

Abstract
This work presents the results of an international interlaboratory comparison on ex situ passive sampling in sediments. The main objectives were to map the state of the science in passively sampling sediments, identify sources of variability, provide recommendations and practical guidance for standardized passive sampling, and advance the use of passive sampling in regulatory decision making by increasing confidence in the use of the technique. The study was performed by a consortium of 11 laboratories and included experiments with 14 passive sampling formats on 3 sediments for 25 target chemicals (PAHs and PCBs). The resulting overall interlaboratory variability was large (a factor of ∼10), but standardization of methods halved this variability. The remaining variability was primarily due to factors not related to passive sampling itself, i.e., sediment heterogeneity and analytical chemistry. Excluding the latter source of variability, by performing all analyses in one laboratory, showed that passive sampling results can have a high precision and a very low intermethod variability (<factor of 1.7). It is concluded that passive sampling, irrespective of the specific method used, is fit for implementation in risk assessment and management of contaminated sediments, provided that method setup and performance, as well as chemical analyses are quality-controlled.
Introduction
Traditional methods for assessing risks and managing contaminated sediments are based on total, solvent-extractable concentrations of sediment-associated chemicals, following normalization to the sediment organic carbon content.1 Within the environmental scientific community it is generally accepted that this approach does not lead to a realistic assessment of actual risks at field-contaminated sites.2 Therefore, several methods for estimating the “bioavailable” concentration or fraction of a chemical have been developed during the past decades. These methods aim to determine the concentration or fraction that is available for causing ecotoxicological effects and more closely reflects actual or potential exposure. Among these methods, partitioning-based, nondepletive extractions with polymers (colloquially referred to as “passive sampling methods”, even though often active mixing of the polymer and the sediment is applied) are considered the best developed and have the most solid scientific basis.3 Through passive sampling, the freely dissolved concentration (Cfree) of a chemical in sediment pore water is determined, which is a good metric of the driving force behind accumulation and toxicological effects in organisms.4 The technique involves direct exposure of a polymer phase to sediment, either in situ or ex situ. Hydrophobic organic chemicals present in the sediment system partition into the polymer and the resulting polymer-sorbed equilibrium concentration is used to calculate Cfree. Several different polymers have been applied as a sampling phase, including polydimethylsiloxane (PDMS), polyethylene (PE), polyoxymethylene (POM), polyacrylate (PAc), and silicone rubber (SR), with the polymers being available in different formats.5
Despite the multitude of sampler formats and application possibilities, passive sampling is currently primarily used for scientific purposes and as an indicator of sediment remediation performance, rather than to design sediment management approaches. Acceptance in the risk assessment and regulatory community has been slow, among other reasons because so many different types of passive samplers are applied and the lack of standardized methods. There is a perception outside the scientific community that no scientific consensus exists on which is the best method to use.2 Although guidelines for selection of specific polymers have been proposed,5 and the application of different passive samplers and (calculation and analysis) methods should theoretically yield identical Cfree values, it is currently unknown if this actually holds true and the intermethod variability has not yet been adequately quantified. This information is crucial, however, when implementing passive sampling in risk assessment practices for contaminated sediments, as recently suggested by the US EPA.6
In November 2012, a SETAC workshop on passive sampling in sediments was held in Costa Mesa (CA, U.S.A.), with the goal of advancing the application of passive sampling in the risk assessment and management of contaminated sediments.2 During the workshop, several research needs and bottlenecks for implementation were identified, including the above-mentioned issue and the necessity for a round-robin interlaboratory study, standardization of methods, and characterization of sources of uncertainty.2,5 In response, an international interlaboratory comparison study was initiated, with the main objectives to (i) map the state of the science in ex situ passive sampling in sediments, and the interlaboratory and intermethod variability in Cfree determinations; (ii) identify the sources of variability in Cfree as determined with passive sampling; (iii) propose measures to reduce variability and to provide practical guidance (standardized methods); and (iv) increase the overall confidence in passive sampling to advance its use outside the scientific domain, i.e., in regulatory decision making. The results of the interlaboratory comparison are presented in this paper. Practical guidance (i.e., a proposed standard method) and polymer–water partition coefficients (Kpw) needed to calculate Cfree will be presented in separate, forthcoming papers.
Study Design
Eleven research laboratories from four different countries (U.S.A., The Netherlands, Norway, and the Czech Republic) participated in the study. The Utrecht University laboratory acted as coordinating laboratory. Each participating laboratory had a proven track record in passive sampling in sediments and contributed to the study by applying its own passive sampling procedures (i.e., format, experimental setup), previously published in the peer-reviewed literature. In total, 14 passive sampling formats were included, which differed in polymer material, source, form (i.e., polymer sheet vs coating on a glass (SPME) fiber), or thickness. Five of the 11 laboratories applied multiple formats. Passive sampling experiments were performed with three sediments, including two field-contaminated sediments and one unpolluted sediment that was spiked in the coordinating laboratory. Target chemicals included 12 polychlorinated biphenyls (PCBs) and 13 polycyclic aromatic hydrocarbons (PAHs). Cfree values of these chemicals were determined in 5-fold for each sediment in the following set of tiered experiments. In the first experiment, each laboratory followed its own procedure(s). The resulting Cfree values were reported to the coordinating laboratory, along with the Kpw values used in the calculations and a description of the methods applied. This experiment mapped the overall variability in passive sampling methods. In the second experiment, participants were asked to redo the measurements, but to strictly apply a “standard” protocol that was dictated by the coordinating laboratory. This experiment was performed in duplicate: one set of sample extracts was analyzed by the respective participant, to quantify the contribution of employing different protocols to the overall variability; the other set was shipped to, and analyzed by the coordinating laboratory, in order to evaluate the contribution of analytical chemistry to the overall variability. All participants were also provided with a standard solution of the target chemicals, of which the reported concentrations yielded a direct measure of the analytical (instrumental calibration) variability. In the third experiment, the coordinating laboratory applied the “standard” protocol to all 14 passive sampling formats (as shared by the participants) to identify the intermethod variability. Finally, supplementary tests were performed to map any additional sources of variation in Cfree, including polymer mass determination, sediment heterogeneity, and sediment storage time.
Materials and Methods
Passive Samplers
An overview of the applied passive samplers (polymer types, thicknesses, suppliers) is given in the Supporting Information (SI Table S1).
Target Chemicals
Target chemicals were the PAHs phenanthrene, anthracene, fluoranthene, pyrene, benz-[a]-anthracene, chrysene, benzo-[e]-pyrene, benzo-[b]-fluoranthene, benzo-[k]-fluoranthene, benzo-[a]-pyrene, benzo-[ghi]-perylene, dibenz-[ah]-anthracene, and indeno-[123,cd]-pyrene; and PCB congeners 18, 28, 52, 66, 77, 101, 118, 138, 153, 170, 180, and 187.
Analytical Standard Solution
A standard solution was prepared for each participant, by adding 50 μL of an acetone spike containing the 25 target chemicals to 950 μL of the participant-specific injection solvent applied during chemical analyses by the respective laboratory (either n-hexane, n-heptane, n-hexane/acetone (1:1), dichloromethane, or acetonitrile). Nominal concentrations (not shared with the participants) were about 50 μg/L for PCBs and 100 μg/L for PAHs.
Sediments
The three testing sediments differed in degree of complexity by passive sampling application. The “least complex” sediment (“SP sediment”) was an unpolluted, sandy sediment, sampled from the small river “Kromme Rijn”, near Werkhoven, The Netherlands. It was sieved through a 1 mm sieve, yielding a 20-kg dry weight (dw) sample, which was intensively mixed for 30 min with a mechanical mixer. Ten 2 kg (dw) portions of the sediment were successively spiked in 5 L glass beakers with relatively high levels of the target chemicals, by adding dropwise 4 mL of an acetone solution containing the target chemicals (PAHs at ∼250 mg/L each; PCBs at ∼150 mg/L each), while intensively mechanically stirring (30 min). All portions were finally pooled in a 110 L concrete mixer, which subsequently mixed this spiked (SP) sediment continuously for 4.5 weeks. The sediment of “intermediate complexity” (“BB sediment”) originated from the “Biesbosch”, a Dutch sedimentation area. This sediment has been used in a previous study in outdoor ditches,7 and the sediment used in the present study was sampled from that site. It contained relatively low native concentrations of the target chemicals, but was known to be homogeneous. Therefore, it was mixed in a concrete mixer for a shorter period of time, i.e., 1.5 week. The most complex sediment (“FD sediment”) was a sediment composed by combining (2:1) a French and a Dutch sediment. The French sediment was sampled from the river Tillet (Aix les Bains, Savoie), was very sandy, and contained hardly any PAHs. PCBs were however present at high concentrations and originated from a former electric transformer manufacturing facility 2 km upstream. The Dutch sediment was sampled from the river Hollandsche IJssel and has been previously studied.8 It contained no detectable PCBs, but PAHs were present at intermediate concentrations, mostly originating from an upstream diesel-powered water pumping station. This sediment also contained nonaqueous phase liquids (NAPLs). The composited sediment was mixed in a concrete mixer for 4 weeks nonstop. Before mixing, a quantity of the biocide sodium azide (NaN3) was added to all sediments, producing a concentration of 100 mg/L water. After mixing, the sediments were divided among amber-colored glass jars in portions sufficient to meet each participant’s requirement to complete the tests (different procedures by different participants required different sediment masses). All jars were closed with aluminum foil-lined lids and shipped in cooled containers to the participants, along with the standard solution and coded autosampler vials and glassware for the standardized experiments. Dry weight and organic carbon content, as well as total concentrations of the target chemicals in the sediments were determined by the coordinating laboratory as previously described.9 The results are provided in Table S2. This information was shared with the participants before initiating the experiments.
Determination of Cfree Based on the Participants’ Own Procedures
In this first experiment, all participants performed Cfree determinations according to their own procedure(s) and analyzed the resulting extracts themselves. Each measurement was performed 5-fold. A summary of the materials used and methods applied by all 11 participants is (anonymously) listed in Table S3. Procedures clearly differed in terms of type of exposure (i.e., static vs dynamic), exposure duration, verification of equilibrium conditions (i.e., use of performance reference compounds (PRCs), multiple sampler thicknesses, or multiple time points), sampler mass, sampler/sediment/water ratio, washing and extraction of samplers, and solvents used.
Determination of Cfree Based on Standardized Procedures
After completing the above experiment, participants received a standardized protocol and were asked to repeat the 5-fold Cfree determinations, strictly adhering to the prescribed procedure. The protocol was method-, sediment-, and participant-specific, but all aspects and steps (except the chemical analysis) were standardized, including sampler/sediment and sediment/water ratio, sampler washing, glassware, composition of the added water, exposure duration (6 weeks), method of shaking and shaking speed, and sampler cleaning and extraction procedures after finishing the exposures. The sampler/sediment ratio was dependent on the sediment properties and the polymer used, and the sampler washing and extraction procedures were specific for different polymers. Furthermore, the sampler extraction was tuned to the solvent used during chemical analysis by the particular participant. A general description of the standardized protocol is presented on pages S20–S21 of the SI. As outlined under “Study Design”, this experiment was performed in duplicate. One set of extracts was analyzed by the participant, the other set was shipped in a cooled container to the coordinating laboratory, where internal standards were added and the extracts were analyzed. The standardized protocol was also applied by the coordinating laboratory to all 14 sampler types (as provided by the participants), in order to quantify the intermethod variability.
Supplementary Tests
Supplementary tests focusing on additional sources of variation in Cfree (polymer mass determination, sediment heterogeneity, and sediment storage time) are described in the SI.
Chemical and Data Analysis
Target chemicals were analyzed by the participants as described in Table S3. GC-MS or GC-ECD was used for PCB quantification, whereas PAHs were analyzed by either GC-MS or HPLC-FLD. Concentrations in the sampler extracts were converted to concentrations in the sampler material (Cs), using the sampler’s mass (sheets) or polymer volume (SPME fibers). Cfree was then obtained by dividing Cs by a polymer- and chemical-specific Kpw. In the first experiment (participants’ own procedures), participants applied their own Kpws (measured themselves or taken from the literature) and some used PRCs in their calculations. In the standardized experiment, a fixed set of Kpw values as measured by the coordinating laboratory according to previously published methods10 was applied. Variability in each experiment was quantified by averaging the 5-fold Cfree measurements of each participant and subsequently calculating a variation factor (VF) for each target chemical. This factor was calculated by assuming the experimentally determined concentrations exhibited normal distributions and then taking the ratio of the 95th percentile (PCTL) value of the averaged Cfree values per target chemical, to the 5th percentile value:
Using this statistic, the range in Cfree was quantified and expressed intuitively as a factor, while excluding outliers. In order to compare experiments and sediments in a simple way, the chemical-specific VF values were averaged per sediment for each experiment (VFav).
Results and Discussion
State of the Science in Passive Sampling Sediment Pore Water
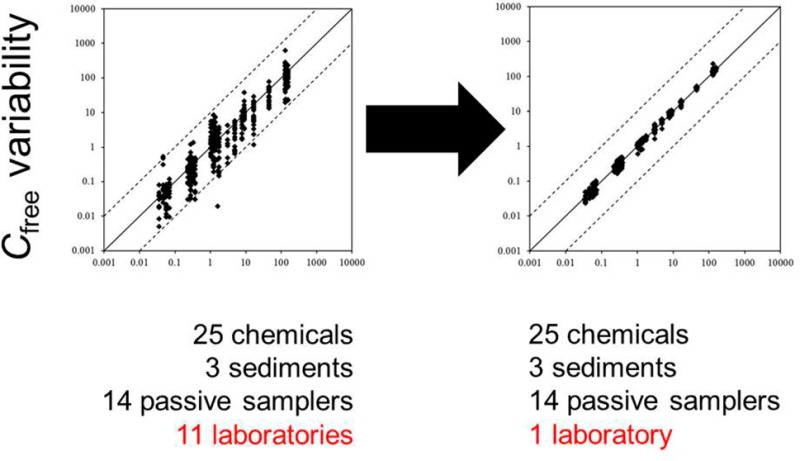
The results of the first experiment, in which all participants performed Cfree determinations according to their own procedures, are presented in panels A1–A3 of Figure 1. In these three figures (one for each sediment), the averaged Cfree data for all target chemicals are plotted against Cfree values obtained by averaging all chemical-specific data produced by the coordinating laboratory (referred to as Lab UU in Figure 1; all passive sampling formats; standardized protocol). (Note that using these values as independent variables does not imply they are the target or actual values; they are solely used as reference.) This way, the data are presented in a straightforward and understandable manner, without any data manipulation, yet clearly demonstrating the data variability. Additionally, in Figure S1, box plots are presented per chemical. Nearly all data points fall within the 10:1 and 1:10 interval, but there is a clear tendency toward under-predicting the averaged data of the coordinating laboratory. Overall, the observed interlaboratory variation is quite large; larger than the variability reported for a previous small-scale interlaboratory passive sampling comparison (i.e., a factor of 2).11 Note, however, that in ref (11) fewer samplers and target analytes (3 and 8, respectively) were tested, using a single sediment. Figure 1 may be also somewhat misleading as the apparent concentration ranges in some cases seem to cover a factor of 100, whereas they are actually composed of data for more than one chemical. The largest variation in the present study was observed for PCB-77 in the BB and FD sediments, where the concentration ranges did indeed span a factor of 100 and even 2400, respectively (see Figure S1 and Table S4, in which ranges for all chemicals are presented). The cause for the deviating behavior of this particular chemical is as follows. PCB-77 was a target chemical, which was added to the SP sediment, but it was not present at detectable concentrations in the field-contaminated BB and FD sediments (Table S2), as revealed by dedicated GC-MS analyses at the coordinating laboratory. Nevertheless, several participants reported considerable Cfree values for the chemical in these sediments. The large concentration ranges observed can thus be explained by the different detection (MS; ECD) and separation (GC columns) approaches applied by different participants, which will have resulted in inconsistencies in interfering/mis-identified peaks. Because the coordinating laboratory did not report a value, the Cfree of PCB-77 as reported by one of the participants was used as x-axis value in Figure 1, in order to be able to visualize the variability of this chemical. Since the data for PCB-77 in the BB and FD sediments obscure the average variability, they were excluded from the data analysis when calculating VFav values. These VFav values are listed in Table 1. Values for the first experiment are 9.7 ± 4.1, 9.4 ± 6.3, and 10.8 ± 4.5 for the BB, FD, and SP sediment, respectively. Apparently, when omitting the PCB-77 data, there are no obvious differences in variability among the three sediments, even though they were selected/composed based on differences in complexity for passive sampling. This may imply that passive sampling produces results, which are independent of the type of sediment studied, but it should be stressed that the overall variability is so large that any subtle differences between results for the various sediments may be fully obscured.
Figure 1.

Variability in freely dissolved concentrations (Cfree) determined in three sediments as measured with passive sampling methods (A) when the participants of the interlaboratory comparison followed their own protocols, (B) after standardization of Kpws and experimental protocols, (C) when, in addition to B, all chemical analyses were performed in one laboratory, and (D) when both experiments and analyses with all samplers were performed in one laboratory. Solid lines represent the 1:1 relationships; dashed lines indicate ± a factor of 10. The n number in each plot indicates the number of data points.
Table 1. Averaged Variation Factors (VFav; ± Standard Deviations) per Sediment and Per Experimenta.
| BB sedimentb | FD sedimentb | SP sediment | |
|---|---|---|---|
| measurements based on own protocols | 9.7 ± 4.1 | 9.4 ± 6.3 | 10.8 ± 4.5 |
| standardizing Kpw values | 8.9 ± 3.6 | 9.3 ± 4.6 | 10.8 ± 5.6 |
| standardizing protocols and Kpw values | 4.4 ± 1.4 | 4.6 ± 2.2 | 4.5 ± 1.2 |
| standardizing and chemical analyses in one lab | 2.4 ± 0.89 | 2.4 ± 0.72 | 2.6 ± 0.82 |
| all work performed in one lab | 1.6 ± 0.35 | 1.7 ± 0.42 | 1.7 ± 0.31 |
The VFav values are calculated by averaging the VF values of all chemicals for one sediment in a specific experiment. The number of chemicals included is 23–25, depending on the sediment and experiment.
Data for PCB-77 are excluded (see text for explanation).
Note that the variation observed in Figure 1 A1–A3 includes variability as introduced by (i) different laboratories, applying different protocols carried out by different people (interlaboratory variability), (ii) the use of different Kpw values by different participants, (iii) different ways of analyzing the chemicals, (iv) potential sediment heterogeneity and contaminant instability; and (v) the use of different passive sampling approaches (intermethod variability). The contribution of each of these sources will be discussed in a semiquantitative manner in the subsequent sections.
Impact of Standardizing Kpw values
Since most of the measurements performed by the participants involved equilibrium passive sampling, and inaccuracies in the Kpw of target analytes under equilibrium conditions are considered “a major source of concern”,12 one would expect a clear contribution to reducing the overall variability by standardizing the Kpws used for calculating Cfree values. After all, the participants applied Kpw values measured in their own laboratory or values taken from the literature. As such, there were considerable differences between the values that were used. For PDMS, the largest difference between the lowest and the highest chemical-specific Kpw values was a factor of 7, whereas for PE and POM this was even a factor of 13 and 20, respectively. The impact of standardizing Kpws was investigated by using Kpw values that had been determined for each sampler/chemical by the coordinating laboratory (manuscript in preparation). Remarkably, the impact of using Kpw values from a single source on the overall variability was negligible, as shown in Figure S2. The VFav values did not significantly change after recalculating the Cfree data as reported by the participants, using Kpw values from the single source (see Table 1). The position of the data points, however, did change in many cases, which makes sense, as Kpw determines the absolute value for Cfree. In other words, standardizing Kpws does not reduce the variability of Cfree measurements, but still is of utmost importance, because of the final accuracy of Cfree data. Using inaccurate Kpws will yield biased Cfree data, which is an unwanted situation when applying passive sampling for assessing risks of contaminated sediments. Therefore, it is recommended that high-quality, accurate (standardized) Kpw values be used by the passive sampling community.5,13
Impact of Standardizing Experimental Protocols
Standardizing the experimental protocols, in addition to the Kpw values, had a clear impact on the Cfree interlaboratory variability. Figures 1 B1–B3, S1, and Table 1 demonstrate that the variability roughly halved, with the VFav values being reduced to between 4 and 5 for all tested sediments. This obviously implies that the methodology of passive sampling measurements influences the outcomes and that standardization of passive sampling methods is definitely desirable. Because multiple issues and steps were standardized in the protocols, it is not possible to attribute the variation reductions to a specific aspect of the protocols; there are several likely candidates. The most important aspects that were standardized (thus changed for certain participants) included the sampler/sediment and sediment/water ratios, sampler washing procedure, glassware used, exposure duration, method of shaking and shaking speed, and the sampler cleaning and extraction procedures after finishing the exposures. Smedes et al.14 showed that the sampler/sediment ratio may influence the equilibrium concentration in the sampler (and thereby the calculated Cfree), as it was observed to be inversely related to this metric, due to depletion of the system. Also for the presently investigated sediments a similar relationship was observed (see Figure S3). In the standardized protocol, the ratio was set such that chemical depletion from the three sediments was always below 2% for all chemicals and samplers.14 However, when performing the measurements according to their own procedure(s), some participants applied (much) higher ratios, which will have resulted in higher depletion ratios (theoretically up to about 70%). Therefore, standardization of this step most probably will have contributed to the variation reduction. Likewise, Smedes et al.14 demonstrated that the sediment/water ratio can affect the system’s kinetics. Higher ratios yielded faster equilibration. Optimization of this ratio, together with a sufficient equilibration time and shaking regime, assured (near) equilibrium in all cases during the standardized experiment, as illustrated in Figure S4. In the first experiment in which the participants followed their own procedures, several participants (presumably) did not achieve equilibrium for all chemicals. PRCs were used to correct for this in several cases, following different calculation approaches, but such a correction may introduce uncertainties and inaccuracies.15,16 This particularly applies to the more hydrophobic chemicals, for which the correction by some participants was based on extrapolation from released fractions of less hydrophobic PRCs only. It should be stressed though that correction for the degree of nonequilibrium based on PRCs does not necessarily introduce substantial error, as demonstrated by the experiments from one participant (Figure S5). Whereas the standardized protocol prescribed thorough mixing and no PRCs, the procedure of this particular participant involved static exposures and included PRC corrections. Results of both approaches agreed within a factor of about 2 for all chemicals and sediments.
Standardization of some of the other aspects of the protocols may also have contributed to the variability reduction, but their contributions are probably less substantial. Sampler extraction after the exposures to sediments may be an exception, as the specific solvent used or the handling of samplers/extracts (e.g., cleanup or evaporation steps) by participants may have introduced variability through, for instance, variable extraction recoveries or losses of contaminants.
Contribution of Analytical Chemistry to the Variability
Even after standardizing Kpw values and experimental protocols, considerable variability in the interlaboratory Cfree data remained (Figure 1 B1–B3). This variability again roughly halved when all passive sampling extracts were analyzed by one laboratory (see Figures 1 C1–C3 and S1). The VFav decreased to about 2.5 for all three sediments (Table 1). As such, chemical analyses had a substantial contribution to the overall variability. A similar conclusion was also drawn for other interlaboratory comparison studies on passive sampling in surface waters,17,18 but certainly is not restricted to passive sampling measurements. Each experiment involving chemical analyses will suffer from errors introduced through inaccuracies in the identification, integration, and calibration of compounds. The case of PCB-77, as discussed above, already demonstrated that identification is the first crucial step and, if not performed correctly, can result in huge interlaboratory variability. Peak area quantification generally may not be considered as the step that contributes most to the overall variability introduced through chemical analysis. However, in complex chromatograms with coeluting peaks, baseline selection is subjective and poor integrations may add from a few percent of error to perhaps a factor of 2 or more. Any error will strongly depend on the sediment, the chemical, the analytical separation power, the selectivity of identification, the integration approach (i.e., quantification based on peak area or height), and the efficacy of any cleanup procedure. The major source of error introduced by analytical chemistry is likely related to calibration. Apart from correct application of internal standards, final concentrations quantified in the analyzed extracts are directly related to the accuracy of calibration standards. Even for PAHs and PCBs, i.e., compounds that are often routinely analyzed, this accuracy may be insufficient. The analysis of the standard solution in the present study demonstrated that the variation in PCB concentrations was characterized by a VF of 2 to 3, while for PAHs it was 3 to 4.5 (see Figure 2). From Figure 2 and the difference between Figure 1B and C, it can thus be concluded that a major part of the present interlaboratory variability in Cfree data originates from a step that basically has nothing to do with passive sampling measurements, but is part of every experiment involving the measure of chemicals, and is often overlooked as a source of error in experimental results. Therefore, including a standard solution in interlaboratory comparison studies involving chemical quantification is a prerequisite.
Figure 2.

Variation factors (95th PCTL/5th PCTL) calculated based on the (range of) concentrations of the target chemicals in the analytical standard, as reported by the participants of the interlaboratory comparison.
Other Sources of Variability
Figure 1C shows the variability in the results of experiments that were standardized and of which the extracts were analyzed by one laboratory. The observed variability will therefore only be caused by (i) intermethod variability, which will be discussed below, (ii) variability in the performance of the standardized procedure by different laboratories, and (iii) other sources of variability. Two other sources of variability were investigated in the present study: the accuracy of sampler mass and fiber coating volume (i.e., analytical weighing and the use of nominal fiber coating thicknesses), and sediment heterogeneity (originating from insufficient mixing and different storage times). Generally, sheet samplers are weighed on a balance and the concentrations quantified in polymers are expressed on a sampler mass basis. Hence, an inaccurate balance or weighing procedure may introduce error and consequently increase data variability. The results of the weighing test, however demonstrated that sampler weights generally were within 1% of the weights recorded by the coordinating laboratory. Only one participant reported weights deviating up to 4.7%. These differences are small and, consequently, weighing did not contribute significantly to the experimental variability in the present study.
When deriving the coating volume of a SPME fiber, product specifications provided by the manufacturer are rarely questioned, although it often remains unsure how these were established. A comparison of coating volumes calculated based on nominal, manufacturer-provided thicknesses versus measured ones (Table S5) demonstrated considerable differences, which amounted up to 16%. As such, fiber coating volumes may be a potential source of variability in Cfree. However, two of the fibers showing the largest deviations (S30-1 and PAc) were used by the coordinating laboratory only, which used actual volumes throughout the different experiments. Therefore, in the present study, the use of nominal coating volumes may only have been a potential source of variability for the S10-1 fiber, albeit not in the experiments where the chemical analyses (and subsequent calculations) were performed by the coordinating laboratory.
Results of the sediment heterogeneity experiment showed that even after mixing for several weeks, sediment heterogeneity may also have contributed to the observed overall variability in Cfree. VFav values of 1.1 to 1.4 for the field-contaminated BB and FD sediments and 1.2 to even 2.4 for the spiked SP sediment were calculated (see Table S6). The VF values are rather chemical-independent for the BB and FD sediments, but for the SP sediment, they increase with chemical hydrophobicity (see Table S6). Apparently, mixing this spiked sediment for up to 4.5 weeks in a concrete mixer was insufficient to allow full chemical homogenization for the most hydrophobic compounds. Note that the results presented here were obtained by analyzing several sediment sub samples (n = 10) taken directly from the concrete mixer. These samples do not necessarily perfectly represent the sediment samples as received by the participants, considering the large sediment volume that was contained in the mixers. After filling all the jars with sediment required by the participants, excess sediment was placed in spare jars. The VFav values thus do not per se exactly quantify the actual variability caused by sediment heterogeneity in the experiments and cannot be directly deduced from the values in Table 1. They do indicate, however, that sediment heterogeneity potentially may have contributed to the variability observed in Figures 1A–C. Apart from that, sediment heterogeneity within a single sediment batch as received by a participant is expected to be much smaller, as will be shown below (intramethod variability).
Measurements performed with sediments stored for 4.5 months in the refrigerator, as compared to measurements initiated directly after sampling from the concrete mixers demonstrated that Cfree of the target PAHs and PCBs decreased with about 20% in the FD sediment and 10% in the BB and SP sediments. This suggests that sediment storage also cannot be excluded as a source of variability. However, the time between the first participant starting the first experiment and the last participant starting this experiment, was only one month. Therefore, it is not very likely that storage time contributed significantly to the variability in Figure 1A. The first and last started standardized experiments were, however, three months apart and storage time thus may have been an additional source of variability in Figure 1B. It should be stressed though that the two measurements (i.e., before and after storage) were performed with two different sediment batches (jars); as such, sediment heterogeneity may also have caused (part of) the difference in Cfree. Assuming the concentration decrease is a real phenomenon, progressive sorption (redistribution) may be the underlying mechanism for the SP and FD sediment. However, for the field-contaminated BB sediment this process is improbable. Degradation is unlikely in all cases (chemicals, sediments).
Intramethod and Intermethod Variability
The last experiment included Cfree measurements with all sampler formats by the coordinating laboratory. From this experiment, both the intra- and intermethod variability could be deduced. As observed before,19 the intramethod variability appeared very low. For sheet samplers (PE, POM, SR), relative standard deviations (RSDs) of the 5-fold measurements were generally <5% and for the (homogeneous) BB sediment, RSDs were often below 2 or even 1%, indicating very high repeatability. Prerequisites for low RSDs are that the measurements are performed by skilled personnel, trained to work with passive samplers and to perform high-quality chemical analyses (including highly consistent integrations). For SPME fibers, RSDs of the 5-fold measurements by the coordinating laboratory were somewhat higher, with the values increasing with decreasing coating thickness: RSDs S10 > S30 > S100 > sheets (see Table S7). The cause of this order most probably relates to (i) the fact that the uncertainty in the sampling phase volume increases with decreasing coating thickness (because of increased uncertainties in the actual coating thickness, inaccurate cutting of the fibers, or coating wear during equilibration) and (ii) the thinner the coating, the higher the probability for artifacts to occur through “fouling” (i.e., particles or NAPLs sticking to the coating), potentially causing overestimation of the polymer-sorbed concentration.8
Owing to the high method precision, it was possible to accurately quantify the intermethod variability. The resulting VFav values (see Table 1, last row) demonstrate that on average the results of all 14 passive sampling formats (both sheets and SPME fibers of different polymers, sources, and thicknesses) match within a factor of 1.7. Thus, differences in Cfree determined with a suite of passive samplers were very small (see also Figures 1D and S1D). The underlying VF values do slightly increase with target chemical hydrophobicity, in particular for the PCBs (Table S8). This increase is probably caused by the fact that Kpw values become more uncertain for very hydrophobic chemicals, due to increasing experimental difficulties related to reduced solubilities and slow kinetics.10 Lower Cfree values for the more hydrophobic chemicals cannot explain the observation, as the underlying measured concentrations in the extracts were not related to chemical hydrophobicity.
The data variability is practically identical for the different sediments, here indicating that passive sampling is a robust technique, with which Cfree can be determined precisely, irrespective of the sediment under study. A comparison of the results of the different samplers shows that the highest Cfree values generally were measured with the S100, S30-2, and S10 SPME fibers, whereas the lowest values generally were determined with POM, PE-6, and SR. However, because the differences are so small, in particular relative to the average (see Figure S1 D), it can be concluded that there are no specific polymers behaving substantially differently to all the others and that their usage should be avoided. Different methods do have their specific “pros” and “cons” though (e.g., practicability of handling, ease of Kpw determination, detection limits, etc.). A detailed discussion of these factors is beyond the scope of the present paper.
Overall, it can be concluded from the present study that passive sampling is ready for implementation in actual risk assessment and management practices of contaminated sediments. The technique is robust, as it produces results that are independent of the sediment studied and sampling polymer or format used. However, standard protocols should be applied (most importantly ensuring nondepletion, taking steps to deduce equilibrium concentrations in the polymers, and full sampler extraction) and the analytical chemistry element be carefully quality-controlled (e.g., by means of (certified) external standards). The preparation and use of a passive sampling reference sediment may also be considered as a quality check in future work. On the basis of the standardized procedure, practical guidance and a proposed standard protocol for passive sampling in sediments will be presented in a follow up paper.
Acknowledgments
The coordination of this study was financially supported by the European Chemical Industry Council’s Long-range Research Initiative program (Cefic-LRI), under contract ECO22-IRAS (Bruno Hubesch), and by ILSI-HESI (Michelle Embry). Prof. Emmanuel Naffrechoux and Nathalie Cottin (University of Savoye, France) are kindly acknowledged for sampling the French sediment. Theo Sinnige (Utrecht University, The Netherlands), Henry Beeltje (TNO, The Netherlands), and R. Kelly (Southern Illinois University, U.S.A.) are thanked for their technical and/or analytical support. J.N.A. and P.M.G. acknowledge support from the U.S. Army Corps of Engineers, Humphreys Engineer Center Support Activity, under contract W912HQ-14-C-0028. M.J.L. and S.A.N. thank the Southern Illinois University Morris Doctoral Fellowship for partial funding. G.M.F., L.A.F., and R.L. acknowledge individual support from the Strategic Environmental Research and Development Program (SERDP, 14 ER03-035/ER-2431/ER-2538). U.G. and M.J. acknowledge support from the National Institute of Environmental Health Sciences (Grant R01ES020941).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.est.7b05752.
Description of supplementary tests; tables listing sampling formats, sediment characteristics, participants’ own procedures, concentration ranges, fiber volumes, VFs characterizing sediment heterogeneity and intermethod variability, and intramethod variability (RSDs); and figures showing box plots, the effects of Kpw standardization and sampler/sediment ratio, uptake kinetics in different samplers, and a static/dynamic exposure comparison (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Di Toro D. M.; Zarba C. S.; Hansen D. J.; Berry W. J.; Swartz R. C.; Cowan C. E.; Pavlou S. P.; Allen H. E.; Thomas N. A.; Paquin P. R. Technical basis for establishing sediment quality criteria for nonionic organic chemicals using equilibrium partitioning. Environ. Toxicol. Chem. 1991, 10 (12), 1541–1583. 10.1002/etc.5620101203. [DOI] [Google Scholar]
- Parkerton T. F.; Maruya K. A. Passive sampling in contaminated sediment assessment: building consensus to improve decision making. Integr. Environ. Assess. Manage. 2014, 10 (2), 163–166. 10.1002/ieam.1488. [DOI] [PubMed] [Google Scholar]
- Mayer P.; Parkerton T. F.; Adams R. G.; Cargill J. G.; Gan J.; Gouin T.; Gschwend P. M.; Hawthorne S. B.; Helm P.; Witt G.; You J.; Escher B. I. Passive sampling methods for contaminated sediments: scientific rationale supporting use of freely dissolved concentrations. Integr. Environ. Assess. Manage. 2014, 10 (2), 197–209. 10.1002/ieam.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydy M. J.; Landrum P. F.; Oen A. M.; Allinson M.; Smedes F.; Harwood A. D.; Li H.; Maruya K. A.; Liu J. Passive sampling methods for contaminated sediments: state of the science for organic contaminants. Integr. Environ. Assess. Manage. 2014, 10 (2), 167–178. 10.1002/ieam.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh U.; Kane Driscoll S.; Burgess R. M.; Jonker M. T.; Reible D.; Gobas F.; Choi Y.; Apitz S. E.; Maruya K. A.; Gala W. R.; Mortimer M.; Beegan C. Passive sampling methods for contaminated sediments: practical guidance for selection, calibration, and implementation. Integr. Environ. Assess. Manage. 2014, 10 (2), 210–223. 10.1002/ieam.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhard L. P.; Mount D. R.; Burgess R. M.. Developing sediment remediation goals at superfund sites based on porewater for the protection of benthic life from direct toxicity to nonionic organic chemicals. U.S Environmental Protection Agency; WA, 2017, 74 pp.
- Kupryianchyk D.; Rakowska M. I.; Roessink I.; Reichman E. P.; Grotenhuis J. T. C.; Koelmans A. A. In situ treatment with activated carbon reduces bioaccumulation in aquatic food chains. Environ. Sci. Technol. 2013, 47 (9), 4563–4571. 10.1021/es305265x. [DOI] [PubMed] [Google Scholar]
- Van der Heijden S. A.; Jonker M. T. O. PAH bioavailability in field sediments: Comparing different methods for predicting in situ bioaccumulation. Environ. Sci. Technol. 2009, 43 (10), 3757–3763. 10.1021/es803329p. [DOI] [PubMed] [Google Scholar]
- Jonker M. T. O.; Smedes F. Preferential sorption of planar contaminants in sediments from Lake Ketelmeer, The Netherlands. Environ. Sci. Technol. 2000, 34 (9), 1620–1626. 10.1021/es9906251. [DOI] [Google Scholar]
- Jonker M. T. O.; Van Der Heijden S. A.; Kotte M.; Smedes F. Quantifying the effects of temperature and salinity on partitioning of hydrophobic organic chemicals to silicone rubber passive samplers. Environ. Sci. Technol. 2015, 49 (11), 6791–6799. 10.1021/acs.est.5b00286. [DOI] [PubMed] [Google Scholar]
- Gschwend P. M.; MacFarlane J. K.; Reible D. D.; Lu X.; Hawthorne S. B.; Nakles D. V.; Thompson T. Comparison of polymeric samplers for accurately assessing PCBs in pore waters. Environ. Toxicol. Chem. 2011, 30 (6), 1288–1296. 10.1002/etc.510. [DOI] [PubMed] [Google Scholar]
- Booij K.; Robinson C. D.; Burgess R. M.; Mayer P.; Roberts C. A.; Ahrens L.; Allan I. J.; Brant J.; Jones L.; Kraus U. R.; Larsen M. M.; Lepom P.; Petersen J.; Pröfrock D.; Roose P.; Schäfer S.; Smedes F.; Tixier C.; Vorkamp K.; Whitehouse P. Passive Sampling in Regulatory Chemical Monitoring of Nonpolar Organic Compounds in the Aquatic Environment. Environ. Sci. Technol. 2016, 50 (1), 3–17. 10.1021/acs.est.5b04050. [DOI] [PubMed] [Google Scholar]
- Booij K.; Smedes F.; Allan I. J.. Guidelines for Determining Polymer-Water and Polymer-Polymer Partition Coefficients of Organic Compounds. ICES Techniques in Marine Environmental Sciences; 2017, 32 pp. [Google Scholar]
- Smedes F.; Van Vliet L. A.; Booij K. Multi-ratio equilibrium passive sampling method to estimate accessible and pore water concentrations of polycyclic aromatic hydrocarbons and polychlorinated biphenyls in sediment. Environ. Sci. Technol. 2013, 47 (1), 510–517. 10.1021/es3040945. [DOI] [PubMed] [Google Scholar]
- Apell J. N.; Gschwend P. M. Validating the use of performance reference compounds in passive samplers to assess porewater concentrations in sediment beds. Environ. Sci. Technol. 2014, 48 (17), 10301–10307. 10.1016/j.trac.2015.10.013. [DOI] [PubMed] [Google Scholar]
- Fernandez L. A.; Harvey C. F.; Gschwend P. M. Using performance reference compounds in polyethylene passive samplers to deduce sediment porewater concentrations for numerous target chemicals. Environ. Sci. Technol. 2009, 43 (23), 8888–8894. 10.1016/j.trac.2015.10.013. [DOI] [PubMed] [Google Scholar]
- Vrana B.; Smedes F.; Prokeš R.; Loos R.; Mazzella N.; Miege C.; Budzinski H.; Vermeirssen E.; Ocelka T.; Gravell A.; Kaserzon S. An interlaboratory study on passive sampling of emerging water pollutants. TrAC, Trends Anal. Chem. 2016, 76, 153–165. 10.1016/j.trac.2015.10.013. [DOI] [Google Scholar]
- Booij K.; Smedes F.; Crum S. Laboratory performance study for passive sampling of nonpolar chemicals in water. Environ. Toxicol. Chem. 2017, 36 (5), 1156–1161. 10.1002/etc.3657. [DOI] [PubMed] [Google Scholar]
- Hawthorne S. B.; Jonker M. T. O.; Van Der Heijden S. A.; Grabanski C. B.; Azzolina N. A.; Miller D. J. Measuring picogram per liter concentrations of freely dissolved parent and alkyl PAHs (PAH-34), using passive sampling with polyoxymethylene. Anal. Chem. 2011, 83 (17), 6754–6761. 10.1021/ac201411v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.