

Abstract
Azo dyes, which are characterized by one or more azo bonds, are a predominant class of colorants used in tattooing, cosmetics, foods, and consumer products. These dyes are mainly metabolized by bacteria to colorless aromatic amines, some of which are carcinogenic, by azoreductases that catalyze a NAD(P)H-dependent reduction. The resulting amines are further degraded aerobically by bacteria. Some bacteria have the ability to degrade azo dyes both aerobically and anaerobically. Plant-degrading white rot fungi can break down azo dyes by utilizing a number of oxidases and peroxidases as well. In yeast, a ferric reductase system participates in the extracellular reduction of azo dyes. Recently, two types of azoreductases have been discovered in bacteria. The first class of azoreductases is monomeric flavin-free enzymes containing a putative NAD(P)H binding motif at their N-termini; the second class is polymeric flavin dependent enzymes which are studied more extensively. Azoreductases from bacteria represent novel families of enzymes with little similarity to other reductases. Dissociation and reconstitution of the flavin dependent azoreductases demonstrate that the non-covalent bound flavin prosthetic group is required for the enzymatic functions. In this review, structures and carcinogenicity of azo colorants, protein structure, enzymatic function, and substrate specificity, as well as application of the azo dyes and azoreductases will be discussed.
Keywords: Azo dyes, Azoreductase, Commensal bacteria, Environmental microorganisms, Biodegradation, Aromatic amines, Carcinogenicity
INTRODUCTION
Dyes are classified as anionic (direct, acid, and reactive dyes), cationic (basic dyes), and nonionic (disperse dyes). Anionic and nonionic dyes mostly contain azo or anthraquinone type chromophores [1]. Azo dyes form a major class of chemically related compounds that are ubiquitous in foods, plastics, textiles, pharmaceuticals, paints, printing inks, and cosmetics, but are also used as biological stains in laboratories and clinics [2, 3]. Azo dyes are characterized by one or more R1-N=N-R2 bonds and are reduced to aromatic amines by enzymatic mechanisms (Fig. 1) [4]. It is known that the degree to which a dye azo group is reduced depends on the electron density around the –N=N– bond. Electron-withdrawing groups such as –NH2 and –OH decrease the electron density around the –N=N– bond and facilitate the reduction of the azo group with a simultaneous release of an aromatic amine. Also, placing an electrodonor substitute in an ortho- position in relation to the azo group causes a reduction through the creation of hydrogen bonds on the nitrogen azo group. A similar effect in a simpler reduction within the –N=N– group is observed for water-soluble dyes [5], i.e., those with groups such as –SO3Na and –COOH in their structure (Fig. 1).
Fig. 1.
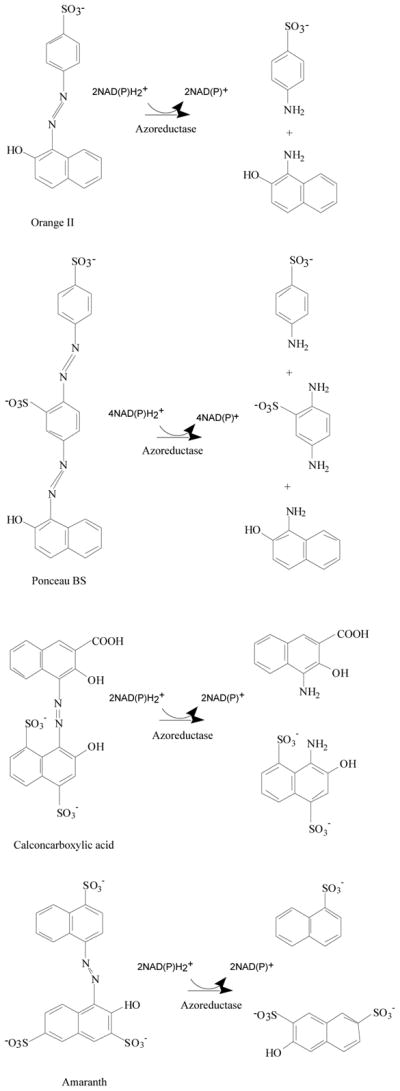
Reductive degradation of azo dyes by azoreductases.
There are thousands of azo dyes on the market, of which more than 500 contain potentially carcinogenic aromatic amines in the chemical formulation [6]. When these compounds enter the human body through ingestion and skin contact or injection, they are metabolized via azoreductases to aromatic amines in the gastrointestinal tract and in the mammalian liver [6–8]. About 90% of 4,000 dyes examined in a survey had LD50 values >2000 mg/kg with highest toxicities being found among basic and diazo dyes [9]. Azo dyes are regarded as relatively persistent pollutants because they are not readily degraded under aerobic conditions [10–12]. Under anaerobic conditions, azo dyes can be reduced by intestinal bacteria and some environmental microorganisms to colorless amines, which may be toxic, mutagenic, and carcinogenic to human and animals [13–18].
There are three basic types of carcinogenic activation mechanisms for azo dyes: (1) reduction and cleavage of the azo bond to produce aromatic amines; (2) oxidation of azo dyes with structures containing free aromatic amine groups; and (3) activation of azo dyes via direct oxidation of the azo linkage to highly reactive electrophilic diazonium salts [18, 19]. For example, using a standard plate test with Salmonella typhimurium TA 1538, it was found that after metabolic activation, Methyl Orange was mutagenic [20]. Methyl Orange was reduced by anaerobic bacteria and then activated by rat liver microsomes to mutagenic products.
Synthetic azo compounds have been identified in tattoo colorants, including derivatives of 3,3′-dichlorobenzidine, azo-derivative of 2-amino-4-nitrotoluene, and phthalocyanine derivatives (Fig. 2) [21, 22]. Aromatic amines, such as benzidine, induce urinary bladder cancer in humans and tumors in some experimental animals [2, 15].
Fig. 2.
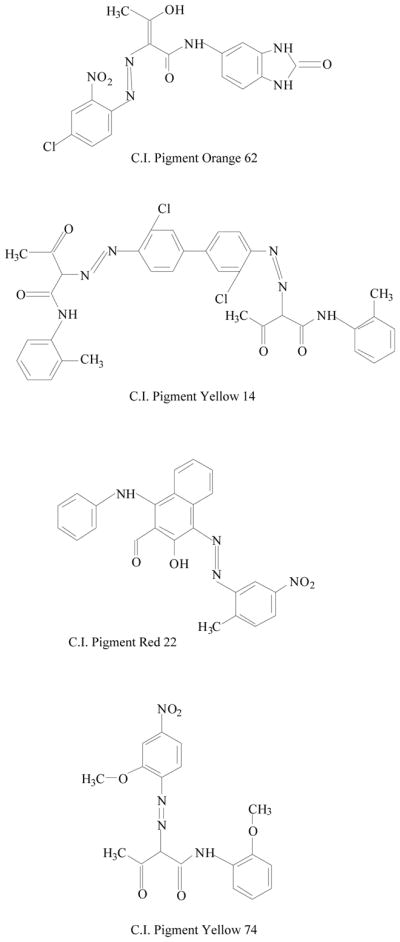
Chemical structures of some azo pigments identified in tattoo inks.
A wide variety of microorganisms, including bacteria and fungi, are capable of decolorizing a diverse range of azo dyes. Some bacteria have the ability to degrade azo dyes both aerobically and anaerobically. Bacterial biodegradation of azo dyes is often initiated by cleavage of azo bonds by azoreductases which are followed by the aerobic degradation of the resulting amines [23]. Fungal degradation of azo dyes mainly occurs from the lignin peroxidase activity under aerobic conditions [24]. In yeast, the ferric reductase system participates in the extracellular reduction of azo dyes [25].
In this review, azo colorants found in food, cosmetics, and the environment will be addressed; as well as the significance of azo reduction by intestinal, skin, and soil microflora. In addition, recent progress on the properties and function of oxygen insensitive aerobic azoreductase from microorganisms will be described.
AZO COLORANTS AND THEIR APPLICATIONS
About 60–70% of dyes applied in textile processing are water-soluble sulfonated azo compounds (Fig. 1) [26, 27]. Azo dyes in these effluents are compounds with low biodegradability. Color removal in generated effluents is an environmentally challenging issue in wastewater treatment [23, 28]. Such wastewater is commonly treated using physicochemical methods such as coagulation, adsorption, and oxidation with ozone. The conventional treatment of colored effluents is expensive and produces significant amounts of sludge and fails to remove all dyes, thus preventing recycling of the treated wastewater. The need for development of a more effective biological system for the treatment of the textile wastewater is of in considerable interest by industry [29–31].
Sudan azo dyes are fat-soluble azo dyes used in industry (Table 1). They are red dyes widely used for coloring hydrocarbon solvents, oils, fats, plastics, printing inks, waxes, petrol, shoe, and floor polishes. They are not permitted in foodstuffs for any purpose. Sudan I (CI Solvent Yellow 14) is a monoazo dye with a chemical formula of 1-phenylazo-2-naphthalenol. Para Red is an industrial azo dye used in printing and chemically similar to Sudan I. Sudan II (CI Solvent Orange 7) is the dimethyl derivative of Sudan I. Sudan III (CI Solvent Red 23) and Sudan IV (CI Solvent Red 24) are diazo dyes [32]. These Sudan dyes are classified as a category 3 carcinogen by the International Agency for Research on Cancer (IARC) [33]. In Europe it has been recently reported that Sudan azo dyes were found in a batch of Worcestershire sauce, garlic curry sauce, and hot chili sauce, palm oils, as well as in other food products [34]. Sudan III has been approved for use in externally applied drug and cosmetic applications, in amounts consistent with good manufacturing practice [35]. Sudan I is a liver and urinary bladder carcinogen in mammals and is considered a possible human mutagen. It forms benzenediazonium ion during cytochrome P-450 catalyzed metabolism. Calf thymus DNA was reacted with Sudan I which was activated by microsomal enzymes or with benzenediazonium ion in vitro [32, 36–38]. Chromatographic analyses of adduct obtained by reaction with DNA or homopolydeoxyribonucleotides showed that the major Sudan I-DNA adduct was formed with deoxyguanosine. This adduct was also found in DNA as a result of reacting with benzenediazonium ion. These results strongly suggest that the benzenediazonium ion derived from Sudan I reacts with DNA in vitro to form the stable 8-(phenylazo) guanine adduct [39]. Sudan II also induced mutation in Salmonella typhimurium TA 1538 in the presence of a rat liver preparation [40]. Concerns about the safety of Sudan III have been raised due to potential metabolic cleavage of Sudan III to yield 4-aminoazobenzene and aniline by skin bacteria [41]. Sudan IV required chemical reduction and microsomal activation to be mutagenic [42].
Table 1.
| Structure | Application | Carcinogenesis |
|---|---|---|
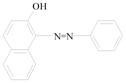 Sudan I |
Coloring solvents, oils, waxes, petrol, printing inks, and shoe. | Liver and urinary bladder carcinogen in mammals and is considered a possible human mutagen. |
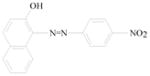 Para Red |
Coloring solvents, oils, waxes, petrol, printing inks, and shoe. | Toxicology not fully investigated. It would be prudent to assume that it, like Sudan I, could be a genotoxic carcinogen. |
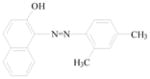 Sudan II |
Coloring solvents, oils, waxes, petrol, printing inks, and shoe. | Tested in mice by bladder implantation, resulting in a high incidence of bladder carcinomas. |
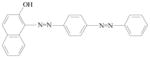 Sudan III |
Externally applied drugs and cosmetics. Used for demonstrating the presence of triglycerides in frozen sections. | Studies were inadequate for evaluation. |
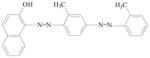 Sudan IV |
Used for demonstrating the presence of triglycerides in frozen sections and commonly for coloring waxes, oils and spirit varnishes. | Tests on laboratory animals indicate it may be mutagenic. |
The art of tattooing has a long history in many countries. Recent social/cultural trends in the United States and other developed countries have resulted in a surge in the popularity of tattooing [43–45]. There has been an apparent change in the chemicals used in tattooing over the past several years. The demand for an increasing number of color shades by customers and reports of toxicity of some of the inorganic salts, has apparently led manufacturers to replace the inorganic salts with organic molecules to achieve the desired colors and possibly reduce some of the toxicity associated with the inorganic salts [21]. Baumler et al. [22] determined the identity of the chemicals in several tattooing colors from five manufacturers and found that only one-third of the tattoo pigments contained single chromophoric compounds, indicating that the remaining two-thirds are complex mixtures of chemicals. Several groups of azo pigments have been identified from the tattoo pigments and some of their chemical structures are shown in Fig. 2. The expanded use of organic pigments in tattoo inks provides a wider range of shades and better color stability, but the safe use of these pigments is in question [46]. Little or no toxicity data have been reported on the pigments identified in tattoos. Although the azo pigments have for the most part been regarded as safe and the bioavailability of aromatic amine compounds from azo pigments based on 3,3′-dichlorobenzidine are relatively low, some pigments have structural alerts for mutagenicity and/or carcinogenicity.
DECOLORIZATION OF AZO COLORANTS BY HUMAN COMMENSAL BACTERIA
Azo colorants reach the human gastrointestinal tract mainly via oral ingestion. The intestinal bacteria are able to degrade various azo dyes [8, 20]. Reduction of azo dyes by the bacteria may produce compounds that are more or less toxic than the parent molecules [47]. Therefore, the azoreduction may increase or decrease any toxic or carcinogenic effects of the azo dyes [7, 47]. The azo dye Direct Black 38 is metabolized to the mutagen benzidine by human intestinal bacteria [48]. The ability of the bacteria residing in the human intestinal tract to reduce azo dyes has led to the development of drugs that are azo-linked to nontherapeutic moieties and polymeric azo prodrugs that provide colon-specific delivery of active metabolites [49–52]. After ingestion, the azoreductases from colonic bacteria cleave the azo bonds, releasing the drugs in their active forms [47, 52, 53]. For example, Sulfazalazine is an azo drug for treatment of inflammatory bowel diseases and the azoreductases cleave the azo linkage in the drug resulting in releasing an anti-inflammatory agent, 5-aminosalicylic acid, and an antibiotic agent, sulfapyridine, in the colon (Fig. 3) [49, 52]. Food azo colorant Amaranth is cleaved by intestinal bacteria to 1-amino-4-naphthalene sulfonic acid and 1-amino-2-hydroxy-3,6-naphthalenedisulfonic acid (Fig. 1), both of which are absorbed through the intestine. In animals, consumption of Amaranth in the diet resulted in constant serum 1-amino-4-naphthalene sulfonic acid levels maximally equivalent to that seen with other routes of administration. Determining exposure to a major Amaranth metabolite is important in evaluating toxicity data and relating it to human exposure as colorant in foods and beverages [54, 55]. Many isolated pure bacterial cultures from the intestine have the ability to convert various azo dyes. Ryan et al. [56] demonstrated that pure cultures of Proteus vulgaris and Escherichia coli could reduce the water-soluble azo dyes Tartrazine, Amaranth, Ponceau SX, Fast Yellow, Naphthalene Fast Orange 2G, Sunset Yellow, Methyl Orange, and Neoprontosil. Chung et al. [20] found that intestinal anaerobes Coprococcus catus, Acidaminococcus fermentans, Bacteroides thetaiotaomicron, Eubacterium biforme, Citrobacter sp., Fusobacterium sp. could reduce Tartraze, Ponceau SX, Sunset Yellow, Methyl Orange, Orange II, and Amaranth. Several strains of anaerobic bacteria capable of reducing azo dyes were isolated from human feces and identified as Eubacterium hadrum, Enbacterium sp., Clostridium clostridiiforme, Butyrivibrio sp., Bacteroides sp., Clostridium paraputrificum, Clostridium nexile, and Clostridium sp [57]. Recently a microarray method was used to identify the intestinal bacterial species involved in azo dye reduction [58]. Seventeen selected bacterial species that showed strong positive microarray results out of 40 predominant human intestinal bacterial species were examined for azo dye reduction activity by pure cultures incubated with Direct Blue 15. Among them, Clostridium perfringens, Clostridium clostridiioforme, Enterococcus faecalis, Ruminococcus obeum, and Bifidobacterium adolescentis showed the highest azo dye reduction activity.
Fig. 3.

Azo linkage in the drug is cleaved by azoreductases resulting in releasing an anti-inflammatory agent, 5-aminosalicylic acid, and an antibiotic agent, sulfapyridine, in the colon (Adapted from [19]).
The skin is the largest organ of the human body. The human skin flora is defined as the microbes present in healthy skin on the skin surface, within the stratum corneum, in the duct (infundibulum) of the sebaceous glands, and in the hair follicles [59]. Almost all parts of human skin are colonized by microorganisms under usual circumstances which do no harm by their presence. The microflora of human skin is diverse with regard to bacterial strains and species. The human skin is inhabited both by aerobic bacteria, predominately Staphylococci and by anaerobic bacteria belonging primarily to the genus Propionibacterium [60]. Platzek et al. [6] reported that representative human skin bacterial strains isolated from healthy human individuals, including Staphylococcus aureus, Staphylococcus epidermidis, Micrococcus luteus, Micrococcus roseus, and Micrococcus varians, can cleave the azo dye Direct Blue 14 to the corresponding aromatic amine o-tolidine. Direct Blue 14 is a typical member of the group of direct dyes which are defined as a water-soluble dye capable of directly dyeing cellulose fibers in textile industry. It was recently demonstrated that when Staphylococcus aureus was grown in the presence of Methyl Red, it rapidly reduced this azo dye. Only 20% of Methyl Red remained after 1 hour incubation, and it was completely degraded in 3 hours. Orange II was reduced by the bacterium as well although at a slower rate than Methyl Red. About 10% and 56% of Orange II dye color disappeared respectively after 3 and 20 h cultivations [61].
DEGRADATION OF AZO DYES BY ENVIRONMENTAL BACTERIA
The isolation of bacteria capable of aerobic decolorization and mineralization of azo dyes has also attracted interest. Several bacterial strains have been described that aerobically decolorize azo dyes by reductive mechanisms in the presence of other carbon or energy sources, indicating that the dyes probably are not used as carbon or energy sources [23, 62, 63]. Bacillus subtilis was able to reductively convert p-aminoazobenzene into aniline and p-phenylenediamine when grown on glucose under aerobic conditions [64]. Many other strains, such as Pseudomonas stutzeri [65], Acetobacter liquefaciens and Klebsiella pneumoniae [66], Citrobacter sp. [67], and Enterobacter agglomerans [68], were able to decolorize Methyl Red when grown on other nutrients under aerobic condition. Also, there were reports that sulfonated azo dyes were reductively cleaved by different bacteria under aerobic conditions with other additional carbon sources in the culture media [69–75]. Blumel et al. [76] isolated an unidentified bacterial strain with the ability to utilize the sulfonated azo compound 4-carboxy-4′-sulfoazobenzene as the sole carbon and energy source. It was suggested that 4-carboxy-4′-sulfoazobenzene was cleaved reductively under aerobic conditions to 4-aminobenzoate and 4′-aminobenzene-sulfonate, which was mineralized by previously established conventional aromatic catabolic pathways [76]. Under anaerobic conditions the sulfonated azo dye Mordant Yellow 3 was reduced by the biomass of a bacterial consortium grown aerobically with 6-aminonaphthalene-2-sulfonic acid. After re-aeration of the culture, the generated aromatic amines 6-aminonaphthalene-2-sulfonate and 5-aminosalicylate were mineralized by different members of the bacterial culture [77]. By using an anaerobic/aerobic treatment process, azo dye Disperse Blue 79 in textile effluents was biotransformed to amines in the anaerobic stage, which were subsequently mineralized in the aerobic phase. An increase in toxicity was observed in the anaerobic stage due to the amines formation, but the wastewater was detoxified after the aerobic treatment resulting in removal of above 90% of Disperse Blue 79 in textile effluents [78].
Streptomyces species are known to produce extracellular peroxidases that have a role in the biodegradation of lignin [79]. Initial work demonstrated that decolorization by Streptomyces chromofuscus A11 was related to the ligninolytic capabilities of the bacterium. The bacterial enzymes responsible for degradation of azo dyes had different substrate specificities than those of the white rot fungus Phanerochaete chrysosporium [80]. Subsequent findings concerning the intermediates and mechanisms of azo dye degradation by ligninolytic peroxidases of Phanerochaete chrysosporium and Streptomyces chromofuscus determined that the enzymes convert the azo dye to a cation radical that is susceptible to nucleophilic attack by water or hydrogen peroxide molecules. The azo linkage is split both asymmetrically and symmetrically. The resulting reactive products undergo several redox reactions that produce a more stable intermediate [79].
Many bacterial strains have been shown to reduce azo compounds under anaerobic conditions [23]. Azo reduction under anaerobic conditions may represent a fortuitous, nonenzymatic reduction by enzymatically generated reduced flavins. Inhibition by oxygen is due to regeneration of the oxidized form of flavins [81]. Previous studies with facultative anaerobic bacteria suggested that reduced flavins generated by cytosolic flavin-dependent reductases were responsible for the unspecific reduction of azo dyes [82, 83]. The ability of cytosolic flavin reductases to act in vitro as azoreductases was demonstrated by experiments using a recombinant flavin reductase in Escherichia coli and Sphingomonas sp. strain BN6 [84]. Cell extracts generally showed much higher rates of anaerobic reduction of azo dyes than did preparations of whole cells. This has been explained by the low permeability of the cell membranes for the highly polar sulfonated azo compounds [84–86]. Sulfonic acid substitution of the azo dye structure apparently blocks effective dye permeation [85]. Treatment of Bacillus cereus cells by toluene removed the block to dye permeation and resulted in the significantly increased passage of sulfonated and carboxylated azo dyes from the external medium into the cell with a concomitant increase in the reduction rate of the dyes [86, 87]. The presence of membrane-bound azo reductase activity was also shown in Sphingomonas sp. strain BN6, indicating that the nonspecific reduction of azo dyes by bacteria does not require transport of the azo dyes or reduced flavins through the cell membrane [88]. It was proposed that, in this system, quinones act as redox mediators which are reduced by a quinone reductase located in the cell membrane of Sphingomonas sp. strain BN6 and that the formed hydroquinones reduce the azo dyes of the culture supernatants in a purely chemical redox reaction. Recently, an oxygen sensitive high molecular mass alkali-thermostable azoreductase was purified and characterized from Bacillus sp. strain SF (Table 2) [29]. The enzyme is an NADH-dependent azoreductase whose activity against azo dyes was enhanced by FAD, implying that it may be a flavin protein.
Table 2.
Properties of Azoreductases from Bacteria
| Species | Molecular mass | Cofactor | Electron donor | pH opti. | Temp. opti. (°C) |
|---|---|---|---|---|---|
| Xenophilus azovorans KF46 [5, 10] | 30 kDa (Monomer) | NRa | NAD(P)H | 6.5 | 45 |
| Pigmentiphaga kullae K24 [89, 90] | 21 kDa (Monomer) | NR | NAD(P)H | 6.2 | 41 |
| Shigella dysenteriae [96] | 28 kDa (AzoI, homodimer) 11 kDa (AzoII) |
FMN FMN |
NAD(P)H NAD(P)H |
7.0 7.2 |
45 45 |
| Escherichia coli K12 [97] | 28 kDa (AzoI, homodimer) 12 kDa (AzoII) |
FMN FMN |
NAD(P)H NAD(P)H |
7.0 7.0 |
45 45 |
| Bacillus sp. OY1-2 [92] | 19 kDa | NAb | NADPH | NA | 70 |
| Escherichia coli [95] | 46 kDa (Monodimer) | FMN | NADH | NA | NA |
| Enterobacter agglomerans [68] | 28 kDa (Monomer) | NA | NADH | 7.0 | 35 |
| Enterococcus faecalis [91] | 46 kDa (Homodimer) | FMN | NADH | 6.6–7.0 | 35–40 |
| Rhodobacter sphaeroides [93] | 19 kDa | NA | NADH | 8.0 | 50 |
| Staphylococcus aureus [61] | 85 kDa (Homotetramer) | FMN | NADPH | 6.0–6.6 | 35–40 |
| Bacillus sp. strain SF [29] | 62 kDa (Monomer)c | NA | NADH | 8–9 | 80 |
Not required;
Not available;
Oxygen sensitive azoreductase.
AEROBIC FLAVIN-FREE AZOREDUCTASES FROM BACTERIA
The aerobic reductive metabolism of azo dyes requires enzymes called “aerobic azoreductases” that catalyze these reactions in the presence of oxygen [23]. The aerobic Orange II and I azoreductases from the carboxy-orange-degrading Xenophilus azovorans KF46 (formerly Pseudomonas sp. strain K46) and Pigmentiphaga kullae K24 (formerly Pseudomonas sp. strain K24) were purified, characterized, and compared. Both enzymes are monomeric flavin-free azoreductases that use NADPH and NADH as cofactors and reductively cleave several sulfonated azo dyes (Table 2) [10, 89]. The enzymes (30 and 21 kDa) show different substrates specificities. For example, the Orange II azoreductase from Xenophilus azovorans KF46 required the presence of a hydroxyl group in the ortho-position of the aromatic ring of the substrate. In contrast, the Orange I azoreductase from Pigmentiphaga kullae K24 required a hydroxyl group in the para-position of the aromatic ring of the substrate. These two enzymes did not exhibit immunological cross-reactivity with each other [89]. Cloning and characterization of the genes coding for the two enzymes revealed that there was no homology between the Orange II azoreductase from Xenophilus azovorans KF46 and the Orange I azoreductase from Pigmentiphaga kullae K24, which indicate that these two enzymes belong to two separate families [5, 90]. The deduced amino acid sequence of the Orange II azoreductase from Xenophilus azovorans KF46 suggests a molecular mass of 30, 278 Da with a presumed NAD(P)H-binding site identified in the amino-terminal region of the enzyme [5]. Similarly, there was a conserved putative NAD(P)H-binding site in the deduced amino-terminal region of the Orange I azoreductase from Pigmentiphaga kullae K24 which has a predicted molecular mass of 20,557 Da [90]. A 28,000 Da aerobic azoreductase was also purified and characterized from an Enterobacter agglomerans strain isolated from dye-contaminated sludge (Table 2). The enzyme is a monomeric flavin-free azoreductase and its activity is NADH dependent [68]. The enzyme utilized all five tested azo dyes including Methyl Red, Disperse Yellow, Trypan Blue, Amaranth, and Orange G with different relative activities. The Enterobacter agglomerans azoreductase is the only enzyme reported so far which is capable of cleaving Orange G. Normally, accessibility of azo dyes to azoreductase is dependent on the chemical structures of the dyes. It was suggested that electron-withdrawing groups on the phenyl ring accelerated reduction of the azo bond, while a charged functional group in proximity to the azo group or the presence of a second polar group interfered with the reaction. This could be the reason that there was no detectable activity against Orange G by the other aerobic azoreductases studied so far [5, 61, 91]. It should be very interesting to determine the mechanism of this Enterobacter agglomerans azoreductase degrading Orange G and analyze its metabolites.
AEROBIC FLAVIN-DEPENDENT AZOREDUCTASES FROM BACTERIA
Suzuki et al. [92] first described the sequence and subsequent characterization of a gene encoding an aerobic azoreductase from the soil isolate, Bacillus sp. OY1-2 (Bacillus cereus, Table 2). The protein consists of 178 amino acids and presumably it is a FMN dependent azoreductase. The Bacillus sp. OY1-2 azoreductase was predicted to be a member of a novel family of reductases. With an increase in the number of the completed bacterial genome sequences, it was found that similar genes encoding putative proteins belonging to the same family are widely present in Gram-positive bacteria (Fig. 4) [61]. A similar gene encoding azoreductase from Rhodobacter sphaeroies AS1.1737 was expressed and partially characterized [93]. Results of HPLC-MS analyses on Methyl Red cleavage by the Rhodobacter sphaeroies AS1.1737 azoreductase indicated the presence of hydrazo-intermediate, which is not stable and reduced quickly once formed.
Fig. 4.

Unrooted phylogenetic tree of NADPH-flavin azoreductases. Based on TBALSTN results, protein sequences showing similarities were aligned using Jutun Hein method. The unrooted tree was generated using MegAlign of DNASTAR software package. Genbank accession numbers are as follows: Archaeoglobus fulgidus, AE000972; Bacillus anthracis, AE017225; Bacillus cereus, AE017005; Bacillus licheniformis, CP000002; Bacillus subtilis, AB071366; Bacillus sp. OY1-2; Bartonella henselae, BX897699; Clostridium perfringens, BA000016; Gloeobacter violaceus, BA000045; Haloarcula marismortui AY596297; Halobacterium sp. NRC-1, AE005072; Lactobacillus plantarum, AL935261; Mesorhizobium loti, BA000045; Mycobacterium bovis, BX248344; Mycobacterium tuberculosis, AE000516; Nocardia farcinica, AP006618; Nostoc sp. PPC 7120, BA000019; Rhodobacter sphaeroides, AY150311; Staphylococcus aureus, AY545994; Staphylococcus epidermidis, AE016745; Streptomyces avermitilis, AP005049; Streptomyces coelicolor, AL939107; Sinorhizobium meliloti, AL595985; Xanthomonas oryzae AE013598 (Adapted from [61]).
By using homology comparison, direct cloning, and overexpression of the genes coding for hypothetical proteins, Chen et al. [61] were able to identify and express a functional azoreductase gene from Staphylococcus aureus in Escherichia coli. RT-PCR results demonstrated that the azoreductase gene was constitutively expressed in Staphylococcus aureus. The Staphylococcus aureus azoreductase was found to be a homotetramer with a native molecular mass of 85 kDa containing four noncovalently bound FMN molecules. The enzyme requires NADPH, but not NADH, as an electron donor for its activity. It was resolved to dimeric apoprotein by removing the flavin prosthetic groups using hydrophobic-interaction chromatography. The dimeric apo-protein was reconstituted on-column and in free stage with FMN resulting in formation of a fully functional native-like tetrameric enzyme (Fig. 5). The Staphylococcus aureus azoreductase was able to metabolize Methyl Red, Orange II, Amaranth, Ponceau BS, and Ponceau S azo dyes.
Fig. 5.

Mechanisms of the dissociation and re-association of azoreductase of Staphylococcus aureus expressed in Escherichia coli (Adapted from [61]).
The Staphylococcus aureus azoreductase was further annotated and used to search for homologs from other organisms in the DNA and protein databases. It was found that the enzyme has the highest similarity (80%) to a hypothetical protein from another closely related skin bacterium, Staphylococcus epidermis. The Staphylococcus aureus azoreductase has 32% similarity to a hypothetical FMN-dependent protein of unknown function (Yhda protein) from Bacillus subtilis, whose crystal structure has been determined (PDB coordinator: 1NNI). More recently, the three dimensional structure of a homodimeric NAD(P)H-dependent FMN-reductase from yeast Saccharomyces cerevisiae (YLR011w gene product), which has minor activity to cleave Ethyl Red, showed structural homology to Bacillus subtilis Yhda protein [94]. Both Bacillus subtilis Yhda and Saccharomyces cerevisiae YLR011wp have conserved amino acid residues at several crucial positions, which contribute to FMN binding. These same residues are conserved in the Staphylococcus aureus azoreductase as well as in other demonstrated and presumed azoreductases from various bacteria (Fig. 4). It was suggested that the azoreductase from Bacillus sp. strain OY1-2 contains an NAD(P)H-binding motif (GXGXXG) at positions 106 to 111 within the protein [92]. This motif was not conserved in the protein sequence of the Staphylococcus aureus azoreductase or other members of this azoreductase family (Fig. 4). The majority of flavodoxin domains, whose structure is known, have a six residue flavodoxin key fingerprint motif (T/S-X-T-G-X-T) in common [94]. However, the hypothetical FMN phosphate binding loop sequence (T/S-X-T-G-X-T) was not found in the Bacillus subtilis Yhda protein, the Saccharomyces cerevisiaeYLR011 wp, or the Staphylococcus aureus azoreductase. Rather, (P-X-Y-H/N-2X-P/S-G/A-X-L-K-N-A/S-D-L/I-D) a different loop between β3 and α3 helix was formed from several amino acid residues in the Saccharomyces cerevisiae YLR011 wp protein that interact with both FMN parts and are conserved in this enzyme family. Thus, this conserved sequence may serve as a new signature sequence for these novel flavin enzymes [61, 94].
An FMN-dependent NADH-azoreductase from Escherichia coli was identified and partially characterized (Table 2) [95]. The Escherichia coli azoreductase is homodimer which is able to reduce Methyl Red into 2-aminobenzoic acid and N,N′-dimethyl-p-phenylenediamine. More recently, a gene encoding a FMN-dependent aerobic azoreductase that shares a 34% amino acid similarity with the Escherichia coli azoreductase has been identified in Enterococcus faecalis. The purified recombinant enzyme is a homodimer with a molecular weight of 43 kDa, containing two molecules of FMN per molecule. The Enterococcus faecalis azoreductase required FMN and NADH, but not NADPH, as a preferred electron donor for its activity. The enzyme was not only able to decolorize Methyl Red, but was also able to convert the sulfonated azo dyes Orange II, Amaranth, Ponceau BS, and Ponceau S [91]. Although there is only a 34% similarity between the deduced amino acid sequences of Escherichia coli and Enterococcus faecalis azoreductases, these two enzymes probably belong to the same family, which do not share any significant similarity with the other azoreductase families. By intensive searching of the GenBank data base, it was found that similar genes encoding putative proteins belonging to the same family are widely present in both Gram-positive and Gram-negative intestinal bacteria.
Table 2 summarizes the characterized azoreductases from soil, intestinal, and skin bacteria, which some of the enzymes are able to degrade various azo dyes. So far, four aerobic azoreductase families with no significant similarity among them have been identified in bacteria. These four enzyme families can be divided into two groups, one is flavin-free [5, 10, 89, 90] and the other is flavin dependent [61, 75, 91, 93, 95]. In addition, an interesting phenomenon exists where all flavin dependent aerobic azoreductases exclusively requires FMN instead of FAD for their activities.
DEGRADATION OF AZO DYES BY FILAMENTOUS FUNGI AND YEASTS
White rot fungi can degrade many complex compounds by producing extracellular hydrolytic enzymes. It has been shown that Phanerochaete chrysosporium, Geotrichum candium, Trametes modesta, Bjerkandera adusta, Penicillium sp., Pleurotus ostreatus, Pycnoporus cinnabarinus, and Pyricularia oryzae are able to degrade complex azo dyes [62, 98–100]. Most of the azo dye degrading enzymes are oxidases and peroxidases with a very high oxidative capacity. It has been suggested that these enzymes could oxidize the azo dyes to form compounds of lower molecular weights [101]. Among these enzymes, manganese peroxidase (MnP), lignin peroxidase (LiP), and laccase are most frequently applied to azo dye degradation [28]. LiP is a H2O2-dependent enzyme which contains a heme prosthetic group [102], and was demonstrated to oxidize a range of lignin model compounds [103]. Some of the peroxidase activity detected in the extracellular fluid of Phanerochaete chrysosporium was later found to be dependent on Mn2+, leading to the discovery of MnP [104]. LiP and MnP show a similar reaction mechanism through initiating free-radical production which oxidizes other chemicals including azo dyes. These enzymes are considered to be the core of the lignin-degrading system of white rot fungi [98]. Both LiP and MnP are produced by Phanerochaete, Bjerkandera, and Trametes species and some other white rot fungi [98, 105]. However, in cultures of other white rot fungi, such as Pleurotus, Phlebia, and Ceriporiopsis species, only LiP activities were detected [98, 105, 106]. A major LiP isoenzyme from Phanerochaete chrysosporium had both LiP and MnP activities [107]. Phanerochaete chrysosporium LiP and MnP were both able to degrade azo dyes with the only differences occurring in substrate specificity [23, 80, 108]. Previous studies have demonstrated that MnP, with its cofactors in adequate concentration, can degrade azo dye Orange II, reaching 90% decolorization in a relatively short period of time [109]. Phanerochaete chrysosporium, Phanerochaete ostreatus, Trametes versicolor, and Aureobasidium pullulans were screened for degradation of sulfonic azo dyes. Trametes versicolor showed the best biodegradation performance among these fungi, which was confirmed by the degradation of various substituted fungal bio-accessible reactive azo dyes [110]. Laccase is a multicopper enzyme which catalyses the oxidation of phenolic and non-phenolic compounds. Electrons received from the substrate are subsequently transferred to oxygen, which is reduced to water [99, 111, 112]. Laccase is able to decolorize several textile azo dyes, which the decolorization efficiency can be improved remarkably in the presence of mediators [99, 112].
Several ascomycete yeast strains display similar decolorizing behaviors [113]. The yeast-mediated process requires an alternative carbon and energy source and is independent of previous exposure to the dyes. When substrate dyes are polar, their reduction is extracellular, strongly suggesting the involvement of an externally directed plasma membrane redox system. In Saccharomyces cerevisiae, the ferric reductase system participates in the extracellular reduction of azo dyes. The ferric reductase I (FRE1) deleted Saccharomyces cerevisiae mutant strain showed much-reduced decolorizing capabilities. The FRE1 gene complemented the phenotype of Saccharomyces cerevisiae FRE1 deleted cells. It restored the ability to grow in medium without externally added iron and to decolorize the dye, following a pattern similar to the one observed in the wild-type strain. This indicated that Fre1p (component of plasma membrane ferric reductase) is a major component of the azo reductase activity [25].
FUTURE DIRECTIONS
Azo dyes are used in a wide variety of products in our daily life. It is known that under certain conditions, azo dyes can be cleaved into aromatic amines, many of which are carcinogenic and mutagenic. There are many factors affecting azoreductase activity. Our knowledge about bacterial azoreductase is still limited. It is essential to know whether the azoreductase enzyme system in bacteria is constitutive, inducible, or repressible. A better understating of azoreductase will lead scientist to search for new azoreductases with broad substrate specificity and high specific activity. Although a number of microorganisms capable of azo dye reduction have been identified, there are only a relatively small portion of bacteria have been screened and studied in detail for azo dye degradation so far. Therefore, it could be important to isolate new bacteria with high capabilities to degrade azo dyes. The biochemical pathways of azo dye mineralization are still poorly understood in bacteria. The complexity of the total degradation of azo dyes indicates the need for more research. There has been considerable progress in molecular cloning and characterization of the azoreductases from various microorganisms. However, the molecular mechanism and function of azoreductases on azo dyes are not well known. Further progress in understanding the function of azoreductases is due in part to X-ray crystallography. This process will add to our knowledge of how flavin, NAD(P)H, and the azo dye bind to the enzymes and provide vital information about the molecular mechanism for switching between oxidized and reduced stages of the azoreductases. Recently, the azoreductase from Enterococcus faecalis was successfully crystallized in the presence of FMN and its structure was resolved (our unpublished data). Site-directed mutagenesis can be used in determining activity and potential binding sites of the azoreductase. It will also be very interesting to elucidate the structures of the flavin free-azoreductases and compare the differences of these two types of azoreductases.
Acknowledgments
Thanks to Dr. Carl E. Cerniglia and Sherryll L. Hopper for helpful discussions. I would also like to thank Drs. Christopher A. Elkins, and Mark E. Hart for their critical review of the manuscript.
References
- 1.Kuhad RC, Sood N, Tripathi KK, Singh A, Ward OP. Adv Appl Microbiol. 2004;56:185–213. doi: 10.1016/S0065-2164(04)56006-9. [DOI] [PubMed] [Google Scholar]
- 2.Chung K-T. Mutat Res. 1983;114:269–281. doi: 10.1016/0165-1110(83)90035-0. [DOI] [PubMed] [Google Scholar]
- 3.Moller P, Wallin H. Mutat Res. 2000;462:13–30. doi: 10.1016/s1383-5742(99)00090-3. [DOI] [PubMed] [Google Scholar]
- 4.Nakayama T, Kimura T, Kodama M, Nagata C. Carcinogenesis. 1983;4:765–769. doi: 10.1093/carcin/4.6.765. [DOI] [PubMed] [Google Scholar]
- 5.Blumel S, Knackmuss HJ, Stolz A. Appl Environ Microbiol. 2002;68:3948–3955. doi: 10.1128/AEM.68.8.3948-3955.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Platzek T, Lang C, Grohmann G, Gi US, Baltes W. Hum Exp Toxicol. 1999;18:552–559. doi: 10.1191/096032799678845061. [DOI] [PubMed] [Google Scholar]
- 7.Levine WG. Drug Metab Rev. 1991;23:253–309. doi: 10.3109/03602539109029761. [DOI] [PubMed] [Google Scholar]
- 8.Chung KT, Stevens SE, Jr, Cerniglia CE. Crit Rev Microbiol. 1992;18:175–190. doi: 10.3109/10408419209114557. [DOI] [PubMed] [Google Scholar]
- 9.Shore J. Ind J Fib Text Res. 1996;21:1–29. [Google Scholar]
- 10.Zimmermann T, Kulla HG, Leisinger T. Eur J Biochem. 1982;129:197–203. doi: 10.1111/j.1432-1033.1982.tb07040.x. [DOI] [PubMed] [Google Scholar]
- 11.Michaels GB, Lewis DL. Environ Toxicol Chem. 1985;4:45–50. [Google Scholar]
- 12.Michaels GB, Lewis DL. Environ Toxicol Chem. 1986;5:161–168. [Google Scholar]
- 13.Chung KT, Fulk GE, Andrews AW. Mutat Res. 1978;58:375–379. doi: 10.1016/0165-1218(78)90033-2. [DOI] [PubMed] [Google Scholar]
- 14.Brown JP. Appl Environ Microbiol. 1981;41:1283–1286. doi: 10.1128/aem.41.5.1283-1286.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Combes RD, Haveland-Smith RB. Mutat Res. 1982;98:101–248. doi: 10.1016/0165-1110(82)90015-x. [DOI] [PubMed] [Google Scholar]
- 16.Cerniglia CE, Freeman JP, Franklin W, Pack LD. Carcinogenesis. 1982;3:1255–1260. doi: 10.1093/carcin/3.11.1255. [DOI] [PubMed] [Google Scholar]
- 17.Levine WG. Biochem Pharmacol. 1985;34:3259–3264. doi: 10.1016/0006-2952(85)90343-0. [DOI] [PubMed] [Google Scholar]
- 18.Chung K-T. Environ Carcino & Ecotox Revs. 2000:C18, 51–74. [Google Scholar]
- 19.Brown MA, Devito SC. Crit Rev Environ Sci Technol. 1993;23:249–324. [Google Scholar]
- 20.Chung KT, Fulk GE, Egan M. Appl Environ Microbiol. 1978;35:558–562. doi: 10.1128/aem.35.3.558-562.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.von Lehmann G, Pierchalla P. Derm Beruf Umwelt. 1988;36:152–156. [PubMed] [Google Scholar]
- 22.Baumler W, Eibler ET, Hohenleutner U, Sens B, Sauer J, Landthaler M. Lasers in Surg Med. 2000;26:13–21. doi: 10.1002/(sici)1096-9101(2000)26:1<13::aid-lsm4>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 23.Stolz A. Appl Microbiol Biotechnol. 2001;56:69–80. doi: 10.1007/s002530100686. [DOI] [PubMed] [Google Scholar]
- 24.Fu Y, Viraraghavan T. Biores Technol. 2001;79:251–262. doi: 10.1016/s0960-8524(01)00028-1. [DOI] [PubMed] [Google Scholar]
- 25.Ramalho PA, Paiva S, Cavaco-Paulo A, Casal M, Cardoso MH, Ramalho MT. Appl Environ Microbiol. 2005;71:3882–3888. doi: 10.1128/AEM.71.7.3882-3888.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carliell CM, Barclay SJ, Naidoo N, Buckley CA, Mulholland DA, Senior E. Water SA. 1995;21:61–69. [Google Scholar]
- 27.Van der Zee FP, Lettinga G, Field JA. Chemosphere. 2001;44:1169–1176. doi: 10.1016/s0045-6535(00)00270-8. [DOI] [PubMed] [Google Scholar]
- 28.Lopez C, Valade AG, Combourieu B, Mielgo I, Bouchon B, Lema JM. Anal Biochem. 2004;335:135–149. doi: 10.1016/j.ab.2004.08.037. [DOI] [PubMed] [Google Scholar]
- 29.Maier J, Kandelbauer A, Erlacher A, Cavaco-Paulo A, Gubitz GM. Appl Environ Microbiol. 2004;70:737–844. doi: 10.1128/AEM.70.2.837-844.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krull R, Hempel DC. Bioproc Eng. 1994;10:229–234. [Google Scholar]
- 31.Krull R, Hemmi M, Otto P, Hempel DC. Water Sci Technol. 1998;38:339–346. [Google Scholar]
- 32.Mazzetti M, Fascioli R, Mazzoncini I, Spinelli G, Morelli I, Bertoli A. Food Addit Contam. 2004;21:935–941. doi: 10.1080/02652030400007252. [DOI] [PubMed] [Google Scholar]
- 33.International Agency for Research on Cancer. Monograph. Vol. 8. Lyon: IARC; 1975. Sudan I; pp. 225–231. [Google Scholar]
- 34.Calbiani F, Careri M, Elviri L, Mangia L, Zagnoni I. J Chromatogr A. 2004;1042:123–130. doi: 10.1016/j.chroma.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 35.Haynes CT, Bronaugh RL, Yourick J. In abstracts of the 11th Annual FDA Science Forum-Advancing Public Health through Innovative Science; Washington D.C: FDA; 2005. p. 102. [Google Scholar]
- 36.NCI. Technical Reports No. 226. US National Cancer Institute; Bethesda, MD: 1982. Carcinogenesis bioassay of C.I. Solvent Yellow 14 in F344/N rats and B6C3F1 mice. [Google Scholar]
- 37.Garner RC, Martin CN, Clayson DB. Chemical Carcinogens. In: Searle CE, editor. ACS Moenograph. Vol. 182. ACS; Washington D.C: 1984. pp. 175–302. [Google Scholar]
- 38.Westmoreland C, Gatehouse DG. Carcinogenesis. 1991;12:1403–1407. doi: 10.1093/carcin/12.8.1403. [DOI] [PubMed] [Google Scholar]
- 39.Stiborova M, Asfaw B, Frei E, Schmeiser HH, Wiessler M. Chem Res Toxicol. 1995;8:489–498. doi: 10.1021/tx00046a002. [DOI] [PubMed] [Google Scholar]
- 40.Garner R, Nutman CA. Mutat Res. 1977;44:9–19. doi: 10.1016/0027-5107(77)90110-5. [DOI] [PubMed] [Google Scholar]
- 41.Pielesz A, Baranowska I, Rybak A, Wlochowicz A. Ecotoxicol Environ Saf. 2002;53:42–47. doi: 10.1006/eesa.2002.2191. [DOI] [PubMed] [Google Scholar]
- 42.Brown JP, Roehm GW, Brown RJ. Mutat Res. 1978;56:249–271. doi: 10.1016/0027-5107(78)90192-6. [DOI] [PubMed] [Google Scholar]
- 43.Brown KM, Perimutter P, McDermott RJ. J School Health. 2000;70:355–360. doi: 10.1111/j.1746-1561.2000.tb07273.x. [DOI] [PubMed] [Google Scholar]
- 44.Drews DR, Allison CK, Probst JR. Psychol Rep. 2000;86:475–481. doi: 10.2466/pr0.2000.86.2.475. [DOI] [PubMed] [Google Scholar]
- 45.Marcoux D. Dermatol Clin. 2000;18:667–673. doi: 10.1016/s0733-8635(05)70218-7. [DOI] [PubMed] [Google Scholar]
- 46.Petigara BR. In abstracts of the 11th Annual FDA Science Forum-Advancing Public Health through Innovative Science; Washington D.C: FDA; 2005. p. 92. [Google Scholar]
- 47.Collier SW, Storm JE, Bronaugh RL. Toxicol Appl Pharmacol. 1993;118:73–79. doi: 10.1006/taap.1993.1011. [DOI] [PubMed] [Google Scholar]
- 48.Cerniglia CE, Zhou Z, Manning BW, Federle TW, Heflich RH. Mutat Res. 1986;175:11–16. doi: 10.1016/0165-7992(86)90138-7. [DOI] [PubMed] [Google Scholar]
- 49.Brown JP, McGarraugh GV, Parkinson TM, Wingard RE., Jr J Med Chem. 1983;26:1300–1307. doi: 10.1021/jm00363a015. [DOI] [PubMed] [Google Scholar]
- 50.Van den Mooter G, Maris B, Samyn C, Augustijns P, Kinget R. J Pharm Sci. 1995;86:1321–1327. doi: 10.1021/js9702630. [DOI] [PubMed] [Google Scholar]
- 51.Shantha KL, Ravichandran P, Rao KP. Biomaterials. 1995;16:1313–1318. doi: 10.1016/0142-9612(95)91046-2. [DOI] [PubMed] [Google Scholar]
- 52.Rain CP, Nobel S, Faulds D. Drugs. 1995;50:137–156. doi: 10.2165/00003495-199550010-00009. [DOI] [PubMed] [Google Scholar]
- 53.Rafii F, Coleman T. J Basic Microbiol. 1999;39:29–35. [PubMed] [Google Scholar]
- 54.Pritchard AB, Holmes PA, Kirschman JC. Toxicol Appl Pharmacol. 1976;35:1–10. doi: 10.1016/0041-008x(76)90105-8. [DOI] [PubMed] [Google Scholar]
- 55.Parkinson TM, Brown JP. Ann Rew Nutr. 1981;1:175–205. doi: 10.1146/annurev.nu.01.070181.001135. [DOI] [PubMed] [Google Scholar]
- 56.Ryan A, Roxon JJ, Sivayavirojana A. Nature. 1968;219:854–856. doi: 10.1038/219854a0. [DOI] [PubMed] [Google Scholar]
- 57.Rafii F, Franklin W, Cerniglia CE. Appl Environ Microbiol. 1990;56:2146–2151. doi: 10.1128/aem.56.7.2146-2151.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang RF, Chen H, Cerniglia CE. Biosens Bioelectron. 2004;20:699–705. doi: 10.1016/j.bios.2004.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roth RR, James WD. J Am Acad Dermatol. 1989;20:367–390. doi: 10.1016/s0190-9622(89)70048-7. [DOI] [PubMed] [Google Scholar]
- 60.Hentges DJ. Clin Infect Dis. 1993;16(Suppl 4):S175–180. doi: 10.1093/clinids/16.supplement_4.s175. [DOI] [PubMed] [Google Scholar]
- 61.Chen H, Hooper S, Cerniglia CE. Microbiology. 2005;151:1433–1441. doi: 10.1099/mic.0.27805-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McMullan G, Meehan C, Conneely A, Kirby N, Robinson T, Nigam P, Banat IM, Merchant R, Smyth WF. App Microbiol Biotechnol. 2001;56:81–87. doi: 10.1007/s002530000587. [DOI] [PubMed] [Google Scholar]
- 63.Banat IM, Nigam P, Singh D, Marchant R. Biores Technol. 1996;58:217–227. [Google Scholar]
- 64.Zissi U, Lyberatos G, Pavlou S. J Ind Microbiol Biotechnol. 1997;19:49–55. [Google Scholar]
- 65.Yatome C, Matsufuru H, Taguchi T, Ogawa T. Appl Microbial Biotechnol. 1993;39:778–181. [Google Scholar]
- 66.Wong PK, Yuen PY. Water Res. 1996;30:1736–1744. [Google Scholar]
- 67.An H, Qian Y, Gu X, Tang WZ. Chemosphere. 1996;33:2533–2542. doi: 10.1016/s0045-6535(96)00349-9. [DOI] [PubMed] [Google Scholar]
- 68.Moutaouakkil A, Zeroual Y, Zzayri FZ, Talbi M, Lee K, Blaghen M. Arch Biochem Biophys. 2003;413:139–146. doi: 10.1016/s0003-9861(03)00096-1. [DOI] [PubMed] [Google Scholar]
- 69.Dykes GA, Timm RG, von Holy A. Appl Environ Microbiol. 1994;60:3027–3029. doi: 10.1128/aem.60.8.3027-3029.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jiang H, Bishop PL. Water Sci Technol. 1994;29:525–530. [Google Scholar]
- 71.Coughlin MF, Kinkle BK, Tepper A, Ishop PL. Water Sci Tech. 1997;36:215–220. [Google Scholar]
- 72.Coughlin MF, Kinkle BK, Bishop PL. Ind Microbiol Biotechnol. 1999;23:341–346. doi: 10.1038/sj.jim.2900746. [DOI] [PubMed] [Google Scholar]
- 73.Coughlin MF, Kinkle BK, Bishop PL. Chemosphere. 2002;46:11–19. doi: 10.1016/s0045-6535(01)00096-0. [DOI] [PubMed] [Google Scholar]
- 74.Sugiura W, Miyashita T, Yakoyama T, Arai M. J Biosci Bioeng. 1999;88:577–581. doi: 10.1016/s1389-1723(00)87680-x. [DOI] [PubMed] [Google Scholar]
- 75.Suzuki T, Timofei S, Kurunczi L, Dietze U, Schuurmann G. Chemosphere. 2001;45:1–9. doi: 10.1016/s0045-6535(01)00074-1. [DOI] [PubMed] [Google Scholar]
- 76.Blumel S, Contzen M, Lutz M, Stolz A, Knackmuss HJ. Appl Environ Microbiol. 1998;64:2315–2317. doi: 10.1128/aem.64.6.2315-2317.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Haug W, Schmidt A, Nortemann B, Hempel DC, Stolz A, Kanckmuss HJ. Appl Environ Microbiol. 1991;57:3144–3149. doi: 10.1128/aem.57.11.3144-3149.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Melgoza RMa, Cruz A, Buitron G. Water Sci Technol. 2004;150:149–155. [PubMed] [Google Scholar]
- 79.Goszczynski S, Paszczynski A, Pasti-Grigsby MB, Crawford RL, Crawford DL. J Bacteriol. 1994;176:1339–1347. doi: 10.1128/jb.176.5.1339-1347.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Paszcyzynski A, Pasti-Grigsby MB, Goszczynski S, Crawford KL, Crawford RL. Enzyme Microb Technol. 1991;13:378–384. [Google Scholar]
- 81.Gingell R, Walker R. Xenobiotica. 1971;1:231–239. doi: 10.3109/00498257109033172. [DOI] [PubMed] [Google Scholar]
- 82.Roxon JJ, Ryan AJ, Wright SE. Food Chem Toxic. 1967;5:645–656. doi: 10.1016/s0015-6264(67)83216-4. [DOI] [PubMed] [Google Scholar]
- 83.Walker R. Food Cosmet Toxicol. 1970;8:659–676. doi: 10.1016/s0015-6264(70)80455-2. [DOI] [PubMed] [Google Scholar]
- 84.Russ R, Rau J, Stolz A. Appl Environ Microbiol. 2000;66:429–1434. doi: 10.1128/aem.66.4.1429-1434.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wuhrmann K, Mechsner K, Kappeler T. Eur J Appl Microbiol Biotechnol. 1980;9:325–338. [Google Scholar]
- 86.Mechsner K, Wuhrmann K. Eur J Appl Microbiol Biotechnol. 1982;15:123–126. [Google Scholar]
- 87.Chung KT, Stevens SE., Jr Environ Toxicol Chem. 1993;12:2121–2132. [Google Scholar]
- 88.Kudlich M, Keck A, Klein J, Stolz A. Appl Environ Microbiol. 1997;63:3691–3694. doi: 10.1128/aem.63.9.3691-3694.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zimmermann T, Gasser F, Kulla HG, Leisinger T. Arch Microbiol. 1984;138:37–43. doi: 10.1007/BF00425404. [DOI] [PubMed] [Google Scholar]
- 90.Blumel S, Stolz A. Appl Microbiol Biotech. 2003;62:186–190. doi: 10.1007/s00253-003-1316-5. [DOI] [PubMed] [Google Scholar]
- 91.Chen H, Wang RF, Cerniglia CE. Protein Expr Purif. 2004;34:302–310. doi: 10.1016/j.pep.2003.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Suzuki Y, Yoda T, Ruhul A, Sugiura W. J Biol Chem. 2001;276:9059–9065. doi: 10.1074/jbc.M008083200. [DOI] [PubMed] [Google Scholar]
- 93.Yan B, Zhou J, Wang J, Du C, Hou H, Song Z, Bao Y. FEMS Microbiol Lett. 2004;236:129–136. [Google Scholar]
- 94.Liger D, Graille M, Zhou CZ, Leulliot N, Quevillon-Cheruel S, Blondeau K, Janin J, van Tilbeurgh H. J Biol Chem. 2004;279:34890–34897. doi: 10.1074/jbc.M405404200. [DOI] [PubMed] [Google Scholar]
- 95.Nakanishi M, Yatome C, Ishida N, Kitade Y. J Biol Chem. 2001;276:46394–46399. doi: 10.1074/jbc.M104483200. [DOI] [PubMed] [Google Scholar]
- 96.Ghosh DK, Mandal A, Chaudhuri J. FEMS Microbiol Lett. 1992;98:229–234. doi: 10.1016/0378-1097(92)90161-g. [DOI] [PubMed] [Google Scholar]
- 97.Ghosh DK, Ghosh S, Sadhukhan P, Mandal A, Chaudhuri J. Indian J Exp Biolo. 1993;31:951–94. [PubMed] [Google Scholar]
- 98.Cameron MD, Timofeevski S, Aust SD. Appl Mirobiol Biotechnol. 2000;54:751–758. doi: 10.1007/s002530000459. [DOI] [PubMed] [Google Scholar]
- 99.Nyanhongo GS, Gomes J, Gubitz GM, Zvauya R, Read J, Steiner W. Water Res. 2002;36:1449–1456. doi: 10.1016/s0043-1354(01)00365-7. [DOI] [PubMed] [Google Scholar]
- 100.He F, Hu W, Li Y. Chemosphere. 2004;57:293–301. doi: 10.1016/j.chemosphere.2004.06.036. [DOI] [PubMed] [Google Scholar]
- 101.Cripps C, Bumpus JA, Aust SD. Appl Environ Microbiol. 1990;56:1114–1118. doi: 10.1128/aem.56.4.1114-1118.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tien M, Kirk TK. Proc Natl Acad Sci USA. 1984;81:2280–2284. doi: 10.1073/pnas.81.8.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Glenn JK, Morgan MA, Mayfield MB, Kuwahara M, Gold MH. Biochem Biophys Res Commun. 1983;114:1077–1083. doi: 10.1016/0006-291x(83)90672-1. [DOI] [PubMed] [Google Scholar]
- 104.Glenn JK, Gold MH. Arch Biochem Biophys. 1985;242:329–341. doi: 10.1016/0003-9861(85)90217-6. [DOI] [PubMed] [Google Scholar]
- 105.Hattakka A. FEMS Microbiol Rev. 1994;13:125–135. [Google Scholar]
- 106.Ruttimann C, Vicuna R, Mozuchi MD, Kirk TK. Appl Environ Microbiol. 1991;57:3652–3655. doi: 10.1128/aem.57.12.3652-3655.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Khindaria A, Barr DP, Aust SD. Biochemistry. 1995;23:7773–7779. doi: 10.1021/bi00023a025. [DOI] [PubMed] [Google Scholar]
- 108.Pasti-Grigsby MB, Paszcyzynski A, Goszczynski S, Crawford KL, Crawford RL. Appl Environ Microbiol. 1992;58:3605–3613. doi: 10.1128/aem.58.11.3605-3613.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mielgo I, Lopez C, Moreira MT, Feijoo G, Lema JM. Biotechnol Prog. 2003;19:325–331. doi: 10.1021/bp020136w. [DOI] [PubMed] [Google Scholar]
- 110.Martins MAM, Lima N, Silvestre AJD, Queiroz MJ. Chemosphere. 2003;52:967–973. doi: 10.1016/S0045-6535(03)00286-8. [DOI] [PubMed] [Google Scholar]
- 111.Eggert C, Temp U, Eriksson KE. Appl Environ Microbiol. 1996;62:1151–1158. doi: 10.1128/aem.62.4.1151-1158.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chivukula M, Renganathan V. Appl Environ Microbiol. 1995;61:4374–4377. doi: 10.1128/aem.61.12.4374-4377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ramalho PA, Cardoso MH, Cavaco-Paulo A, Ramalho MT. Appl Environ Microbiol. 2004;70:2279–2288. doi: 10.1128/AEM.70.4.2279-2288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]