 |
As mentioned in the introduction, the most prominent ectopeptidases that cleave at the Tyr-Gly bond with high affinity for enkephalins in the rat hippocampus are APN and PSA42. Autoradiographic profiling showed moderate to high labeling of APN in the hippocampus54. PSA and APN have similar Km for YGGFL (32 µM, 45 µM, respectively)41, 55. PSA is highly sensitive to both puromycin (Ki = 1 µM) and bestatin (Ki = 0.5 µM) while APN is significantly more inhibited by bestatin (Ki = 4 µM) than puromycin (Ki = 100 µM)41. Previous work in our group showed little to no effect of puromycin on hydrolysis of exogenously applied YGGFL in whole OHSCs40. We demonstrate here that the Km of the bestatin-sensitive aminopeptidase activity in both CA3 and CA1 are comparable to that of APN and the Ki we derive from inhibitor experiments for both regions are comparable to that of bestatin for APN, as reported in the literature. This combination of results suggests that a significant contribution to the activity we see in both hippocampal regions is from APN.
Fitting integrated Michaelis Menten equation to complementary S/S0 data yielded interesting results (see Table SI-1). In contrast to the results based on P/S0, which showed a ~three-fold higher activity in the CA1 than the CA3, the activity in CA1 is ~two-fold higher than that of CA3 (p = 0.0045). This is due to the fact that the three-fold difference in aminopeptidase activity when basing the measurement on the product of aminopeptidase hydrolysis is “diluted” by the activity of other enzymes present that hydrolyze the substrate at a different peptide bond. Vmax in both regions are significantly higher than those derived using P/S0 in Table 2 (p < 0.0001 for CA3 and p = 0.0037 for CA1). This is consistent with the hypothesis that there is an additional enzyme or enzymes hydrolyzing YGGFL at sites other than the Tyr-Gly bond. Km values were not significantly different from those derived from P/S0 data (p = 0.48 for CA3 and p = 0.88 for CA1); however, Km values are significantly different (p < 0.0001) between the two regions. Based on this data, we hypothesize that the “missing” 30% in the (P+S)/S0 ratio may be due to membrane-bound neutral endopeptidase (NEP, enkephalinase, neprilysin, CD10, EC 3.4.24.11) and/or angiotensin converting enzyme (ACE, EC 3.4.15.1). NEP has moderate expression in the hippocampus56, has good affinity for YGGFL (Km = 74–200 µM)57, cleaves it and its product GGFL at the Gly-Phe bond, and is bestatin-insensitive. Similarly, ACE also cleaves at the Gly-Phe bond. EO sampling data from Xu et al. semi-quantitatively showed that a cocktail of thiorphan (selective NEP inhibitor), captopril (ACE inhibitor), and GEMSA (carboxypeptidase inhibitor) increased the GGFL peak by 2-fold relative to the IS40. Since carboxypeptidases have low affinity for peptides with C-terminal leucines according to the MEROPS database, this is indirect evidence NEP and/or ACE may contribute to the missing 30%. We focus here on membrane-bound peptidases because we demonstrated before40 that incubation of the collected sample at room temperature for 45 min did not change, quantitatively or qualitatively, the outcome compared to samples quenched with acid immediately after collection. This makes it unlikely that there is hydrolysis by soluble enzymes contributing to the observed outcome.
Neuroprotection from aminopeptidase inhibition
After we observed higher aminopeptidase activity in the CA1, we wondered whether or not this higher activity contributes to the selective damage of the CA1 to ischemia. Recall that enkephalin acting through the DOR are neuroprotective during ischemia. It has been suggested that ectopeptidases are required to maintain a tight control of the level of opioid peptides58. Although literature on the neuroprotective effects of DOR and enkephalins is extensive, there is little discussion on the role of the ectopeptidases that hydrolyze enkephalins in neuroprotection. Ansorge et al. showed that the use of synthetic APN inhibitor actinonin significantly reduced cell death of the CA1 pyramidal neurons59. They did not examine effects of APN inhibition on the other subregions of the hippocampus nor did they explore why this neuroprotection occurred. Moreover, opiorphin, a naturally-occurring antidepressant in humans, protects enkephalins from degradation by APN60, 61 and elicits antidepressant-like effects by activating endogenous δ-opioidergic pathways61. Thus, we questioned 1) whether the higher aminopeptidase activity that we observed in the CA1 may contribute to selective CA1 vulnerability to ischemic damage because of faster hydrolysis (and deactivation) of neuroprotective peptides in that region, and 2) if so, whether the neuroprotection occurs through enkephalin-dependent pathway. We thus performed a series of imaging experiments to explore whether inhibiting aminopeptidase activity protects CA1 and/or CA3 against ischemic damage following 20-, 30-, and 40-min OGD. Fluorescence from the DNA intercalator propidium iodide (PI) was used as a cell death marker. All cell death is reported as (as a %), where F is the fluorescence intensity of samples, F+ is the fluorescence intensity of NMDA-treated cultures, and F− is the fluorescence intensity of negative controls (no treatment).
First of all, in agreement with literature62, there is higher cell death in the CA1 than the CA3 for longer OGD times (p = 0.038, 0.011 for 30-min and 40-min OGD respectively, one-tail t-test, Figure 3F–G). There is no significant difference in cell death between the two regions at 20-min OGD (p = 0.15, one-tail t-test, Figure 3E). We found that addition of bestatin immediately after OGD significantly decreased cell death in the CA1 for all OGD durations (p = 0.032, 0.035, 0.032 for 20-, 30-, and 40-min OGD, respectively, one-tail, Figure 3E–G). These results support the first hypothesis that higher aminopeptidase activity in the CA1 may contribute to selective CA1 vulnerability to ischemic damage because of faster hydrolysis (and deactivation) of neuroprotective peptides in that region. We did not observe any effect of bestatin administration on CA3 neuronal cell death compared to OGD treatment alone (p = 0.11, 0.32, 0.18 for 20-, 30-, and 40-min OGD, one-tail). Literature searching revealed no reports of enkephalins or enkephalin-derivatives rescuing the CA3 region from ischemic damage, which is consistent with our findings.
Figure 3.

A) Propidium iodide (PI) fluorescence for positive control (200 µM N-methyl-D-aspartic acid, NMDA) and negative control (NEG), B) PI fluorescence after 20-min oxygen-glucose deprivation alone (OGD), OGD and incubation with 100 µM bestatin immediately after (BEST), OGD and incubation of cultures with 10 µM naltrindole hydrochloride 20 min prior to OGD as well as during OGD (NAL), and OGD with both bestatin and naltrindole incubation (BEST + NAL), C) same as B) except for 30-min OGD condition, D) same as B) except for 40-min OGD condition, E–G) % cell death for the different conditions mentioned previously for 20-, 30-, and 40-min OGD conditions, respectively. All images were taken on the Leica TCS SP5 confocal and multiphoton microscope. *p < 0.05, **p < 0.01, ***p < 0.001.
Secondly, because aminopeptidase N is not a selective enzyme and has numerous natural substrates, including angiotensin III, both Leu- and Met-enkephalin, substance P, and bradykinin, we wanted to determine whether or not the neuroprotection from bestatin administration is from an increase in enkephalin lifetime. Both Leu- and Met-enkephalin have high affinities for DOR, a ten-times lower affinity for the mu opioid receptor and negligible affinity for the kappa opioid receptor63. An enkephalin analog [D-Ala2, D-Leu5] administered post-transient forebrain ischemia is neuroprotective to hippocampal CA1 neurons and this neuroprotection was reversed using naltrindole64, a selective DOR antagonist65. We hypothesize that the bestatin-induced decrease in cell death from OGD in the CA1 occurs through an enkephalin-dependent pathway, thus a selective DOR antagonist should reverse this neuroprotective effect. Co-administration of naltrindole with bestatin increased the cell death observed in the CA1 neurons relative to those treated with bestatin alone for all three OGD durations (p = 0.0001, 0.038, 0.0012 for 20-, 30-, and 40-min OGD, respectively, one-tail). We conclude that the effect of bestatin is most likely caused by an increase in enkephalin steady-state concentration in the extracellular space in CA1. An important caveat is that we measured enzyme activity pre-OGD and determined cell death 24 hours post-OGD. Indeed, Röhnert et al. demonstrated using immunohistochemistry, RT-PCR, and protease activity assays that aminopeptidase N is upregulated in the frontal, temporal, and occipital cortices from 6 hrs to 7 days post ischemia66. However, this study did not look at subregional differences in the hippocampus.
Interestingly, administration of naltrindole without co-administratrion of bestatin increased cell death relative to OGD only for 30-min OGD (p = 0.042, one-tail, Figure 3F) but not for shorter (20-min, p = 0.11) or longer (40-min, p = 0.80) ischemic insults. The time dependence of this naltrindole effect is unexpected. Several factors contribute to the observed effect of the antagonist. After shorter (20-min) ischemic insult, which is considered a mild form of ischemia for OHSCs and is often used as a preconditioning paradigm27, there are increases in both DOR mRNA and protein as well as Leu enkephalin levels27. On the other hand, Leu-enkephalin levels27, DOR mRNA and protein27, 67, 68 are suppressed after severe ischemia. The latter seems at odds with the observations that enkephalin administration post-ischemia rescues the CA1 pyramidal neurons64. The DOR system is both dynamic and responsive to its environment. Agonist binding to DORs results in internalization of the ligand/receptor complex, which leads to enhanced receptor recycling and resensitization (receptor reactivation) in an agonist-specific manner69 (reviewed by Gendron et al.25). Clearly, more work is required to fully understand the time-dependence of the effect of a DOR antagonist on neuroprotection in the hippocampal formation25.
Based on these results, we propose that the three-fold higher aminopeptidase activity in the CA1 results in significantly shorter lifetime of neuroprotective endogenous opioid peptides in the ECS of the CA1 region compared to that in the CA3. The shorter lifetime of these peptides in the ECS decreases their effectiveness to activate the DOR reducing neuroprotection. The inhibitor bestatin reduces the aminopeptidase activity in the CA1 such that the steady-state concentration of enkephalins are higher in the ECS, increasing activation of DORs, consequently reducing cell death in the CA1 following OGD.
CONCLUSIONS
We have measured for the first time quantitative differences in membrane-bound aminopeptidase activity in different regions of organotypic hippocampal cultures using a novel EOPPP-cLC method. The difference in aminopeptidase activity was the motivation to carry out further experiments to determine whether this difference might contribute to the differential susceptibility of CA1 and CA3 pyramidal neurons to OGD. Indeed, it does. Bestatin at all three OGD durations decreases neuronal damage in the CA1 region but has no effect in the CA3. This effect is likely due to the increase in steady-state extracellular concentration of enkephalins which leads to neuroprotection via the DOR. The method not only opens doors to understanding membrane-bound enzyme activities in their native environment but also allows for answers to some important, long-standing neurochemical and biochemical questions that have been unanswered due to lack of appropriate tools.
METHODS
Organotypic hippocampal slice cultures
Dissection and culturing techniques were adapted from Gogolla et al.70 and approved by Institutional Animal Care and Use Committee (IACUC protocol #14021579) at the University of Pittsburgh. Briefly, the freshly decapitated heads of postnatal 7-day-old Sprague Dawley pups were disinfected by submersion in 70% ethanol prior to dissection. The hippocampi were removed and chopped along the septotemporal axis at a thickness of 350 µm (McIlwain, model TC752). Slices were immediately transferred to a Petri dish and incubated at 4 °C for 30 min prior to plating. The slices were separated under a dissection microscope using microspatulas. Slices with intact laminar structures were chosen for plating. To plate, 2–4 slices were placed onto a single porous (0.4 µm) modified PTFE insert membranes (Millipore) and cultured in a six-well plate (Sarstedt) with 95% air/5% CO2 maintained at 36.5 °C. Culture medium is 50% opti-MEM (Gibco), 25% horse serum (Gibco), 25% Hank’s balanced salt solution with phenol red (Life Technologies, 14025076), and 1% D-(+)-glucose (Sigma Aldrich). The medium was changed every 2–3 days (every other day for 4 cultures/well). The cultures can be kept alive ex vivo for up to 4 weeks (Figure 1A).
Capillary liquid chromatography
High-pressure pumps, autosampler, column oven, and UV-Vis detector are part of the Ultimate 3000 Nano LC system (Thermo Fisher). After samples were transferred to the autosampler vial (see sampling experimental section), they were injected onto a homemade capillary column (100 µm inner diameter (i.d.) × 1.7 µm particles, CSH-C18) and separated at 50 °C. Pump flow rate was 1.0 µL/min under isocratic conditions. Mobile phase composition was 20% acetonitrile (Fisher Scientific) in HPLC-grade water (Fisher Scientific) with 0.1% TFA. Absorbance measurements were made at 214 nm. Peak areas were measured from chromatograms in Chromeleon (Thermo Fisher). Using calibration curves, we converted the peak areas to concentrations.
Analysis of cLC data
Calibration standards of YGGFL, IS, GGFM, and GGFL (1–10 µM for all except for GGFL, for which the concentration range was 0.1–1 µM) were injected between every sample for quality control and for quantitative analysis (Figure 1C). The degree of dilution of GGFM concentration was used to calculate the volume ejected from the sampling capillary. From there, we calculated the concentration of GGFL, substrate, and IS in the sampling capillary. The calculation is summarized by the following equation (using product GGFL as an example):
| [2] |
where P is the concentration of GGFL in the tissue after perfusion (not to be confused with P*, which is the concentration of GGFL in the sampling capillary), mGGFL is the moles of GGFL collected, Vt is the volume of ECS collected, Vs is the volume of the sampling capillary, Ueo is the flow rate during perfusion (L/min), ts is the total sampling time (in min), AGGFL is the peak area of GGFL from chromatogram (in mAU ×min), fGGFL is the calibration factor for GGFL peak (in ), AGGFM is the peak area of GGFM (in mAU ×min), and fGGFM is the calibration factor for GGFM (in ). GGFMi and Vi are, respectively, the concentration and volume of GGFM solution in the autosampler vial prior to adding the sample from the sampling capillary. The concentration of substrate and IS in the tissue could also be calculated in a similar fashion. The final quantity reported was a concentration ratio of GGFL (or YGGFL) to IS in order to normalize for differences in collection efficiency (see next section).
Fitting to integrated Michaelis-Menten equation
S*0 was varied (0.2, 0.4, 0.8, 1.2, 2.2 mM) while ‘t’ was held constant. Because the substrate is diluted in the tissue, we perfuse at the same time an internal standard at the same values of concentration as the substrate (IS = S0). From cLC chromatograms we obtain concentrations of YGGFL, GGFL, and IS collected at each S*0. We previously53 showed that normalizing all collected quantities to the IS allows for correction for collection efficiency due to tissue-to-tissue variations and, if the IS has similar diffusion coefficient to the substrate of interest (as it applies here), then it can also be treated as a surrogate for the amount of substrate that could have been collected if no reaction had occurred. Thus we divide the measured concentration of YGGFL and GGFL by the concentration of IS collected in the sampling capillary to obtain S/S0 and P/S0, respectively. Ou et al.53 details how to fit the integrated form of the Michaelis Menten (reproduced below) to the perfusion data P/S0 to obtain the best estimates of Michaelis Menten kinetics.
| [3] |
All t-tests were done using the QuickCalcs software from GraphPad Software, Inc.
Electroosmotic push-pull perfusion
Prior to sampling, cultures were incubated in medium with 7 µM propidium iodide (PI, Sigma Aldrich) for 24 hours to assess their health. Only healthy cultures were used for sampling. Sampling was performed as previously described (refer to Figure 1)50. In brief, the source fused silica capillary (200 µm i.d. × 30 cm, Polymicro Technologies) was pulled to a bee-stinger tip using a laser-based capillary puller (Sutter Instruments, Inc., model P-2000) and then cut to 15–20 µm i.d. The source and sampling (100 µm i.d. × 30 cm) capillaries (Polymicro Technologies) were filled with Hanks’ balanced salt solution (HBSS, with calcium, with magnesium, no phenol red, Sigma Aldrich), mounted, and positioned using micromanipulators (Sutter Instruments, Inc., model MP-285) above a clean Petri dish (Corning Life Sciences) upon which the insert membrane and OHSC were placed. The other ends of the two capillaries (not in contact with OHSC) were submerged into two separate Petri dishes, each with 1.2 mL buffer and a Pt wire (Alfa Aesar). The Pt wires were connected via alligator clips to a high-voltage source (Stanford Research Systems, model PS350). The source capillary was filled with the substrate (YGGFL, American Peptide Company), internal standard (DYDAGDFDL, IS, GL Biochem Ltd), and Texas Red dextran 3kDa (Life Technologies). The source capillary was placed 60 µm below the OHSC surface at a 45° angle. The sampling capillary was placed 20 µm above the surface. We showed that the diagonal depth of source capillary into the tissue (40–125 µm) and distance of sampling capillary above the tissue (14–60 µm) have no effect on the flow rate. Perfusate consisted of fluorescent TR3 dye, YGGFL (0.2–2.2 mM), and IS (0.2–2.2 mM). Perfusate solutions were made so that concentration of YGGFL and IS were the same for any given experiment. The current was turned on at t = 0 (Figure 1B). After 10 min of sampling the current was turned off, the sampling capillary was removed from the micromanipulator and the contents were pushed via insulin syringe (Fisher Scientific) into 10 µL HBSS solution of 16 µM GGFM (BACHEM) with 0.1% trifluoroacetic acid (TFA, Sigma Aldrich). The collected sample was pipetted into a conical glass insert (Thermo Fisher Scientific) and placed into a glass autosampler vial (Chrom Tech, Inc.) for injection onto the cLC column. For initial substrate concentrations less than 400 µM, samples from 8 experiments were combined into one injection. For initial substrate concentration greater than or equal to 400 µM, 4 experiments were combined. The sampling capillary was rinsed with 0.1 M NaOH between each sampling experiment to clean the capillary walls of any adsorbed residues. The base was rinsed out with additional HBSS. For inhibitor sampling experiments, all experimental conditions were the same as above, except 100 µM bestatin hydrochloride (Sigma Aldrich) was added to the bath underneath the cultures the night prior to sampling. This inhibitor was chosen based on OHSC sampling data from Xu et al.40. In the determination of Michaelis-Menten parameters with inhibitor, we only performed experiments at S*0 = 0.4, 0.8, and 1.2 mM.
Oxygen-glucose deprivation and neuronal survival
Oxygen-glucose deprivation was done by submersion of OHSCs (up to four insert membranes at a time) in a nitrogen-purged, glucose-free buffer. A submersion chamber was fabricated in-house (Chemistry & Physics Machine Shop, University of Pittsburgh) that allows for perfusion of up to four insert membranes in a single experiment (maximum 16 individual OHSCs). Glucose-free HBSS (125 mM NaCl (Fisher Scientific), 5 mM KCl, 2 mM CaCl2 (Fisher Scientific), 1 mM MgSO4 (Fisher Scientific), 25 mM NaHCO3 (Fisher Scientific), 1.25 mM NaH2PO4 (Sigma Aldrich), 25 mM sucrose (Sigma Aldrich), pH 7.40) was prepared the day before experiment and warmed to 36 °C before use. Solution was poured into the submersion chamber and circulated using a variable-flow peristaltic pump (Fisher Scientific). The buffer was bubbled with N2 (Matheson). Dissolved O2 levels were monitored using DO110 portable dissolved O2 meter (Oakton Instruments). All components of the OGD chamber were placed into the humidified CO2 incubator to maintain the temperature throughout the experiment. Once O2 levels reached 0% (with 100% calibrated to O2 level in the ambient air), the OHSCs were secured into a 4-well platform (built in-house) and submerged in the bubbling buffer solution for 20, 30, or 40 min (depending on the condition). This time frame was chosen based on previous work by others59, 71–73. Tissues that were treated with naltrindole were pre-incubated for 20 min and naltrindole was also added in the OGD solution. Immediately after the treatment, the dishes were returned to 6-well plate containing warm 36 °C culture medium with or without bestatin. Cultures were returned to the incubator for 2 hours and then exchanged for new medium containing PI. Cultures were imaged using the Leica TCS SP5 confocal and multi-photon microscope 24 hours later. The Argon and DPSS561 lasers were used. The Argon laser was set to 25% power. The visible 561-nm wavelength laser was set to 15% power. Images were taken as 512×512 at 100 Hz with 5 line averages and pinhole size set to 74.25 µm.
Regions of interest were drawn around CA3 and CA1 areas of the hippocampus. Area of the regions was determined by area of visible damage (PI fluorescence) in positive controls, which were exposed to 200 µM NMDA (Sigma Aldrich) for 40 min. The same areas were drawn for all experimental groups as well as negative controls. Exposure time was determined by the positive control fluorescence. Negative controls were not subjected to any treatment and were kept in the same incubator as the positive controls and OGD chamber. The incubator was maintained at 5% CO2 in air and 36 °C. The mean fluorescence intensity was measured. Cell death was measured as a percent .
Supplementary Material
Acknowledgments
We would also like to thank Prof. Germán Barrionuevo from the Department of Neuroscience (University of Pittsburgh) for useful discussions as well as Bocheng Yin and Jenna DeVivo for help with preliminary experiments. We would also like to thank the Chemistry & Physics Machine Shop for making plastic discs for the tissue chopper as well as the OGD-chamber that was built in-house. We would also like to thank Dr. Ed Bouvier at Waters Corporation for the chromatographic particles.
Funding
This work was funded by the NIH (R01 GM044842) and the University of Pittsburgh (Arts & Sciences and Andrew Mellon Predoctoral Fellowships for Y.O.).
ABBREVIATIONS
- ACE
angiotensin converting enzyme
- APN
aminopeptidase N
- CA1/3
cornu ammonis 1/3
- cLC
capillary liquid chromatography
- DOR
delta-opioid receptor
- ECS
extracellular space
- IS
internal standard
- L-VGCC
L-type voltage gated calcium channel
- NEP
neutral endopeptidase
- OGD
oxygen-glucose deprivation
- OHSCs
organotypic hippocampal slice cultures
- P
product
- PSA
puromycin-sensitive aminopeptidase
- S
substrate
- TR3
Texas Red dextran 3k molecular weight
Footnotes
Table summarizing Vmax and Km derived from fitting integrated Michaelis-Menten to complementary S/S0 perfusion data, table summarizing the % cell death for all experimental groups in both regions of the hippocampus, control cell death (propidium iodide) experiments with 10 µM naltrindole.
The authors declare no conflict of interest.
References
- 1.Bartsch T, Döhring J, Reuter S, Finke C, Rohr A, Brauer H, Deuschl G, Jansen O. Selective neuronal vulnerability of human hippocampal CA1 neurons: Lesion evolution, temporal course, and pattern of hippocampal damage in diffusion-weighted MR imaging. J. Cereb. Blood Flow Metab. 2015;35:1836–1845. doi: 10.1038/jcbfm.2015.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brierley JB, Cooper JE. Cerebral complications of hypotensive anaesthesia in a healthy adult. J. Neurol., Neurosurg. Psychiatry. 1962;25:24–30. doi: 10.1136/jnnp.25.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann. Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 4.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 5.Allard J, Paci P, Vander Elst L, Ris L. Regional and time-dependent neuroprotective effect of hypothermia following oxygen-glucose deprivation. Hippocampus. 2015;25:197–207. doi: 10.1002/hipo.22364. [DOI] [PubMed] [Google Scholar]
- 6.Yin B, Barrionuevo G, Batinic-Haberle I, Sandberg M, Weber SG. Differences in reperfusion-induced mitochondrial oxidative stress and cell death between hippocampal CA1 and CA3 subfields are due to the mitochondrial thioredoxin system. Antioxid. Redox Signaling. 2017;27:534–549. doi: 10.1089/ars.2016.6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt-Kastner R, Freund TF. Selective vulnerability of the hippocampus in brain ischemia. Neurosci. 1991;40:599–636. doi: 10.1016/0306-4522(91)90001-5. [DOI] [PubMed] [Google Scholar]
- 9.Widmann R, Kuroiwa T, Bonnekoh P, Hossmann KA. [14C] Leucine incorporation into brain proteins in gerbils after transient ischemia: Relationship to selective vulnerability of hippocampus. J. Neurochem. 1991;56:789–796. doi: 10.1111/j.1471-4159.1991.tb01993.x. [DOI] [PubMed] [Google Scholar]
- 10.Paschen W. Shutdown of translation: Lethal or protective? Unfolded protein response versus apoptosis. J. Cereb. Blood Flow Metab. 2003;23:773–779. doi: 10.1097/01.WCB.0000075009.47474.F9. [DOI] [PubMed] [Google Scholar]
- 11.DeGracia DJ, Hu BR. Irreversible translation arrest in the reperfused brain. J. Cereb. Blood Flow Metab. 2007;27:875–893. doi: 10.1038/sj.jcbfm.9600388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Magnusson K, Wieloch T. Impairment of protein ubiquitination may cause delayed neuronal death. Neurosci. Lett. 1989;96:264–270. doi: 10.1016/0304-3940(89)90389-3. [DOI] [PubMed] [Google Scholar]
- 13.Oikawa S, Yamada T, Minohata T, Kobayashi H, Furukawa A, Tada-Oikawa S, Hiraku Y, Murata M, Kikuchi M, Yamashima T. Proteomic identification of carbonylated proteins in the monkey hippocampus after ischemia–reperfusion. Free Radical Biol. Med. 2009;46:1472–1477. doi: 10.1016/j.freeradbiomed.2009.02.029. [DOI] [PubMed] [Google Scholar]
- 14.Yang WJ, Hu J, Uemura A, Tetzlaff F, Augustin HG, Fischer A. Semaphorin-3C signals through Neuropilin-1 and PlexinD1 receptors to inhibit pathological angiogenesis. EMBO Mol. Med. 2015:e201404922. doi: 10.15252/emmm.201404922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bartsch T, Alfke K, Stingele R, Rohr A, Freitag-Wolf S, Jansen O, Deuschl G. Selective affection of hippocampal CA1 neurons in patients with transient global amnesia without long-term sequelae. Brain. 2006;129:2874–2884. doi: 10.1093/brain/awl248. [DOI] [PubMed] [Google Scholar]
- 16.Bartsch T, Alfke K, Wolff S, Rohr A, Jansen O, Deuschl G. Focal MR spectroscopy of hippocampal CA1 lesions in transient global amnesia. Neurol. 2008;70:1030–1035. doi: 10.1212/01.wnl.0000306633.06027.33. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Michaelis EK. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010;2:12. doi: 10.3389/fnagi.2010.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartsch T, Wulff P. The hippocampus in aging and disease: From plasticity to vulnerability. Neurosci. 2015;309:1–16. doi: 10.1016/j.neuroscience.2015.07.084. [DOI] [PubMed] [Google Scholar]
- 19.Kubo T, Yokoi T, Hagiwara Y, Fukumori R, Goshima Y, Misu Y. Characteristics of protective effects of NMDA antagonist and calcium channel antagonist on ischemic calcium accumulation in rat hippocampal CA1 region. Brain Res. Bull. 2001;54:413–419. doi: 10.1016/s0361-9230(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 20.Mitani A, Kadoya F, Nakamura Y, Kataoka K. Visualization of hypoxia-induced glutamate release in gerbil hippocampal slice. Neurosci. Lett. 1991;122:167–170. doi: 10.1016/0304-3940(91)90849-o. [DOI] [PubMed] [Google Scholar]
- 21.Thorlin T, Eriksson P, Nilsson M, Hansson E, Roennback L. Opioid receptor stimulation modulate intracellular Ca2+ in cultured neurons and astroglial cells. Regul. Pept. 1994:S15–S16. [Google Scholar]
- 22.Chao D, Bazzy-Asaad A, Balboni G, Xia Y. delta-, but not mu-, opioid receptor stabilizes K+ homeostasis by reducing Ca2+ influx in the cortex during acute hypoxia. J. Cell. Physiol. 2007;212:60–67. doi: 10.1002/jcp.21000. [DOI] [PubMed] [Google Scholar]
- 23.Commons KG, Milner TA. Localization of delta opioid receptor immunoreactivity in interneurons and pyramidal cells in the rat hippocampus. J. Comp. Neurol. 1997;381:373–387. [PubMed] [Google Scholar]
- 24.Erbs E, Faget L, Scherrer G, Kessler P, Hentsch D, Vonesch JL, Matifas A, Kieffer BL, Massotte D. Distribution of delta opioid receptor-expressing neurons in the mouse hippocampus. Neurosci. 2012;221:203–213. doi: 10.1016/j.neuroscience.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gendron L, Mittal N, Beaudry H, Walwyn W. Recent advances on the δ opioid receptor: from trafficking to function. Br. J. Pharmacol. 2015;172:403–419. doi: 10.1111/bph.12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Gibney GT, Zhao P, Xia Y. Neuroprotective role of δ-opioid receptors in cortical neurons. Am. J. Physiol. - Cell Physiol. 2002;282:C1225–C1234. doi: 10.1152/ajpcell.00226.2001. [DOI] [PubMed] [Google Scholar]
- 27.Ma M-C, Qian H, Ghassemi F, Zhao P, Xia Y. Oxygen-sensitive δ-opioid receptor-regulated survival and death Signals: Novel insights into neuronal preconditioning and protection. J. Biol. Chem. 2005;280:16208–16218. doi: 10.1074/jbc.M408055200. [DOI] [PubMed] [Google Scholar]
- 28.Gao CJ, Niu L, Ren PC, Wang W, Zhu C, Li YQ, Chai W, Sun XD. Hypoxic preconditioning attenuates global cerebral ischemic injury following asphyxial cardiac arrest through regulation of delta opioid receptor system. Neurosci. 2012;202:352–362. doi: 10.1016/j.neuroscience.2011.11.060. [DOI] [PubMed] [Google Scholar]
- 29.Zhao P, Huang Y, Zuo Z. Opioid preconditioning induces opioid receptor-dependent delayed neuroprotection against ischemia in rats. J. Neuropathol. Exp. Neurol. 2006;65:945–952. doi: 10.1097/01.jnen.0000235123.05677.4b. [DOI] [PubMed] [Google Scholar]
- 30.Maslov LN, Naryzhnaia NV, Tsibulnikov SY, Kolar F, Zhang Y, Wang H, Gusakova AM, Lishmanov YB. Role of endogenous opioid peptides in the infarct size-limiting effect of adaptation to chronic continuous hypoxia. Life Sci. 2013;93:373–379. doi: 10.1016/j.lfs.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 31.He X, Sandhu HK, Yang Y, Hua F, Belser N, Kim DH, Xia Y. Neuroprotection against hypoxia/ischemia: δ-opioid receptor-mediated cellular/molecular events. Cell. Mol. Life Sci. 2013;70:2291–2303. doi: 10.1007/s00018-012-1167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu PC, Thureson-Klein Å, Klein RL. Exocytosis from large dense cored vesicles outside the active synaptic zones of terminals within the trigeminal subnucleus caudalis: a possible mechanism for neuropeptide release. Neurosci. 1986;19:43–54. doi: 10.1016/0306-4522(86)90004-7. [DOI] [PubMed] [Google Scholar]
- 33.Herkenham M. Mismatches between neurotransmitter and receptor localizations in brain: observations and implications. Neurosci. 1987;23:1–38. doi: 10.1016/0306-4522(87)90268-5. [DOI] [PubMed] [Google Scholar]
- 34.Thureson-Klein Å, Klein RL. Exocytosis from neuronal large dense-cored vesicles. Int. Rev. Cytol. 1990;121:67. doi: 10.1016/s0074-7696(08)60659-2. [DOI] [PubMed] [Google Scholar]
- 35.Mentlein R. Cell-surface peptidases. Int. Rev. Cytol. 2004;235:165–213. doi: 10.1016/S0074-7696(04)35004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Konkoy CS, Davis TP. Ectoenzymes as sites of peptide regulation. Trends Pharmacol. Sci. 1996;17:288–294. doi: 10.1016/0165-6147(96)10036-5. [DOI] [PubMed] [Google Scholar]
- 37.Ou Y, Wu J, Sandberg M, Weber S. Electroosmotic perfusion of tissue: sampling the extracellular space and quantitative assessment of membrane-bound enzyme activity in organotypic hippocampal slice cultures. Anal. Bioanal. Chem. 2014;406:6455–6468. doi: 10.1007/s00216-014-8067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hernandez J, Segarra AB, Ramirez M, Banegas I, de Gasparo M, Alba F, Vives F, Duran R, Prieto I. Stress influences brain enkephalinase, oxytocinase and angiotensinase activities: A new hypothesis. Neuropsychobiology. 2009;59:184–189. doi: 10.1159/000219306. [DOI] [PubMed] [Google Scholar]
- 39.Nalivaevaa NN, Fisk L, Kochkina EG, Plesneva SA, Zhuravin IA, Babusikova EVA, Dobrota D, Turner AJ. Effect of hypoxia/ischemia and hypoxic preconditioning/reperfusion on expression of some amyloid-degrading enzymes. Ann. N. Y. Acad. Sci. 2004;1035:21–33. doi: 10.1196/annals.1332.002. [DOI] [PubMed] [Google Scholar]
- 40.Xu H, Guy Y, Hamsher A, Shi G, Sandberg M, Weber SG. Electroosmotic sampling. Application to determination of ectopeptidase activity in organotypic hippocampal slice cultures. Anal. Chem. 2010;82:6377–6383. doi: 10.1021/ac1012706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gros C, Giros B, Schwartz JC. Identification of aminopeptidase M as an enkephalin-inactivating enzyme in rat cerebral membranes. Biochem. 1985;24:2179–2185. doi: 10.1021/bi00330a011. [DOI] [PubMed] [Google Scholar]
- 42.Irazusta J, Larrinaga G, Agirregoitia N, Varona A, Casis L. Effects of morphine administration and its withdrawal on rat brain aminopeptidase activities. Reg. Pept. 2003;110:225–230. doi: 10.1016/s0167-0115(02)00218-5. [DOI] [PubMed] [Google Scholar]
- 43.George SJ, Johnson JL. In situ zymography. In: Clark IM, editor. Matrix Metalloproteinase Protoc. Springer; New York, NY: 2001. pp. 271–277. [Google Scholar]
- 44.Gawlak M, Górkiewicz T, Gorlewicz A, Konopacki FA, Kaczmarek L, Wilczynski GM. High resolution in situ zymography reveals matrix metalloproteinase activity at glutamatergic synapses. Neurosci. 2009;158:167–176. doi: 10.1016/j.neuroscience.2008.05.045. [DOI] [PubMed] [Google Scholar]
- 45.Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: Inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29:1020–1030. doi: 10.1161/01.str.29.5.1020. [DOI] [PubMed] [Google Scholar]
- 46.Rosell A, Ortega-Aznar A, Alvarez-Sabín J, Fernández-Cadenas I, Ribó M, Molina CA, Lo EH, Montaner J. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic stroke. Stroke. 2006;37:1399–1406. doi: 10.1161/01.STR.0000223001.06264.af. [DOI] [PubMed] [Google Scholar]
- 47.Kurschat P, Wickenhauser C, Groth W, Krieg T, Mauch C. Identification of activated matrix metalloproteinase-2 (MMP-2) as the main gelatinolytic enzyme in malignant melanoma by in situ zymography. J. Pathol. 2002;197:179–187. doi: 10.1002/path.1080. [DOI] [PubMed] [Google Scholar]
- 48.Hamsher AE, Xu H, Guy Y, Sandberg M, Weber SG. Minimizing tissue damage in electroosmotic sampling. Anal. Chem. 2010;82:6370–6376. doi: 10.1021/ac101271r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu J, Sandberg M, Weber SG. Integrated electroosmotic perfusion of tissue with online microfluidic analysis to track the metabolism of cystamine, pantethine, and coenzyme A. Anal. Chem. 2013;85:12020–12027. doi: 10.1021/ac403005z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rupert AE, Ou Y, Sandberg M, Weber SG. Electroosmotic push-pull perfusion: Description and application to qualitative analysis of the hydrolysis of exogenous galanin in organotypic hippocampal slice cultures. ACS Chem. Neurosci. 2013;4:838–848. doi: 10.1021/cn400082d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rupert AE, Ou Y, Sandberg M, Weber SG. Assessment of tissue viability following electroosmotic push–pull perfusion from organotypic hippocampal slice cultures. ACS Chem. Neurosci. 2013;4:849–857. doi: 10.1021/cn4000814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de la Baume S, Yi CC, Schwartz JC, Chaillet P, Marcais-Collado H, Costentin J. Participation of both ‘enkephalinase’ and aminopeptidase activities in the metabolism of endogenous enkephalins. Neurosci. 1983;8:143–151. doi: 10.1016/0306-4522(83)90033-7. [DOI] [PubMed] [Google Scholar]
- 53.Ou Y, Weber SG. Numerical modeling of electroosmotic push-pull perfusion and assessment of its application to quantitative determination of enzymatic activity in the extracellular space of mammalian tissue. Anal. Chem. 2017;89:5864–5873. doi: 10.1021/acs.analchem.7b00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Noble F, Banisadr G, Jardinaud F, Popovici T, Lai-Kuen R, Chen H, Bischoff L, Parsadaniantz SM, Fournie-Zaluski MC, Roques BP. First discrete autoradiographic distribution of aminopeptidase N in various structures of rat brain and spinal cord using the selective iodinated inhibitor [125I] RB 129. Neurosci. 2001;105:479–488. doi: 10.1016/S0306-4522(01)00185-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Safavi A, Hersh LB. Degradation of dynorphin-related peptides by the puromycin-sensitive aminopeptidase and aminopeptidase M. J. Neurochem. 1995;65:389–395. doi: 10.1046/j.1471-4159.1995.65010389.x. [DOI] [PubMed] [Google Scholar]
- 56.Waksman G, Hamel E, Fournié-Zaluski M-C, Roques BP. Autoradiographic comparison of the distribution of the neutral endopeptidase" enkephalinase" and of mu and delta opioid receptors in rat brain. Proc. Natl. Acad. Sci. U. S. A. 1986;83:1523–1527. doi: 10.1073/pnas.83.5.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Welches WR, Brosnihan KB, Ferrario CM. A comparison of the properties and enzymatic activities of three angiotensin processing enzymes: angiotensin converting enzyme, prolyl endopeptidase and neutral endopeptidase 24.11. Life Sci. 1993;52:1461–1480. doi: 10.1016/0024-3205(93)90108-f. [DOI] [PubMed] [Google Scholar]
- 58.Larrinaga G, Gil J, Meana JJ, Ruiz F, Callado LF, Irazusta J. Aminopeptidase activity in the postmortem brain of human heroin addicts. Neurochem. Int. 2005;46:213–219. doi: 10.1016/j.neuint.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 59.Ansorge S, Lendeckel U, Striggow F, Neubert K, Reymann K, Kahne T. Use of dipeptidypeptidase IV inhibitors and aminopeptidase N inhibitors individually or in combination for the prevention and/or therapy of ischemia-caused acute and chronic neurodegenerative diseases, and pharmaceutical compositions. Keyneurotek A.-G. I.G.; Germany: 2002. p. 16. [Google Scholar]
- 60.Wisner A, Dufour E, Messaoudi M, Nejdi A, Marcel A, Ungeeheuer M-N, Rougeot C. Human opiorphin, a natural antinociceptive modulator of opioid-dependent pathways. Proc. Natl. Acad. Sci. U. S. A. 2006;103:17979–17984. doi: 10.1073/pnas.0605865103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Javelot H, Messaoudi M, Garnier S, Rougeot C. Human opiorphin is a naturally occurring antidepressant acting selectively on enkephalin-dependent delta-opioid pathways. J. Physiol. Pharmacol. 2010;61:355–362. [PubMed] [Google Scholar]
- 62.Noraberg J, Poulsen FR, Blaabjerg M, Kristensen BW, Bonde C, Montero M, Meyer M, Gramsbergen JB, Zimmer J. Organotypic hippocampal slice cultures for studies of brain damage, neuroprotection and neurorepair. Curr. drug Targets: CNS Neurol. Disord. 2005;4:435–452. doi: 10.2174/1568007054546108. [DOI] [PubMed] [Google Scholar]
- 63.Janecka A, Fichna J, Janecki T. Opioid receptors and their ligands. Curr. Top. Med. Chem. 2004;4:1–17. doi: 10.2174/1568026043451618. [DOI] [PubMed] [Google Scholar]
- 64.Wang S, Duan Y, Su D, Li W, Tan J, Yang D, Wang W, Zhao Z, Wang X. Delta opioid peptide [D-Ala2, D-Leu5] enkephalin (DADLE) triggers postconditioning against transient forebrain ischemia. Eur. J. Pharmacol. 2011;658:140–144. doi: 10.1016/j.ejphar.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 65.Portoghese PS, Sultana M, Nagase H, Takemori AE. Application of the message-address concept in the design of highly potent and selective non-peptide delta opioid receptor antagonists. J. Med. Chem. 1988;31:281–282. doi: 10.1021/jm00397a001. [DOI] [PubMed] [Google Scholar]
- 66.Röhnert P, Schmidt W, Emmerlich P, Goihl A, Wrenger S, Bank U, Nordhoff K, Täger M, Ansorge S, Reinhold D. Dipeptidyl peptidase IV, aminopeptidase N and DPIV/APN-like proteases in cerebral ischemia. J. Neuroinflammation. 2012;9:44. doi: 10.1186/1742-2094-9-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mayfield KP, Kozak W, Malvin GM, Porreca F. Hypoxia decreases opioid delta receptor expression in mouse brain. Neurosci. 1996;72:785–789. doi: 10.1016/0306-4522(95)00585-4. [DOI] [PubMed] [Google Scholar]
- 68.Boutin H, Dauphin F, MacKenzie ET, Jauzac P. Differential time-course decreases in nonselective, μ-, δ-, and κ-opioid receptors after focal cerebral ischemia in mice. Stroke. 1999;30:1271–1278. doi: 10.1161/01.str.30.6.1271. [DOI] [PubMed] [Google Scholar]
- 69.Audet N, Charfi I, Mnie-Filali O, Amraei M, Chabot-Doré A-J, Millecamps M, Stone LS, Pineyro G. Differential Association of Receptor-Gβγ Complexes with β-Arrestin2 Determines Recycling Bias and Potential for Tolerance of Delta Opioid Receptor Agonists. J. Neurosci. 2012;32:4827–4840. doi: 10.1523/JNEUROSCI.3734-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gogolla N, Galimberti I, DePaola V, Caroni P. Preparation of organotypic hippocampal slice cultures for long-term live imaging. Nature Protoc. 2006;1:1165–1171. doi: 10.1038/nprot.2006.168. [DOI] [PubMed] [Google Scholar]
- 71.Son D, Lee P, Lee J, Kim H, Kim SY. Neuroprotective effect of wogonin in hippocampal slice culture exposed to oxygen and glucose deprivation. Eur. J. Pharmacol. 2004;493:99–102. doi: 10.1016/j.ejphar.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 72.Martinez-Sanchez M, Striggow F, Schröder UH, Kahlert S, Reymann KG, Reiser G. Na+ and Ca2+ homeostasis pathways, cell death and protection after oxygen–glucose-deprivation in organotypic hippocampal slice cultures. Neurosci. 2004;128:729–740. doi: 10.1016/j.neuroscience.2004.06.074. [DOI] [PubMed] [Google Scholar]
- 73.Ziemka-Nalecz M, Stanaszek L, Zalewska T. Oxygen-glucose deprivation promotes gliogenesis and microglia activation in organotypic hippocampal slice culture: involvement of metalloproteinases. Acta Neurobiol. Exp. (Wars) 2013;73:130–142. doi: 10.55782/ane-2013-1927. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.